

Abstract
The main feedback loop driving circadian rhythm in mice is controlled in part by the genes encoding the cryptochromes Cry1 and Cry2. Targeted mutation of both Cry1 and Cry2 delay the early onset of tumor formation in p53 null mutant mice. Furthermore, Ras-transformed p53 and Cry null mouse skin fibroblasts are more sensitive than p53 mutants to apoptotic cell death initiated by agents that activate either the intrinsic or extrinsic apoptosis pathways. Here we investigated the effect of Cry1 and Cry2 mutations on cell death by other genotoxic agents that generate alkylated bases, interstrand crosslinks, DNA-protein crosslinks and double-strand breaks. Both UV and the UV mimetic compound oxaliplatin and the radiomimetic compound doxorubicin promoted apoptosis by upregulating the tumor suppressor p73. However, only the UV and oxaliplatin-induced upregulation of p73 mediated by the transcription factor Egr1, but not the doxorubicin-induced upregulation mediated by the transcription factor E2F1, was enhanced by Cry1/Cry2 double mutation. Accordingly, Egr1 downregulation reduced oxaliplatin-induced apoptosis whereas E2F1 downregulation reduced doxorubicin-induced apoptosis. Our findings establish distinct roles for cryptochromes in intrinsic apoptosis induced by UV-mimetic and radiomimetic agents.
Introduction
Cryptochrome (Cry) is the main repressor in the transcription-translation feedback loop that generates circadian rhythmicity in mice (1). Recently we reported that Cry mutation in Ras-transformed p53 null mouse skin fibroblasts sensitized the cells to apoptosis by UV and UV-mimetic agents such as oxaliplatin (2, 3). Further analysis revealed that this enhanced apoptosis was caused by the amplified upregulation of the p73 tumor suppressor due to the high level of the clock controlled transcription factor Egr1 in Cry null cells (3). Since p73 is a DNA damage inducible gene (4, 5) we wished to find out the effect of other DNA damaging agents on p73 induction and apoptosis in cells with and without a functional circadian clock. The p73 gene has a complex regulatory mechanism that includes E2F1, Egr1, and C-EBPα transcription factors. There are 1 C-EBPα, 3 E2F1, and 5 Egr1 binding sites in the p73 promoter (4–8). The p73 protein level is mainly regulated by transcription that is activated by E2F1 and Egr1 and is repressed by C-EBPα. DNA damage by doxorubicin or cisplatin causes dissociation of C-EBPα from the promoter and its nuclear export enabling the transactivators to upregulate p73 transcription (4). Interestingly, of these two transcriptional activators Egr1 is a circadian clock controlled gene with a robust rhythmicity (9) whereas E2F1 is not controlled by the circadian clock (10). While investigating the effect of circadian clock disruption on cellular response to DNA damage we discovered that circadian clock disruption by Cry mutation led to elevated level of expression of Egr1 which resulted in high level of expression of p73 upon DNA damage by UV or oxaliplatin and enhanced apoptosis resulting in increased clonogenic cell death by these genotoxicants (3).
Here, we wished to investigate other DNA damaging agents that generate DNA lesions that encompass the entire spectrum of repair pathways for their effects on p73-medaited apoptosis in the absence or presence of Cry in p53 null cells in order to find out whether Cry mutation sensitizes these cells to all genotoxicants. We used UV and oxaliplatin (UV-mimetic) whose major lesions are repaired by nucleotide excision repair, ethylmethane sulfonate (EMS) that induces base alkylation repaired by base excision repair, doxorubicin that induces double-strand breaks repaired by non-homologous end-joining and homologous recombination, mitomycin C that induces interstrand crosslinks repaired by recombination and translesion synthesis, and camptothecin that induces DNA-protein crosslinks repaired by proteolysis and recombination (11, 12). We found that at equitoxic doses only the UV-mimetic (UV and oxaliplatin) and the radiomimetic (doxorubin and camptothecin) agents induced p73 expression. Furthermore, surprisingly, we found that while oxaliplatin induces p73 through Egr1-mediated transcriptional upregulation, doxorubicin induces p73 through E2F1-mediated transcriptional upregulation. As a consequence the clonogenic killing and apoptotic effects of oxaliplatin are enhanced by Cry mutation that causes constitutive upregulation of Egr1 but the cell killing and apoptotic effects of doxorubicin are not affected by Cry mutation. As oxaliplatin plus doxorubicin combination is used in various chemotherapeutic regimens (13) these findings may help in designing more effective drug delivery protocols for cancer therapy.
Materials and Methods
Establishment of p53−/− and p53−/−Cry1/2−/− cell lines
Wild-type and p53−/− mice of C57BL/6J background were obtained from The Jackson Laboratory and p53−/− Cry1/2−/− mice in C57BL/6J background were generated in our laboratory (2). These cell lines are authenticated by microarray analysis using the whole genome oligo microarrays (Agilent Technologies) for mouse specific gene expression analysis. The Ras-transformed p53−/− and p53−/− Cry1/2−/− cell lines were generated in our laboratory as described previously (3). These cells were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with penicillin, streptomycin, and 10% fetal bovine serum (FBS). The cells were maintained in an incubator at 37 °C under 5% CO2.
Chemicals and siRNA
Oxaliplatin, doxorubicin, camptothecin, mitomycin C, and ethyl methanesulphonate (EMS) were purchased from Sigma. DharmaFECT (Dharmacon) reagent was used according to the manufacturer’s instruction for transcfection of ON-TARGET plus SMARTpool or single siRNA duplexes obtained from Dharmacon [Egr1 (L-040286-00-0005, J-040286-08-005, J-040286-07-005), E2F1 (L-044993-00-0005, J-044993-16-0005, J-044993-15-0005) or nontargeting siRNA (D-001810-10-05) as a control]. To ensure specificity of Egr1 and E2F1 downregulation by pooled siRNAs, we also performed downregulation by two single-siRNAs targeting the relevant genes and obtained essentially the same results. The effects of downregulation of Egr1 and E2F1 by siRNA are specific, because downregulation of one does not affect the level of the other transcription factors (Fig S1).
Clonogenic cell survival assay
The clonogenic cell survival assay was done as described previously with some modifications (2). Cells were seeded at low density to ensure the formation of 200 colonies per six-well plate. Following plating, cells were kept in growth medium for 10–14 hrs and treated with indicated ranges of UV given at a dose rate of 0.32 J/m2/sec or incubated with indicated concentrations of chemicals for the duration of the experiment. Readily visible colonies (over 50 cells per colony) formed after a 9- to 10-d incubation were stained with 5% methylane blue and counted.
Protein analysis
The following commercial antibodies and reagents were used: E2F1 and Actin (Santa Cruz Biotechnology); cleaved-caspase3, cleaved-PARP1, and Egr1 (Cell Signaling Technology); p73 (Upstate Biotechnology); acetyl-lysine (Millipore); phospho-E2F1 (Rockland); IgM (Thermo Scientific). Cry1 antibodies produced in our laboratory have been described previously (2, 3). For immunoblot analysis, cells were lysed in lysed in a RIPA buffer (50 mM Tris-HCl pH 7.6, 1% Nonidet P40, 140 mM NaCl, 0.1% SDS) supplemented with complete protease inhibitor cocktail (Roche), and 50 μg of lysate were loaded to pre-cast 4–12% gradient SDS/PAGE (Invitrogen).
Immunoprecipitation
Cells were lysed in RIPA buffer supplemented with complete protease inhibitor cocktail (Roche). After pre-clearing with protein A/G agarose plus (Santa Cruz) for 1 hr at 4°C, whole-cell lysates were used for immunoprecipitation with the indicated antibody. one μg of the commercial antibody was added to 1 ml of the cell lysate and incubated at 4°C for 8–12 hrs. After adding protein A/G agarose beads, incubation was continued for an additional 1 hr. The immunoprecipitates were then washed extensively with RIPA buffer and eluted by boiling them with an SDS loading buffer for 5 min.
Statistical analysis
Values are shown as mean ± SD of at least 3 experiments calculated using a two-tailed student t test.
Results
Clonogenic killing by various genotoxicants of p53−/− and p53−/−Cry1/2−/− cells
We employed genotoxic treatments that generate DNA lesions repaired by all of the major repair pathways (base excision, nucleotide excision, double-strand break repair, interstrand crosslink repair, DNA-protein crosslink repair) to examine the effects of clock disruption by Cry mutation on these repair pathways. Fig. 1 shows the result of clonogenic assays for all these treatments. In agreement with our previous report, the killing by UV (Fig. 1A) and oxaliplatin (UV-mimetic agent, Fig. 1B), which produce bulky DNA adducts that are repaired exclusively by nucleotide excision repair (11), is enhanced by Cry mutation. Interestingly, the killing effect of none of the other DNA damaging agents is influenced by Cry (Fig. 1C–F). This suggests that Cry mutation specifically affects the lethality of UV-mimetic agents that produce bulky DNA adducts. We have previously shown that Cry mutation enhanced the clonogenic cell death of p53 null cells by UV and oxaliplatin by amplifying the intrinsic apoptotic response to these agents through increased transcription of p73 tumor suppressor (3). Therefore, we decided to analyze the response of p53−/− and p53−/−Cry1/2−/− cells to these various DNA damaging agents in order to find out if the apoptosis enhancement by Cry mutation was restricted to apoptosis induced by UV-mimetic agents.
Figure 1. Effect of cryptochrome on clonogenic cell death by various genoxoticants.

Cells of the indicated genotypes were irradiated or treated with the indicated dose of (A) UV, (B) oxaliplatin, (C) doxorubicin, (D) camptothecin, (E) mitomycin C, or (F) EMS, and then incubated for 9–10 days until colonies were readily visible. Colonies were stained with 5% methylene blue and then counted to obtain the UV survival curves. Results represent the means of 3 independent experiments (± SD).
Effect of Cry mutation on apoptosis induced by full spectrum of genotoxicants
We performed apoptosis assays at equitoxic doses of all 6 genotoxicants used in the survival assay by employing doses that gave 50% survival in the clonogenic assay. We probed for apoptosis by measuring PARP and caspase 3 cleavage (14) over a period of 12 hrs from the start of UV irradiation or drug treatment. The results are shown in Fig. 2. Several points emerge from this figure. First, even though equitoxic doses were used for all genotoxicants the extent of apoptosis at 12 hrs in p53−/− cells are quite different for the various agents. It is remarkably high in doxorubicin-treated cells and negligible in EMS-treated cells (Fig. 2A–D), indicating that, dependent on the genotoxicant, different apoptosis kinetics or other modes of cell death are responsible for clonogenic killing. Second, and most importantly, within the examined time frame, apoptosis is enhanced by Cry mutation only in cells treated with UV and oxaliplatin, in agreement with the clonogenic survival data that show that Cry mutation enhances only the killing effects of these two agents (Fig. 1A, B). Finally, even though under our experimental conditions, the doxorubicin-induced apoptosis is not amplified by Cry mutation the extent of apoptosis induced by doxorubicin in p53−/− cells is comparable to that seen in p53−/−Cry1/2−/− cells treated with UV or oxaliplatin. In conclusion, the survival data combined with the apoptosis data indicate UV, oxaliplatin, camptothecin, and doxorubicin induce apoptosis-associated cell killing in the absence of p53. Because we had previously determined that the apoptosis induced in p53−/− cells was mostly mediated by p73 and that Cry mutation enhanced this response by upregulating p73 (3) we examined the effect of doxorubicin and UV/oxaliplatin along with those of other genotoxicants on p73 induction.
Figure 2. Effect of cryptochrome on apoptosis induced by various genotoxic agents.

Cells were irradiated or treated with 5 Jm−2 UV, 10 μM oxaliplatin, 5 μM doxorubicin, 1 μM camptothecin, 2.5 μM mitomycin C, or 2.5 mM EMS for the indicated times. Cell lysates were probed for cleaved PARP (c-PARP) (A) and cleaved caspase 3 (c-Casp3) (C) by immunoblotting. Actin served as a loading control. Actin for EMS-treated cells was shown as a representative image. Quantifications of the levels of c-PARP (B) and c-Casp3 (D) normalized to actin are shown. The data are the mean of three independent experiments ±SD.
Mechanisms of p73 upregulation by radiomimetic and UV-mimetic agents
We treated cells with the 6 genotoxic agents used in the clonogenic survival assay and followed p73 induction for 12 hrs. The results are shown in Fig. 3A. In agreement with the apoptosis data, UV-mimetic treatments (UV and oxaliplatin)-caused p73 transcriptional upregulation in both p53−/− (3) and p53+/+ (Fig S2) background is amplified by Cry mutation. In contrast, the extent of p73 induction by doxorubicin and camptothecin is comparable to that achieved by UV-mimetic treatments but is independent of the Cry status of the cell line (Fig. 3A, B). Thus, it appears that while UV/oxaliplatin and doxorubicin/camptothecin induce p73 efficiently there is a difference between the induction mechanisms because one is affected by the status of Cry and the other is not.
Figure 3. Differential p73 induction by cryptochrome in p53 null background.
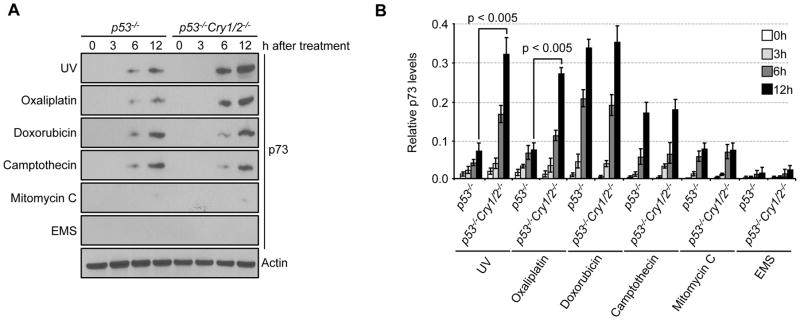
(A) Cells were irradiated with or exposed to 5 Jm−2 UV, 10 μM oxaliplatin, 5 μM doxorubicin, 1 μM camptothecin, 2.5 μM mitomycin C, or 2.5 mM EMS for the indicated times, after which, cell lysates were analyzed by immunoblotting for p73. (B) Results of p73 quantification are normalized to the expression of actin and shown. Actin for EMS-treated cells was shown as a representative image. Error bars indicate one standard deviation from the mean (n=3).
To find out the cause of this differential effect of Cry on cellular response to the two types of DNA lesions (bulky adducts vs. double-strand breaks) known to induce p73 we examined the effects of these two types of DNA damaging agents on two transcription factors known to be involved in DNA damage-induced upregulation of p73, the E2F1 and Egr1 transcription factors (4–8) (camptothecin behaves quite similarly to doxorubicin but since both act by similar mechanisms, double-strand break for doxorubicin and double-strand break associated with DNA-protein crosslink for camptothecin, we chose to pursue only one in further experiments). The results are shown in Fig. 4. As seen in Fig. 4A, both UV and doxorubicin fail to induce Egr1 in the p53−/− genetic background and Egr1 is constitutively elevated in p53−/−Cry1/2−/− cells regardless of DNA damage by either genotoxicants. Importantly, however, doxorubicin upregulates E2F1 and causes its phosphorylation and acetylation. These are necessary for E2F1 activity but UV has no effect on E2F1 levels and posttranslational modification (15–18) (Fig. 4A). Of special relevance the effects of doxorubicin on E2F1 is independent of the Cry status of the p53−/− cell lines, consistent with the clonogenic and apoptosis assays that the clock does not affect cellular response to double-strand breaks as determined by these two endpoints.
Figure 4. Differential effect of cryptochrome on the p73-mediated apoptosis.

(A) Cells were irradiated with 5 Jm−2 UV or treated with 5 μM doxorubicin. Cell lysates were probed for Egr1 or total and phosphorylated forms of E2F1. Actin was used as a loading control. To probe acetylated forms of E2F1 cell lysates were immunoprecipitated with anti-E2F1 antibody. IB, immunoblotting; IP, immunoprecipitation. The immunoprecipitates were subjected to immunoblotting with anti-E2F1 and with antibody against acetylated lysines. (B–E) Effect of cryptochrome on the two types of p73 induction. Mouse fibroblasts of the indicated genotypes were transfected with Egr1 (B, C) or E2F1 (D, E) siRNA, treated with 10μM oxaliplatin (B, D) or 5 μM doxorubicin (C, E) and then analyzed for p73 expression directly by immunoblotting, and for apoptosis by immunoblotting for cleaved PARP and caspase 3.
We investigated the effects of E2F1 and Egr1 in more detail using doxorubicin (double-strand breaks) and oxaliplatin (bulky adducts) as models for clock effects on cellular response to the two types of DNA damage and also because these two drugs are component of several frontline cancer therapy regimens. The findings in Fig. 4A provide a plausible explanation for the differential effects of Cry mutation on cellular response to damage by UV-mimetic and radiomimetic agents: The upregulation of p73 by E2F1 following DNA damage by radiomimetics is the predominant regulatory mechanism (15–18) and hence the elevated Egr1 does not contribute to doxorubicin-induced p73 upregulation. In contrast, it appears that with UV-mimetic treatment the Egr1-mediated transcription is the dominant mechanism and since Egr1 (but not E2F1) is a clock-controlled protein, in p53−/−Cry1/2−/− cells the elevated Egr1 leads to enhanced p73 induction by UV-mimetics and enhanced apoptosis in p53−/−Cry1/2−/− cells compared to p53−/− cells. The enhanced apoptosis is due to the upregulation of p73, because knockdown of p73 by siRNA drastically reduced apoptosis in both p53−/− and p53−/− Cry1/2−/− cells. (Fig S3). To further test this model we downregulated either Egr1 or E2F1 in p53−/− and p53−/−Cry1/2−/− backgrounds and tested the cell lines for p73 induction and apoptosis. Fig. 4B, C shows that downregulation of Egr1 abolished the enhanced p73 induction and apoptosis after oxaliplatin treatment seen in p53−/−Cry1/2−/− cells relative to the p53−/− cells. In contrast, and rather strikingly, Egr1 downregulation has no significant effect on p73 induction and apoptosis by doxorubicin in either background. As predicted by the model, downregulation of E2F1 has the opposite effect (Fig. 4D, E): Reducing E2F1 does not affect either the basal level or the amplified apoptosis induced by oxaliplatin (Fig. 4D) but reduces the doxorubicin-induced p73 induction and apoptosis in both p53−/− and p53−/−Cry1/2−/−background (Fig. 4E). To recapitulate, double-strand breaks upregulate p73 through E2F1 and since E2F1 is not a clock-controlled gene, clock disruption through Cry mutation does not affect apoptosis induced by doxorubicin. In contrast, oxaliplatin upregulates p73 through Egr1 which has a clock-controlled promoter and therefore the high level overproduced Egr1 in Cry mutant cells strongly enhances oxaliplatin-induced apoptosis in p53 mutant cells.
Discussion
We recently reported that cryptochrome mutation rendered the p53 null and the Ras-transformed p53 null cells, but not cells with wild-type p53, more susceptible to killing by agents that activate either the intrinsic or extrinsic apoptosis pathways (3, 19). In those studies UV or oxaliplatin were used to initiate intrinsic apoptosis (3) and TNFα to initiate extrinsic apoptosis (19). Since multiple agents that damage DNA can initiate intrinsic apoptosis and similarly multiple extracellular factors that influence cellular homeostasis can initiate extrinsic apoptosis (14) it is of interest to determine whether intrinsic and extrinsic apoptosis or other death forms induced by a variety of agents are similarly potentiated by Cry mutation. Here we have investigated the contribution of Cry to clonogenic cell death and to intrinsic apoptosis induced by a set of genotoxicants that generate DNA lesions that are repaired by each of the major DNA repair pathways including base excision repair, nucleotide excision repair, double-strand break repair, and crosslink repair. We find that apoptosis and clonogenic cell death by genotoxicants in p53 null background is enhanced by Cry mutation exclusively for UV-mimetics-induced DNA damage and no other. Further, we present evidence indicating that the enhanced apoptosis and clonogenic death by UV-mimetic agents in Cry mutant background is due to the upregulation of the clock-controlled transcription factor Egr1 in Cry mutant cells which lead to high level of induced expression of p73 tumor suppressor/apoptosis promoter.
In cells with functional p53, apoptosis by genotoxic agents is mainly controlled by this tumor suppressor (20). Only in p53 mutant cells p73 becomes the arbitrator of cell fate by intrinsic apoptosis (21). In contrast to p53, DNA damage-induced upregulation of p73 is mainly by transcriptional induction, although post-transcriptional modifications also make some contribution (4–8). The transcriptional regulation of p73 has not been investigated in great detail. Current knowledge may be summarized as follows: The p73 promoter (~2000 bp preceding transcriptional start site) contains, in the 5′ to 3′ direction, 5 Egr1, 1 C-EBPα, and 3 E2F1 binding sites (Fig. 5). Egr1 and E2F1 are transcriptional activators while C-EBPα functions as a repressor. In most cell types in the absence of DNA damage the p73 expression is rather low (8). However, all 3 transcription factors regulating p73 expression are subject to regulatory mechanisms themselves: Egr1 is controlled by the circadian clock (3, 9), C-EBPα is controlled by post-translational modification and nucleo-cytoplasmic shuttling and E2F1 is controlled by the cell cycle and by DNA damage-induced phosphorylation and acetylation which stabilize the protein and enhance its activity (15–18, 22). Upon DNA damage, C-EBPα is phosphorylated, released from the p73 promoter, and excluded from the nucleus; E2F1 is acetylated and stabilized, leading to 10–30 fold induction of p73 transcription from thepromoter which is no longer repressed by C-EBPα. Of the two positive transcription factors E2F1 appears to have the predominant effect. However, the phosphorylation and acetylation of E2F1 are induced by IR and radiomimetic agents such as doxorubicin but not by UV or UV-mimetic agents such platinum-based drugs. As a consequence it might be expected that in our experimental system doxorubicin-induced p73 upregulation which is predominantly controlled by E2F1 would not be affected by the presence or absence of a functional circadian clock because E2F1 is not controlled by the clock. In contrast, upregulation of p73 by UV and UV-mimetic agents seems to be mediated primarily by Egr1 because UV does not affect posttranslational modification of E2F1 and its activity (16–18). Furthermore, since Egr1 is under robust clock control (3, 9), in Cry mutants in which the inhibitory arm of the transcription-translation feedback loop is non-functional Egr1 is highly elevated leading to significantly enhanced DNA damage-inducible p73 transcription even in the absence of contribution from E2F1. Because doxorubicin plus oxaliplatin combination is included in several chemotherapy regimens these findings should be taken into account for optimal efficacy with minimal side effects.
Figure 5. Model for clock effects on DNA damage-induced upregulation of p73 tumor suppressor.
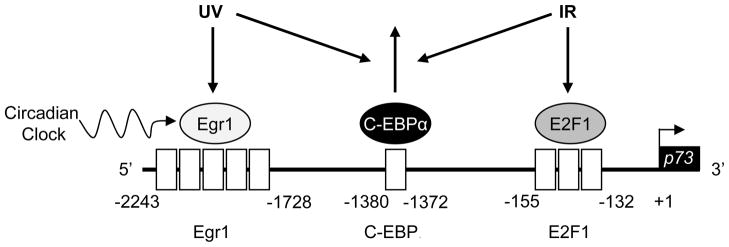
The 2 kb of the upstream sequence of the p73 promoter is shown in the direction 5′–3′, with the transcription start site indicated with a bent arrow. Functional transcription factor binding sites are indicated by open boxes, and the transcription factors are shown as circles. Transcription factor binding sites include: 5 early growth response-1 (Egr1) sites, 1 CCAAT/enhancer-binding protein (C-EBPα) site, and 3 E2F1 sites. Egr1 and E2F1 are transactivators, and C-EBPα functions as a repressor. Both UV and ionizing radiation (IR) cause dissociation of C-EBPα from the promoter. UV-induced upregulation of p73 is mediated by the clock controlled transcription factor Egr1. In contrast, IR-induced p73 upregulation is predominantly controlled by E2F1 which is not be affected by the circadian clock.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants (GM31082 and GM32833) to A. Sancar.
Footnotes
Conflict of interest: NONE
References
- 1.Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–41. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- 2.Ozturk N, Lee JH, Gaddameedhi S, Sancar A. Loss of cryptochrome reduces cancer risk in p53 mutant mice. Proc Natl Acad Sci U S A. 2009;106:2841–6. doi: 10.1073/pnas.0813028106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee JH, Sancar A. Circadian clock disruption improves the efficacy of chemotherapy through p73-mediated apoptosis. Proc Natl Acad Sci U S A. 2011;108:10668–72. doi: 10.1073/pnas.1106284108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marabese M, Vikhanskaya F, Rainelli C, Sakai T, Broggini M. DNA damage induces transcriptional activation of p73 by removing C-EBPalpha repression on E2F1. Nucleic Acids Res. 2003;31:6624–32. doi: 10.1093/nar/gkg869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urist M, Tanaka T, Poyurovsky MV, Prives C. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev. 2004;18:3041–54. doi: 10.1101/gad.1221004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seelan RS, Irwin M, van der Stoop P, Qian C, Kaelin WG, Jr, Liu W. The human p73 promoter: characterization and identification of functional E2F binding sites. Neoplasia. 2002;4:195–203. doi: 10.1038/sj.neo.7900237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pignatelli M, Luna-Medina R, Perez-Rendon A, Santos A, Perez-Castillo A. The transcription factor early growth response factor-1 (EGR-1) promotes apoptosis of neuroblastoma cells. Biochem J. 2003;373:739–46. doi: 10.1042/BJ20021918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu J, Baron V, Mercola D, Mustelin T, Adamson ED. A network of p73, p53 and Egr1 is required for efficient apoptosis in tumor cells. Cell Death Differ. 2007;14:436–46. doi: 10.1038/sj.cdd.4402029. [DOI] [PubMed] [Google Scholar]
- 9.Bai L, Zimmer S, Rickes O, Rohleder N, Holthues H, Engel L, et al. Daily oscillation of gene expression in the retina is phase-advanced with respect to the pineal gland. Brain Res. 2008;1203:89–96. doi: 10.1016/j.brainres.2008.01.073. [DOI] [PubMed] [Google Scholar]
- 10.Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, et al. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009;5:e1000442. doi: 10.1371/journal.pgen.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 12.Antoch MP, Kondratov RV. Circadian proteins and genotoxic stress response. Circ Res. 2010;106:68–78. doi: 10.1161/CIRCRESAHA.109.207076. [DOI] [PubMed] [Google Scholar]
- 13.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–84. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 14.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–83. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin WC, Lin FT, Nevins JR. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001;15:1833–44. [PMC free article] [PubMed] [Google Scholar]
- 16.Pediconi N, Guerrieri F, Vossio S, Bruno T, Belloni L, Schinzari V, et al. hSirT1-dependent regulation of the PCAF-E2F1-p73 apoptotic pathway in response to DNA damage. Mol Cell Biol. 2009;29:1989–98. doi: 10.1128/MCB.00552-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carnevale J, Palander O, Seifried LA, Dick FA. DNA damage signals through differentially modified E2F1 molecules to induce apoptosis. Mol Cell Biol. 2012;32:900–12. doi: 10.1128/MCB.06286-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biswas AK, Johnson DG. Transcriptional and nontranscriptional functions of E2F1 in response to DNA damage. Cancer Res. 2012;72:13–7. doi: 10.1158/0008-5472.CAN-11-2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JH, Sancar A. Regulation of apoptosis by the circadian clock through NF-kappaB signaling. Proc Natl Acad Sci U S A. 2011;108:12036–41. doi: 10.1073/pnas.1108125108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–67. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 21.Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG., Jr Chemosensitivity linked to p73 function. Cancer Cell. 2003;3:403–10. doi: 10.1016/s1535-6108(03)00078-3. [DOI] [PubMed] [Google Scholar]
- 22.Nevins JR. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science. 1992;258:424–9. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.