

Abstract
Peroxiredoxins (Prx) are a family of enzymes which reduce peroxides using a peroxidatic cysteine residue; among these, the PrxQ subfamily members are proposed to be the most ancestral-like yet are among the least characterized. In many PrxQ enzymes, a second “resolving” cysteine is located six residues downstream from the peroxidatic Cys, and these residues form a disulfide during the catalytic cycle. Here, we describe three hyperthermophilic PrxQ crystal structures originally solved by the RIKEN structural genomics group. We reprocessed the diffraction data and carried out further refinement to yield models with Rfree lowered by 2.3–7.2% and resolution extended by 0.2–0.3 Å, making one, at 1.4 Å, the best resolved peroxiredoxin to date. Comparisons of two matched thiol and disulfide forms reveal that the active site conformational change required for disulfide formation involves a transition of about 20 residues from a pair of α-helices to a β-hairpin and 310-helix. Each conformation has about 10 residues with high disorder providing slack that enables the dramatic shift, and the two conformations are anchored to the protein core by distinct non-polar side chains that fill three hydrophobic pockets. Sequence conservation patterns confirm the importance of these and a few additional residues for function. From a broader perspective, this study raises the provocative question of how to make use of the valuable information in the protein data bank generated by structural genomics projects but not described in the literature, perhaps remaining unrecognized and certainly underutilized.
Aerobic organisms must cope with reactive oxygen species (ROS) which can damage DNA, proteins, and lipids, potentially causing loss of membrane integrity and cell death1,2,3,4. Meeting this challenge are a variety of antioxidant defenses, including enzymes of the ubiquitous and highly expressed peroxiredoxin (Prx) family, which specialize in reducing hydrogen peroxide and organic peroxides, and can also reduce peroxynitrite5,6,7,8. Studies of Prxs have given insight into their catalytic mechanism9 and their critical functions within the cell, including possible roles in eukaryotes in cell signaling and tumor suppression10,11. Prxs are also potential drug targets5, as for instance, it has been demonstrated that Prx knockout strains of the causative agent of malaria, Plasmodium falciparum, cope poorly with oxidative stress12,13.
Prxs are divided by sequence similarity into six subfamilies: Prx1, Prx6, Prx5, Tpx, AhpE, and BCP-PrxQ1,14, all of which share a globular tertiary structure, with a conserved core of five α-helices and seven β-strands1,7. Their enzymatic activity requires an active site peroxidatic Cys residue (designated as CP or SPH for the thiol), located within a strictly conserved PXXX(T/S)XXCP motif14. The catalytic cycle involves three chemical steps (Fig 1a). First, the SP− thiolate attacks a peroxide substrate in an SN2 reaction to form sulfenic acid (R-SPOH)9. Second, to recycle the CP, and to prevent over-oxidation from a second peroxide substrate, the subset called 2-Cys Prxs possess a second Cys residue (designated CR for resolving) which reacts with the sulfenic acid to form a disulfide bond15. The disulfide is then reduced by an external agent, such as thioredoxin, to regenerate the substrate-ready form1,5,9,15.
Figure 1.
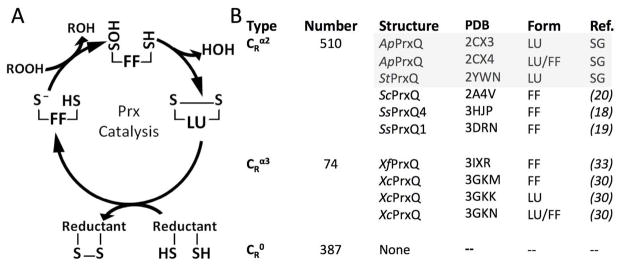
The Prx catalytic cycle and structural knowledge of PrxQ enzymes. (A) The catalytic cycle of 2-Cys Prxs indicating the protein conformation (FF or LU) dominant at each redox state. (B) Listed for each PrxQ subtype are the number of sequences14,42 and the crystal structures solved and conformations seen. Grey denotes the structures in this study, with “SG” standing for structural genomics. The structures include: ApPrxQ - Aeropyrum pernix PrxQ, StPrxQ - Sulfolobus tokodaii PrxQ, ScPrxQ - Saccharomyces cerevisiae PrxQ, SsPrxQ4 - Sulfolobus solfataricus PrxQ4, SsPrxQ1 - Sulfolobus solfataricus PrxQ1, XfPrxQ - Xylella fastidiosa PrxQ, and XcPrxQ - Xanthomonas campestris PrxQ.
Importantly, disulfide formation in all Prxs requires a local unfolding of the active site. The substrate-ready conformation with the CP thiolate at the bottom of an active site pocket is called fully folded (FF) and the disulfide form is called locally unfolded (LU)1. As reviewed by Hall et al1, each type of 2-Cys Prx has unique structural features that effectively stabilize discrete FF and LU conformations and these have been described for the Prx, Tpx, and PrxV subfamilies as well as the BCP-PrxQ subgroup that has the CR residue in helix α3. The one 2-Cys Prx subgroup not yet characterized in this way is the BCP-PrxQ subgroup that has CR in helix α2, with just four residues separating it and the CP. It is this common but little characterized1,5,16 subgroup that is the subject of this work.
Because the name BCP (Bacterioferritin Commigratory Protein) is a historical holdover based on gel migration, we favor simply using the more informative PrxQ name for this subfamily and its members. This both clearly identifies them as Prxs, and matches our recently proposed nomenclature for Prxs from parasites17. Here, we will use PrxQ, even for proteins that have been previously referred to as ‘BCP’. For instance, Escherichia coli BCP will be referred to as E. coli PrxQ.
The PrxQ group includes monomers and dimers18 and has been proposed to represent the subfamily most like the ancestral Prx1. Among PrxQs, the CR position may vary, being absent (~38%) or being located either in α2 (~55%) or α3 (~7%)1. These subtypes are here designated as CR0, CRα2, and CRα3. E. coli PrxQ (a CRα2 enzyme) is the founding and best studied member of the subfamily16 but has yet to be crystallized. To date, seven PrxQ structures have been reported in the literature (Fig 1b), which include both FF and LU conformations of a CRα3 type PrxQ, but only the FF conformation of a CRα2 PrxQ. Among these structures only one, Sulfolobus solfataricus PrxQ4 (SsPrxQ4), is a dimer18.
Fortuitously, although the CRα2 LU conformation is missing among the described structures, it actually has been solved in three crystal structures of dimeric PrxQs from the hyperthermophilic aerobes Sulfolobus tokodaii (StPrxQ), and Aeropyrum pernix (ApPrxQ). StPrxQ has 54% sequence identity with ApPrxQ and 78% identity with SsPrxQ4, the previously described CRα2 PrxQ in the FF conformation. The three structures, one of StPrxQ and two of ApPrxQ, were deposited over five years ago by the RIKEN Structural Genomics Group and have been referred to in a few papers1,4, 16,18,19,20, but have never been carefully interpreted and described in a primary publication.
Here, we report such an analysis of the StPrxQ and ApPrxQ structures. With support from scientists at RIKEN, we have reprocessed the diffraction data and improved the previously deposited structures by additional rounds of polishing refinement. We provide a description of these structures and a detailed analysis of the conformational changes associated with local unfolding in the CRα2 PrxQ enzymes.
EXPERIMENTAL PROCEDURES
Polishing refinements of 2YWN, 2CX3, and 2CX4
Original diffraction data for the structures of StPrxQ (Protein Data Bank code 2YWN), ApPrxQ-LU (PDB code 2CX3) and ApPrxQ-FF/LU (PDB code 2CX4) were obtained from the RIKEN Structural Genomics/Proteomics Initiative and processed and scaled using XDS21. The resolution cutoffs were based on the new criteria of CC1/2 > 0 with P-value < 0.00122, except that for the StPrxQ dataset the high resolution cutoff of 1.4 Å was limited by completeness (dropping below 50%) rather than signal strength (Table 1). We found using Pointless23 that the data for 2CX4 could be processed in space group P4122 and showed no evidence of twinning, contrasting with the previous assignment (according to the PDB file header) of space group P41 with a twinning fraction of 0.5, and twin operator K, H, -L. Similarly, the 2CX3 data, previously handled as space group P64, processed well in P6422. No change was made to the space group of the 2YWN data. To generate initial electron density maps for the structures with new space groups, molecular replacement was carried out with Phaser24 using as search models four and two chains from the deposited 2CX4 and 2CX3 models, respectively.
Table 1.
Data collection and refinement statistics for new and previously deposited models
| Data Collection | ||||||
|---|---|---|---|---|---|---|
| Structure | StPrxQ-LUa | ApPrxQ-FF/LUb | ApPrxQ-LUc | |||
| Space group | C 2 2 21 | P 41 2 2 | P 64 2 2 | |||
| Unit cell a, b, c (Å) | 69.34, 78.65, 61.96 | 132.39, 132.39, 106.53 | 127.00, 127.00, 104.87 | |||
| Resolution (Å) | 50.00-1.40 (1.42-1.40)d | 50.00-2.00 (2.05-2.00)e | 50.00-2.30 (2.34-2.30)f | |||
| Completeness | 99.6 (42.0) | 95.5 (93.5) | 99.8 (99.5) | |||
| Unique reflections | 29551 | 63966 | 22182 | |||
| Multiplicity | 5.8 (2.2) | 12.1 (6.7) | 13.0 (8.1) | |||
| <I/σs> | 29.4 (1.8) | 38.0 (0.6) | 20.7 (0.4) | |||
| CC1/2 | 0.999 (0.809) | 0.999 (0.236) | 1.000 (0.162) | |||
| Refinement | 2YWN | Polished | 2CX4 | Polishedg | 2CX3 | Polishedh |
| Resolution range | 1.6 Å | 1.4 Å | 2.3 Å | 2.0 Å | 2.6 Å | 2.3 Å |
| R-factor (%) | 20.0 | 12.0 | 20.3 | 19.5 | 20.6 | 18.6 |
| R-free (%) | 22.1 | 14.9 | 26.3 | 23.4 | 24.7 | 22.4 |
| Molecules in AU | 1 | 1 | 8 | 4 | 4 | 2 |
| Protein residues | 150 | 151 | 1284 | 642 | 641 | 321 |
| Water molecules | 155 | 305 | 386 | 272 | 196 | 69 |
| Total non-H atoms | 1361 | 1573 | 10743 | 5694 | 5364 | 2714 |
| RMSD lengths (Å) | 0.005 | 0.008 | 0.007 | 0.01 | 0.009 | 0.010 |
| RMSD angles (°) | 1.2 | 1.1 | 1.1 | 1.1 | 1.2 | 1.15 |
| Ramachandran plot | ||||||
| ϕ, ψ-Preferred (%) | 98.6 | 99.3 | 84.7 | 96.7 | 89.1 | 94.3 |
| ϕ, ψ-Allowed (%) | 1.4 | 0.7 | 11.7 | 3.1 | 9.5 | 4.8 |
| ϕ, ψ-Outliers (%) | 0.0 | 0.0 | 3.6 | 0.2i | 1.4 | 1.0j |
| <B factors> (Å2)k | iso | aniso | iso | TLS | iso | TLS |
| Main chain | 14 | 15 | 49 | 55 | 65 | 77 |
| Side chains & waters | 19 | 21 | 49 | 66 | 65 | 84 |
| New PDB Code | -- | 4G2E | -- | 4GQC | -- | 4GQF |
Crystallization conditions as reported in the PDB header: 0.2M Ammonium Acetate, 0.1M HEPES, 25% PEG 3350, pH7.5, Vapor diffusion, Sitting drop, Temperature 293K.
Crystallization conditions as reported in the PDB header: Ammonium Sulfate, MES, TRIS, Sodium Chloride, Dithiothreitol, pH 7, Vapor diffusion, Sitting drop, Temperature 285K.
Crystallization conditions as reported in the PDB header: Ammonium Sulfate, 2-Propanol, TRIS Sodium Chloride, Dithiothreitol, pH 8, Vapor diffusion, Hanging drop, Temperature 293K.
Resolution cut-off previously 1.6 Å with R-meas in the highest resolution bin 12%; R-meas at 1.4 Å is 51%.
Resolution cut-off was previously 2.30 Å with R-meas ~120%; R-meas at 2.0 Å is 350%.
Resolution cut-off was previously 2.60 Å with R-meas ~160%; R-meas at 2.3 Å is 480%.
The polished version also contained 30 sulfate molecules.
The polished version also contained nine sulfate molecules.
Gly3, Thr50, and Glu52 in chain A which all have weak density.
Ala 91 in chain D with ϕ, ψ = −73.4, 26.7 is very close to an allowed region.
Individual atomic B-factors were refined anisotropically (aniso), isotropically (iso), or isotropically plus using TLS with one group per monomer (TLS).
Refinements were carried out using BUSTER25 and/or PHENIX26 without the use of non-crystallographic symmetry restraints, and with manual rebuilding done in Coot27. Ordered waters were added manually and automatically to difference map peaks >3.3 ρ rms also having >1.0 ρ rms density in the 2Fo−Fc map and at least one hydrogen bonding neighbor within 2.4–3.5 Å. In the final models, waters were sorted and renumbered in order of decreasing electron density in the final 2Fo−Fc maps. During the final refinement rounds, geometry restraint weights were adjusted to minimize Rfree, and Molprobity28 was used to find steric problems. Each refinement is briefly described below; the refinement statistics, as well as those of the original PDB entries, are listed in Table 1.
For StPrxQ, minimization of the 2YWN coordinates at 1.6 Å resolution by BUSTER led to R/Rfree values of 19.0/22.5%, and an initial difference map with most peaks being un-modeled water sites. Little density was present for the backbones of residues 44–49, so these were left un-modeled as in the original PDB entry, even though there was weak density at the expected position of the CP-CR disulfide (inferred from a structural overlay of deposited 2CX4/3). A few rounds of refinement brought R/Rfree to 15.7/18.2%, and the resolution was extended to 1.5 Å and then to 1.4 Å. The higher resolution revealed an alternate conformation of the catalytic Arg118. At this stage, refinement with PHENIX with riding hydrogens and individual anisotropic B-factors dropped R/Rfree to 12.7/15.8%. Electron density near Arg118, modeled as waters 17, 45, and 52, has some continuity and may be an acetate. The highest remaining difference peak, at 6.1 ρrms, is the un-modeled CP-CR disulfide and the final R/Rfree values are 12.0/14.9% (Table 1).
ApPrxQ-LU (originally PDB entry 2CX3) contains two half-dimers in the asymmetric unit, both in the LU conformation with a CP-CR disulfide. Initial BUSTER minimization at 2.6 Å of the molecular replacement solution (with no waters) resulted in R/Rfree values of 21.1/24.4%. Manual changes included mostly adjustments of some side chain rotomers and the addition of many waters. TLS refinement using one group per monomer lowered the Rfree by 3.3%. Extension of the resolution to 2.3 Å allowed the modeling of nine sulfate molecules as well as two glycerol molecules. The final R/Rfree values were 18.6/22.4% (Table 1). ApPrxQ-FF/LU (originally PDB entry 2CX4), has two dimers in the asymmetric unit, each containing one subunit in the FF conformation and one in the LU conformation with a CP-CR disulfide. Initial BUSTER minimization at 2.3 Å resolution of the molecular replacement solution led to R/Rfree values of 21.3/24.5%. Main manual changes included removal of a Cys80-Cys80′ (prime denotes the second chain of a dimer) intermolecular disulfide bond, and the addition of many waters. As refinement progressed, 31 sulfates were added with occupancies ranging from 0.3 to 0.9. TLS refinement using one group per monomer dropped the Rfree ~1.0%. Extending the resolution to 2.0 Å allowed identification of three glycerol molecules and further water sites. During the final stages, an oxidized dithiothreitol was modeled into a large oblate electron density peak in the active sites of the FF chains. During refinement, residual negative difference density at the DTT sulfur atoms occurred, which we attributed as due to restraints tethering their B-factors to be close to those of the much better ordered hydroxyls; this was addressed by setting the occupancy for the sulfur atoms to 0.7 which led to a clean difference map in the area. Also, compared to 2CX4, the chains were renamed so that the two asymmetric dimers are composed of chains AC and BD with chains A and B adopting the LU conformation and the chains C and D adopting the FF conformation. The final R/Rfree values were 19.5/23.4% (Table 1).
RESULTS AND DISCUSSION
As expected, the PrxQ chains are all compact domains with the Prx fold and associate to form Atype dimers (further discussed below). In the combined asymmetric units of the three crystal structures, a total of seven chains were modeled, including five with LU conformations and two with FF conformations. StPrxQ (PDB code 4G2E), at 1.4 Å resolution, is the highest resolution structure—actually the highest resolution of any Prx structure1—and its single chain (a half-dimer) in the asymmetric unit adopts the LU conformation. In this structure, the active site loop (residues 44–49) has little density and has not been modeled. The better diffracting of the ApPrxQ crystal forms, at 2.0 Å resolution, has two dual-conformation (FF/LU paired) dimers in the asymmetric unit and thus we refer to it as ApPrxQ-FF/LU (PDB code 4GQC). The terminal residues 1–3 and 164 are not modeled in the LU chains and 1–2 and 164 are not modeled in the FF chains. The other ApPrxQ crystal form, at 2.3 Å, has two LU half-dimers in the asymmetric unit and will be referred to as ApPrxQ-LU (PDB code 4GQF). These chains are missing residues 1 (chain A) and 1–2 (chain B). The non-crystallographic symmetry-related chains overlay within ~0.2–0.3 Å and between the two ApPrxQ crystal forms, the LU chains excluding residues 45–55 (see below) agree within ~0.4 Å. Comparisons of the StPrxQ (LU) with the ApPrxQ LU structures give Cα rmsd of ~1.2–1.6 Å, depending on the chain compared. A DALI29 search of the PDB shows that SsPrxQ4 (PDB entry 3HJP) is the closest other structure to both ApPrxQ and StPrxQ; the sequence identities are 46% and 78%, respectively.
Compared with the previously deposited structures, the reprocessing of the diffraction data and individualized refinements has led to improved models for all three PrxQ crystal structures. The resolutions were extended 0.2–0.3 Å, and refinement lowered the overall Rfree values by 2 to 7%, even using the higher resolution limit (Table 1). The further refinement changed some key aspects of the structure, including removing interchain Cys80-Cys80′ disulfides from the ApPrxQ-FF/LU structure, adding a dithiothreitol (DTT) ligand into the active sites of the FF chains, identifying alternate conformations for residues important in catalysis, and identifying many sulfate ions. Additional major changes were correcting the space groups of the two ApPrxQ crystal forms (see Experimental Procedures). In our subsequent discussion of PrxQ structures, ApPrxQ numbering will be used unless otherwise specified.
Overall structures of ApPrxQ and StPrxQ
Fully folded ApPrxQ
The FF structures (ApPrxQ chains C and D) have the standard Prx secondary structures of five α-helices and seven β-strands (Fig. 2a), with the CP-containing helix α2 sitting in a cradle with β-strands β4, β3, β6, and β7 providing a base, and helices α3 and α5 on the two sides1,15. Notable variations on the standard Prx fold are that helix α2 has a short bent extension we designate α2B, and both the ApPrxQ and StPrxQ structures have an additional β-hairpin between α4 and β6, also present in SsPrxQ418. Also, StPrxQ substitutes a 310 helix in place of α1, as was observed in SsPrxQ418 (Fig. 2b and c).
Figure 2.
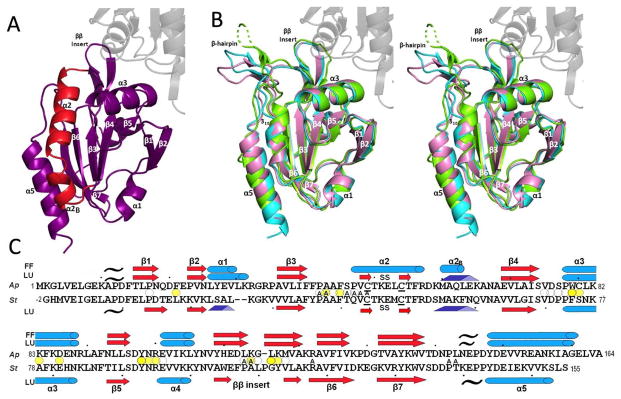
Overall structures of ApPrxQ and StPrxQ. (A) The FF subunit of the ApPrxQ-FF/LU structure is shown with the main segment involved in the conformational transition (residues 46–67) highlighted in red. The universally conserved Prx secondary structures are labeled (except α4 which is behind β5). (B) Representative LU chains are superimposed for ApPrxQ-FF/LU (violet), ApPrxQ-LU (cyan), and StPrxQ (green). The un-modeled part of the StPrxQ β-hairpin is shown as a dashed line. The area of transition highlighted in panel A becomes a β-hairpin and 310 helix (both labeled). For perspective, here and in panel A the second subunit of the dimer is shown for ApPrxQ-LU (ghost gray). (C) Structure-based sequence alignment of ApPrxQ and StPrxQ indicating placements of α-helices (cyan cylinders), β-strands (red arrows and pale red arrows for the inferred StPrxQ β-hairpin), 310 helices (blue triangular rods), and PII spirals (black tildes). Ellipsoids mark residues with surface area buried at the dimer interface of >100 Å2 (dark yellow), 25 to 100 Å2 (medium yellow) and <25 Å2 (white). ApPrxQ residues having surface area buried by the modeled DTT inhibitor are noted with A for Active-Site.
In terms of the active site, the FF conformation tends to be well-conserved among Prxs1,9, and ApPrxQ is no exception with the CP (Cys49 in the initial turn of α2) along with Arg122 and Ser46 at the bottom of the active site pocket (Fig. 3). As was seen for Tpx15 and Prx5 subfamily enzymes, the upper part of the ApPrxQ active site pocket is lined with a collar of non-polar side chains (Fig. 3) and seems well-suited to preferentially reduce organic peroxides, with Trp79′ from the other subunit of the dimer contributing to this collar, suggesting it is part of the complete active site. Indeed, persistent pancake-like electron density was observed (Fig. S1a) that we eventually modeled as an oxidized dithiothreitol (DTT). Although the density was not definitive on its own, the interpretation was made because DTT was present in the crystallization buffer and the density was a good match for how DTT was seen bound to the FF conformation of Human PrxV (PDB code 3MNG)9 with its two hydroxyls mimicking a peroxide substrate (Fig. S1b). We rationalize that the rather weak density for the disulfide of DTT occurs because it has much more variation in position than the much better localized hydroxyls.
Figure 3.

The ApPrxQ FF active site hydrophobic collar. Shown is the active site molecular surface annotated with residue numbers and showing the bound DTT (sticks). The surface is colored by element, distinguishing carbons from the main monomer (dark gray) or the other subunit (light gray), nitrogens (blue), oxygens (red), and sulfurs (yellow). The peroxidatic Cys thiolate surface is marked by a white asterisk (*). The catalytic Arg122 contributes the blue nitrogen surface left of the asterisk, and barely visible red surface right of the asterisk is from the Ser46 hydroxyl.
DTT is the first substrate-mimicking ligand to be identified in a FF CRα2 subtype, (FF SsPrxQ4 was modeled with a chloride ion in the active site)18 and also represents the only such ligand for a dimeric PrxQ. Examples of ligands in monomeric PrxQs include a formate in FF XcPrxQ (3GKM)30 and a citrate molecule in SsPrxQ119, though their exact positions overlay only loosely with the expected H2O2 binding site.9 None of the monomeric PrxQs have as pronounced a hydrophobic collar as the dimeric forms, but have active sites that are much shallower and more polar. Whereas it was proposed for the Tpx enzymes that the active site hydrophobic collar lends specificity for organic peroxides1, no PrxQ enzymes have yet been shown to have such a preference16,31,32,33. Indeed, the monomeric E. coli PrxQ has essentially equivalent kcat/Km values for both hydrogen peroxide and the bulky organic cumene hydroperoxide, although it is less active with t-butyl hydroperoxide.16 Nevertheless, because Trp79′ makes a substantial contribution of non-polar surface area to the CRα2 active site, it could allow dimer formation to influence substrate specificity and catalytic activity.
The locally unfolded conformation of CRα2 PrxQ enzymes
The CRα2 LU conformation is seen in four ApPrxQ chains (two from each crystal form) and the single StPrxQ chain. Compared with the FF conformation, the only substantive backbone changes involve residues 46–68, with residues 46–56 forming a β-hairpin extending out from the protein core, and residues 59–65 forming a 310 helix (Fig. 2). A striking feature is that the direction at which the hairpin extends varies among the independent LU chains (Fig. 2b), even between the ApPrxQ chains, in which case the variation cannot be due to sequence differences. At their tips, the hairpins span a range of ~15 Å, though this is an estimate, because density was so weak for the terminal residues of the StPrxQ hairpin that they were not modeled.
Despite the variation in position, the internal conformations of the hairpin are nearly identical (Fig. 4). The β-hairpin is internally stabilized by three β-strand hydrogen bonds and the disulfide bridge, as well as hydrogen bonds at the tip of the hairpin between the Thr50 side chain hydroxyl and the backbone nitrogens of Glu52 and Leu53. Additionally, the CP backbone nitrogen exhibits a transient hydrogen bond with the carbonyl of Leu143 that is only present in the ApPrxQ-FF/LU, providing the single external stabilizing interaction for the hairpin position. Because the turn is highly mobile, with B-factors over 80 Å2 in the ApPrxQ-FF/LU structure (see Fig. 6a), the conformational details must be considered tentative.
Figure 4.

Consistent internal conformation of the ApPrxQ and StPrxQ β-hairpins. The overlaid β-hairpins of all five LU chains are shown: two from ApPrxQ-FF/LU (cyan), two from ApPrxQ-LU (violet), and one from StPrxQ (green). Residues are labeled and H-bonds (dashed lines) internal to the hairpin and to the carbonyl of residue 142 are shown. As modeled, the conformation is a type I-like turn with ϕ, ψ (Lys51) ≈ −70, −35 and ϕ, ψ (Glu52) ≈ −130, −30.
Figure 6.

Changes in disorder associated with the FF – LU transition. (A) B-factors of the FF (purple) and LU (salmon) chains from the ApPrxQ-FF/LU crystal form. Secondary structure along the chain is indicated. (B) Same as panel A, but for the SsPrxQ4 FF chain (orange) and the StPrxQ LU chain (green) using StPrxQ numbering. The left y-axis is the StPrxQ B-factor and the right y-axis is the SsPrxQ4 B-factor. In StPrxQ α1 is a 310-helix (as shown), but in SsPrxQ4 it is an α-helix as for most other Prxs. This change is not FF/LU related, but due to a one residue insertion in SsPrxQ4.
Sulfate binding
An additional noteworthy feature of the ApPrxQ structures, which were crystallized using ammonium sulfate as a precipitant, is the presence of many ordered sulfate molecules. Although ammonium sulfate is a common crystallizing agent, such extensive localization of sulfates does not generally occur. Interestingly, only one sulfate site, found near His110, was present in both crystal forms and both conformations, however, implying that most are not discrete well-defined sulfate binding sites. It has been suggested in studies of halophiles that their proteins have evolved to be stable in the high concentrations of sodium partly through an increase in surface acidic residues which coordinate the positively-charged sodium ions34. Similarly, because the environment of the hyperthermophile A. pernix contains high concentrations of sulfate species, especially thiosulfate35, the organism’s proteins may have evolved to take advantage of interactions with these solutes to achieve thermostability.
Transition between FF and LU conformations of CRα2 PrxQs
A naïve prediction might be that disulfide bond formation between Cys residues separated by just four amino acids might involve only modest structural changes, as is true, for example, in the active site of glutathione reductase36. However, an overlay of the FF and LU ApPrxQ structures shows that the transition between FF and LU conformations involves very large (4–17 Å) backbone shifts of residues 47–67 and smaller (0.5–2 Å) shifts of residues 80–90 and 140–160 (Fig 5a). The substantial rearrangement of residues 47–67 involves the unfolding of helix α2 to make a β-hairpin and the extension of helix α2B to become a 310 helix (Fig 5b); the smaller shifts are adaptations that accommodate the changes in residues 47–67. In this striking transition, in order to allow the eleven residues in the β-hairpin to stick out like a tongue from the protein surface, it can be seen that helix α2B plays the role of excess slack that can be spooled out in the form of a 310 helix that is more tightly associated with the protein core to cover about 8.5 Å of the gap that would otherwise have been opened (Fig. 5b).
Figure 5.

Essential structural features of the PrxQ CRα2 FF↔LU transition. (A) Cα shifts between the FF and LU conformations are given for the ApPrxQ pair (purple) and the StPrxQ/SsPrxQ4 pair (green). ApPrxQ numbering is used and shifts <4 Å are shown in the inset. Black bars indicate the regions undergoing intermediate exchange in the AtPrxQ NMR study37. Peaks near residues 25 and 110 in the StPrxQ/SsPrxQ4 pair are not related to the FF/LU transition, but due, respectively, to a one residue insertion in StPrxQ4 and the influence of crystal contacts. (B) Orthogonal views of the backbone ribbon for residues 46 – 68 in the FF (purple) and LU (salmon) conformations. Side chains for the hydrophobic anchors are shown as sticks (labeled in the lower view) and regions in each conformation not well anchored and providing slack that extends as part of the conformational change are indicated in the upper view. (C) Schematic model for how the hydrophobic anchors shift interactions with the protein core during the conformation change.
The ability of α2B to transition to a 310 helix is made possible by key non-polar side chains that can interact with three hydrophobic pockets in the protein core (Fig. 5b and c). In FF ApPrxQ, the anchoring side chains are Leu53, Phe56, and Leu63, and are replaced in the LU form by Met60, Leu63, and Ala66. Because the side chain positions do not match perfectly, the core residues lining the pocket (such as Ile39 of β3, Phe84 of α3, and Tyr148 of α5) adjust, explaining the smaller backbone shifts in residues 80–90 and 140–160. Also, the side chains of Glu145 and Asp58 aid the transition by making N-cap hydrogen bonds with the backbone NH of residues 59 and 60 in the first turn of the 310 helix. The movements near residue 140 also reflect shifts in the backbone carbonyl interactions with Arg122 and the gain of the Leu143 carbonyl H-bond with the β-hairpin (Fig 4). The small shifts in α3 are partly related to the movement of Arg57, which in the FF form links α2 with the α3-β5 loop by H-bonding with the Asn88 carbonyl. The transition is also helped by the relatively smooth, non-polar faces of the β-strands 3, 4, and 5, with few “sticky” polar interactions, as was similarly described for Tpx subfamily enzymes15.
Because of the high sequence similarity between FF SsPrxQ4 and LU StPrxQ (with only two differences in the 21-residue transition segment: StPrxQ Gln42→Ser and Gln60→Glu), this pair provides a second view of the FF↔LU conformation transition. Other than differences near residues 25 and 110 that are not correlated to the transition, this comparison also shows large backbone shifts of residues 47–67 and smaller shifts near residues 85–90 and 140–150 (Fig 5a, green line). For this pair, all of the features described above are present, including the two 310 helix N-capping interactions and the existence of three key hydrophobic anchor positions, which in this case involve FF Met53, Phe56, and Phe63 being replaced by LU Met60, Phe63, and Val66.
Equally dramatic are changes in chain disorder that are associated with the FF-LU transition of residues 47–67 (Fig 6). In the FF active site, residues 47–57, corresponding to helix α2, are as ordered as the protein core, but are much less ordered when adopting the LU β-hairpin. In contrast, the α2B residues 58–67 are rather disordered in the FF form but become much more ordered in LU as the single weak anchoring provided by residue 63 is replaced by strong anchoring of residues 60, 63, and 66 (Fig. 5c). The high B-factors of α2B in the FF form can be understood in that the whole helix is only tethered to the protein core by one non-polar side chain (position 63) and that tethering involves a larger side chain (Leu in ApPrxQ and Phe in StPrxQ) sticking only its tip into a small pocket that is sized more optimally for an Ala (as in ApPrxQ) or a Val (as in StPrxQ).
An interesting question for understanding the Prx FF↔LU transition is whether there is always an ongoing rapid interchange between the two conformations, or whether the transition only occurs when triggered by a redox change in the active site CP residue. Protein crystallography cannot provide data to answer this question, but NMR can. Fortuitously, an extensive NMR study of a monomeric CRα2 PrxQ, Arabidopsis thaliana PrxQ (AtPrxQ)37 with ~40–50% sequence identity to ApPrxQ and StPrxQ, gives further insight. The dynamics of both the reduced (dithiol) and oxidized (disulfide) forms were studied. Of the 140 non-Proline residues in the protein, 132 could be assigned in the disulfide form, but only 63 for the reduced form. The unassigned residues suffered from extensive line-broadening due to conformational exchange dynamics, meaning that by this measure the FF conformation is more disordered than the LU. The regions involved were in four segments that correlate reasonably well with the regions that undergo conformational/environmental change in the FF↔LU transition (Fig. 5a). Among the residues without large Cα shifts, residues 35–45 pack against α2B and residues 110–125 form part of the active site collar, so in both cases their environment becomes much more solvent-exposed in the LU conformation.
These data indicate that even though fully reduced PrxQ molecules are largely populating the FF conformations, they are also constantly sampling the LU conformation—according to model-free analysis at a rate of ~1650 times per second37. For Prx1 subfamily enzymes38, proteins with a disrupted A-type interface had a less stabilized and less restricted FF conformation39, implying that dimeric PrxQ proteins may have a slightly more restricted conformational dynamics than the monomer of the NMR study. That the disulfide form is on the whole less dynamic, despite the floppy β-hairpin (which in fact are the main unassigned residues in this form), can be understood in that the formation of the disulfide “locks” the protein into the LU conformation by removing its ability to sample the FF conformation until the disulfide is reduced. These studies support the view that local unfolding of even the reduced form of the protein occurs sufficiently frequently to support catalysis, and that formation of SPOH does not trigger the conformational change but simply allows the chemistry of disulfide formation to trap the protein in the LU conformation.
The facile local unfolding of the reduced protein implies that the energy barrier between conformations is small, with neither state being highly stabilized. We speculate that whereas the FF conformation has many interactions stabilizing it (e.g. much buried surface and salt bridges spanning from Lys59-Asp149 and Lys51-Glu45), α2B is relatively poorly stabilized. In contrast, in the LU conformation the α2B residues are interacting well with the protein, but the α2 residues (in the β-hairpin) are poorly stabilized. Together, this leads to neither state being highly stable and levels the energy landscape, aiding the process of constant transition between conformations.
Dimer Interface
As previously mentioned, all chains we have analyzed are A-type dimers, with interfaces involving about 30 residues from strands β1 and β2, and the loops prior to α2, α3, and α4 (Fig. 7a)12,38. These interacting regions group well into the defined sequence regions numbered 0 through 4 that were used for other Prxs12,38. As was noted in the SsPrxQ4 description18, the ApPrxQ and StPrxQ interfaces involve residues from a ββ insertion (here designated as Region 4) absent in the monomeric AtPrxQ37, and ScPrxQ20. However, the monomeric XcPrxQ and XfPrxQ have such an insertion (though it is extended and contains a higher portion of residues with polar side chains), meaning this insertion cannot be strictly used as a marker for dimeric PrxQs.
Figure 7.
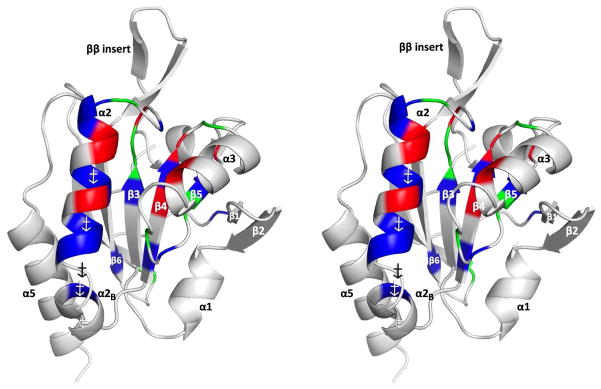
The PrxQ dimer interface is unperturbed during the catalytic cycle. (A) ApPrxQ-LU/LU (cyan/cyan), ApPrxQ-FF/LU (purple/pink), StPrxQ-LU/LU (green/green), and SsPrxQ4-FF/FF (gold/gold) dimers overlaid based on the top subunit. (B) Residues with > 3 Å2 surface buried at the ApPrxQ-LU/LU dimer interface grouped into sequence regions 0 through 438 and colored to correspond with C. (C) Close up of interactions of the ApPrxQ-FF/LU interface twofold. The coordination of conserved buried Water81 is shown, as well the primary conformation and 2Fo−Fc electron density at 2.0 ρrms for Cys80 and Cys80′, which do not form a disulfide.
Despite the dramatic shift between FF and LU conformations, we find that the dimer interface remains virtually identical between the FF/LU, LU/LU, and FF/FF (from SsPrxQ4) dimers (Fig. 7a). As all are essentially equivalent, we focus our description on the ApPrxQ interface, noting differences where needed. The dimerization interface buries about 2000 Å2 of surface (1050 and 915 Å2 per chain in ApPrxQ, and StPrxQ, respectively). Most of the buried area is non-polar with the four most buried positions being Phe45 (70–75 Å2), Trp79 (150 Å2), Tyr98 (120 Å2), and Leu116 (105–110 Å2) one each from Regions 1 to 4 (Fig. 7b). Leu116 of Region 4 is part of the ββ insertion and its role is conserved between the PrxQ dimers as a tight packing interaction into a non-polar pocket formed by the side chains of Tyr98′, Trp79′, Pro78′, and Phe21′. The insertion is also stabilized by variable hydrogen bonds depending on sequence differences. An important point is that the close Cys80–Cys80′ pair (unique to ApPrxQ) pack together but do not form a disulfide (Fig. 7c).
In terms of well-buried polar interactions, there is one water site (Water 81) at the dimer interface conserved between ApPrxQ, StPrxQ, and SsPrxQ4. It is among the more highly ordered water sites in the structures (the waters are ordered based on decreasing electron density). Water 81 makes bridging H-bonds between the Ser77 hydroxyl, the Asp76′ carboxylate and the Ala41′ backbone oxygen (Fig. 7c). Additional polar interactions that are not in common occur near the periphery of the interfaces, such as the symmetric intermolecular salt bridges in ApPrxQ between Lys83-Glu87′, and Glu87-Lys83′.
As was noted above, the interfaces of ApPrxQ and StPrxQ contribute to the active site (Fig. 2a and Fig. 3). In fact, in addition to Trp79′, six other residues interacting with the DTT ligand (Fig. 2c) are also involved in the dimer interface. An interesting question is how the FF/LU mixed dimers came to be stabilized in the crystal form. The systematic heterogeneity seems related to crystal packing interactions: in the FF/LU crystal form it is impossible for the FF chains to be LU because the LU disulfide loop would collide with Lys31 of the adjacent FF chain. We hypothesize that crystals are nucleated by the FF/LU heterodimers that form as the DTT present becomes oxidized, and then the crystal can grow as more protein oxidizes. Once an FF/LU dimer is incorporated into the crystal lattice, the crystal packing interactions and the DTT binding hinder the LU transition of the FF subunit, so it cannot be oxidized via disulfide exchange. As long as some reduced DTT remains, disulfide exchange can occur to allow remaining soluble dimers to adopt the crystallizable FF/LU form. We emphasize that the existence of the FF/LU dimers should not be taken as evidence for half-of-the-sites reactivity or another more general allosteric relationship.
As a final thought on the relevance of the dimer interface, a characterization of SsPrxQs31 found that the dimeric SsPrxQ4 was substantially more heat stable, displayed optimal activity levels at higher temperatures, and retained a higher relative activity when incubated at 95° C, compared to the monomeric SsPrxQ1 and SsPrxQ3. SsPrxQ4 was also found to be exceptionally resistant to chemical denaturation, retaining 50% activity after 30 min in 6 M urea31. The authors proposed that the dimeric forms of PrxQs would allow these hyperthermophiles to temporarily endure a larger range of temperature fluctuation by providing protection from reactive oxygen species at temperatures that would denature the organism’s other PrxQ enzymes.
Sequence Conservation among CRα2, CRα3, and CR0 PrxQs
The PrxQ subfamily contains a surprising diversity in the mechanism by which the sulfenic acid form is resolved, containing CRα2, CRα3, and CR0 Prxs. Whereas the CRα2 PrxQs undergo the drastic unraveling of α2 documented here, CRα3 PrxQ conformations change little between redox states, requiring only the inward-bending of the α2-α3 loop and the flipping of the CP and CR side chains to form the disulfide30. To see what sequence features are conserved with the CRα2 subtype, conservation patterns were assessed for 511 CRα2, 74 CRα3, and 387 CR0 PrxQ sequences, and the roles of each highly conserved position was identified (Table 2 and Fig. 8). By also assessing conservation at these positions among the other subtypes we are able to comment on which patterns appear to be unique to the CRα 2 subtype and which are conserved in PrxQs in general. We consider three tiers of highly conserved residues: ≥98% identical, ≥90% identical, or ≥90% identical between two residue types, all for residues present in at least 95% of the sequences.
Table 2.
Conservation of PrxQ CRα2 Positionsa
| # | CRα2 | CRα3 | CR0 | Conf.b | Functionc |
|---|---|---|---|---|---|
| Phe 14 |

|

|
FF-LU | Structure | |
| lle 39 |

|

|

|
FF-LU | Cradle |
| Phe 40 |
|
|
|
FF-LU | Structure |
| Phe 41 |

|
|

|
FF-LU | Cradle |
| Pro 42 |
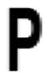
|
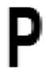
|
|
FF-LU | Catalysis |
| Ala 44 |

|
|
|
FF-LU | Structure |
| Ser 46 |

|

|

|
FF-LU | Catalysis |
| Pro 47 |

|
|
|
FF LU |
Collar Structure |
| Val 48 |

|
|
|
FF LU |
Collar Structure |
| Cys 49 |

|

|

|
FF LU |
CP CP |
| Thr 50 |
|
|

|
FF LU |
Hbond→46bb Hbond→52bb, 53bb |
| Glu 52 |

|
|
|
FF LU |
Hbond→R122 Structure |
| Leu 53 |
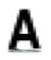
|
|
|
FF LU |
Anchor Structure |
| Cys 54 |

|
FF LU |
CR CR |
||
| Phe 56 |
|
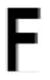
|
|
FF LU |
Anchor Structure |
| Arg 57 |

|
FF LU |
Hbond→D58, 88bb Structure |
||
| Asp 58 |

|
FF LU |
Hbond→R57 Hbond→60bb, R122d |
||
| Leu 63 |

|

|
|
FF LU |
Anchor A (310 N-cap) |
| Val 70 |

|
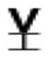
|
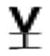
|
FF-LU | Cradle |
| Ala 72 |
|
|
|
FF-LU | Structure |
| lle 73 |
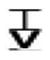
|
|
|
FF-LU | Structure |
| Ser 74 |
|
|

|
FF-LU | Hbond→D76 |
| Asp 76 |

|
|
|
FF-LU | Hbond→S74 |
| Phe 84 |
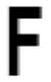
|
|
|
FF-LU | Cradle |
| Leu 90 |
|
|
|
FF-LU | Cradle |
| Phe 92 |

|

|
FF-LU | Cradle | |
| Leu 94 |
|
|
|
FF-LU | Cradle |
| Leu 95 |
|

|
FF-LU | Structure | |
| Asp 97 |

|
|
|
FF-LU | Hbond to 101bb, 102bb |
| Arg 122 |

|
|

|
FF-LU | Catalysis |
| Val1 27 |

|

|
|
FF-LU | Structure |
| Gly 131 |
|
|
|
FF | Structuree |
The sequence relative to ApPrxQ is given, with conservation of positions among subgroups expressed as a standard SeqLogo of 511 CRα2 sequences, 74 CRα3 sequences, and 387 CR0 sequences. Those positions that exhibited conservation patterns of ≥90% or ≥90% between two residues are shown, with blanks indicating a lack of conservation for that subtype. The overall size of the SeqLogo image corresponds to the number of total sequences that aligned at this position and the relative height of the single letter residue abbreviation corresponds to the percentage of conservation, with a minimum sequence alignment cutoff of 95%. For example, position Cys49 represents 100% of sequences aligning and 100% conservation of a Cys.
The conformation(s) in which the function occurs: fully folded (FF), locally unfolded (LU) or both (FF-LU).
General function and important interactions are defined as: Catalysis; involved in catalysis and/or substrate positioning, Collar; part of the hydrophobic collar of the active site, Anchor; one of the hydrophobic anchors involved in conformation change, Cradle; packs against α2, α2B, or the active site, Hbond; makes important structural interactions through hydrogen bonding, with the residue specified if the bond is to another side chain, or “bb” if to a backbone atom, Structure; maintains general protein structure.
The distance between Asp58 and Arg122 is variable, and in some chains is too great to form a salt-bridge.
Forms a type I turn with Φ, ψ Gly-specific at 90, 0.
The data was visualized using WebLogo 3.3 (http://weblogo.threeplusone.com/).43
Figure 8.

Positions highly conserved in CRα2 PrxQ enzymes. Stereo-view backbone ribbon of the FF subunit from the ApPrxQ-FF/LU structure colored to highlight conservation, distinguishing positions with ≥98% identity (red), ≥90% identity (green), and ≥90% identity between two amino acids (blue). The remaining residues are grey.
The highly conserved residues are mostly centered around the FF active site, especially positions 38–58, which encompasses the end of β3, the loop before helix α2 (the CP loop), and most of α2. This can be interpreted as a conservation of redox function, given that these proteins have an identical mode and site for catalysis, making the structure and stability of the α2 region paramount. Closely related to this are the side chains that line the cradle in which helices α2/α2B sit and this accounts for most of the other residues that have similar conservation across all three PrxQ subtypes. In contrast to what was seen for the Tpx subfamily of Prxs15, residues at the dimer interface are not well-conserved. This is not surprising given that the CRα 2 subtype includes both dimers and monomers. In fact, only five dimer interface positions are highly conserved (Pro42, Ser46, Asp76, Pro47, Phe56) and none of these have more than 25 Å2 surface buried at the interface in (Fig. 7b), consistent with the conservation of these positions being related to functions distinct from dimerization. The ββ insertion (positions 113–117) common to all known PrxQ dimers is present in only ~30 of the 511 sequences, suggesting CRα 2 PrxQs are predominantly monomers (or form dimers of a different type) and even that number may be an over-estimate, because as mentioned above, some monomers contain this insertion.
Particularly interesting are positions with conservation patterns unique to the CRα 2 subtype. These include the completely unique Cys54 (CR), Arg57, and Asp58, and the somewhat unique Leu53, Phe56, and Leu63. Each of these positions has been mentioned above as having specific roles in the FF-LU transition (Table 2). The conservation of these positions provides further evidence that their specific roles are rather important for facilitating the transition. That there are no further residues with a distinct CRα 2-specific conservation pattern supports the idea that we have a handle on the important structure-function features of the CRα 2 PrxQs.
Challenge and opportunity of unpublished structural genomics results
Structural genomics groups have contributed greatly to our knowledge of protein structures. Already by 2006 their work accounted for ~20% of the total number of structures added to the PDB40. However, less well-documented is what fraction of these structures have never received the individual attention and detail-oriented analyses needed for a primary literature report. Our analyses here of the three PrxQ structures illustrates that such unpublished structures can contain a wealth of information useful to a fielde.g. 41, but also that they can have real shortcomings—errors and incomplete interpretations—that detract from their reliability. This is of course a significant problem when, as for the previously deposited ApPrxQ and StPrxQ entries, the un-vetted structures get used “as is” by others for structure-based sequence alignments1,18,19,30,14, assigning residue functionality14, threading37, protein-protein docking studies18, homology modeling18, overlays30, and other structure-based comparisons1,30. For unpublished structural genomics structures to provide maximal benefit, they must be carefully analyzed and described, and how this will be done is an important unresolved issue that raises novel ethical and policy questions that need to be explicitly addressed by the structural biology community.
Supplementary Material
Acknowledgments
For carrying out this study, we are grateful to Professors S. Yokoyama and S. Kuramitsu of RIKEN Systems and Structural Biology Center who provided the original diffraction data for the structures in this study and whose support allayed our concerns about the appropriateness of describing unpublished work carried out by others. This research was funded in part by National Institutes of Health grant GM050389 to LBP and PAK.
Abbreviations
- Prx
peroxiredoxin
- CP
peroxidatic cysteine
- CR
resolving cysteine
- FF
fully folded
- LU
locally unfolded
- BCP
Bacterioferritin Comigratory Protein
- CRα2
PrxQ enzymes with CR located in α2
- CRα3
PrxQ enzymes with CR located in α3
- CR0
PrxQ enzymes lacking CR
- SsPrxQ1
Sulfolobus solfataricus PrxQ1
- SsPrxQ4
Sulfolobus solfataricus PrxQ4
- ScPrxQ
Saccharomyces cerevisiae PrxQ
- XfPrxQ
Xylella fastidiosa PrxQ
- XcPrxQ
Xanthomonas campestris PrxQ
- AtPrxQ
Arabidopsis thaliana PrxQ
- ApPrxQ
Aeropyrum pernix PrxQ
- StPrxQ
Sulfolobus tokodaii PrxQ
- PDB
Protein Data Bank
- DTT
1,4-dithiothreitol
- NMR
nuclear magnetic resonance
Footnotes
This study was supported by a grant from the National Institutes of Health to L.B.P. RO1 GM050389.
SUPPORTING INFORMATION AVAILABLE
Figure S1 shows the evidence for the modeled DTT. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hall A, Nelson K, Poole LB, Karplus PA. Structure-based Insights into the Catalytic Power and Conformational Dexterity of Peroxiredoxins. Antioxid Redox Signaling. 2011;15:795–815. doi: 10.1089/ars.2010.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 3.Niki E, Yamamoto Y, Komuro E, Sato K. Membrane Damage Due to Lipid Oxidation. J Clin Nutr. 1991;53:201S–205S. doi: 10.1093/ajcn/53.1.201S. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura T, Kado Y, Yamaguchi T, Matsumura H, Ishikawa K, Inoue T. Crystal Structure of Peroxiredoxin from Aeropyrum Pernix K1 Complexed with Its Substrate, Hydrogen Peroxide. J Biochem. 2010;147:109–115. doi: 10.1093/jb/mvp154. [DOI] [PubMed] [Google Scholar]
- 5.Karplus PA, Hall A. Structural Survey of the PeroxiredoxinsPeroxiredoxin Systems. Subcell Biochem. 2007;44:41–60. doi: 10.1007/978-1-4020-6051-9_3. [DOI] [PubMed] [Google Scholar]
- 6.Hofmann B, Hecht HJ, Flohé L. Peroxiredoxins. Biol Chem. 2002;383:347– 364. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- 7.Knoops B, Loumaye E, Eecken V. Evolution of the PeroxiredoxinsPeroxiredoxin Systems. Subcell Biochem. 2007;44:27–40. doi: 10.1007/978-1-4020-6051-9_2. [DOI] [PubMed] [Google Scholar]
- 8.Trujillo M, Ferrer-Sueta G, Thomson L, Flohé L, Radi R. Kinetics of Peroxiredoxins and their Role in the Decomposition of PeroxynitritePeroxiredoxin Systems. Subcell Biochem. 2007;44:83–113. doi: 10.1007/978-1-4020-6051-9_5. [DOI] [PubMed] [Google Scholar]
- 9.Hall A, Parsonage D, Poole LB, Karplus PA. Structural Evidence that Peroxiredoxin Catalytic Power Is Based on Transition-State Stabilization. J Mol Biol. 2010;402:194–209. doi: 10.1016/j.jmb.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin Evolution and the Regulation of Hydrogen Peroxide Signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 11.Hall A, Karplus PA, Poole LB. Typical 2-Cys peroxiredoxins – structures, mechanisms and functions. FEBS Journal. 2009;276:2469–2477. doi: 10.1111/j.1742-4658.2009.06985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarma GN, Nickel C, Rahlfs S, Fischer M, Becker K, Karplus PA. Crystal Structure of a Novel Plasmodium falciparum 1-Cys Peroxiredoxin. J Mol Biol. 2005;346:1021–1034. doi: 10.1016/j.jmb.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 13.Krnajski Z, Gilberger TW, Walter RD, Cowman AF, Müller S. Thioredoxin Reductase Is Essential for the Survival of Plasmodium falciparum Erythrocytic Stages. J Biol Chem. 2002;277:25970–25975. doi: 10.1074/jbc.M203539200. [DOI] [PubMed] [Google Scholar]
- 14.Nelson KJ, Knutson ST, Soito L, Klomsiri C, Poole LB, Fetrow JS. Analysis of the peroxiredoxin family: Using active-site structure and sequence information for global classification and residue analysis. Proteins: Struct, Funct, and Bioinf. 2010;79:947–964. doi: 10.1002/prot.22936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hall A, Sankaran B, Poole LB, Karplus PA. Structural Changes Common to Catalysis in the Tpx Peroxiredoxin Subfamily. J Mol Biol. 2009;393:867–881. doi: 10.1016/j.jmb.2009.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reeves SA, Parsonage D, Nelson KJ, Poole LB. Kinetic and Thermodynamic Features Reveal That Escherichia coli BCP Is an Unusually Versatile Peroxiredoxin. Biochemistry. 2011;50:8970–8981. doi: 10.1021/bi200935d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gretes MC, Poole LB, Karplus PA. Peroxiredoxins in Parasites. Antioxid Redox Signaling. 2012;17:608–633. doi: 10.1089/ars.2011.4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Limauro D, D’Ambrosio K, Langella E, De Simone G, Galdi I, Pedone C, Pedone E, Bartolucci S. Exploring the catalytic mechanism of the first dimeric Bcp: Functional, structural and docking analyses of Bcp4 from Sulfolobus solfataricus. Biochimie. 2010;92:1435–1444. doi: 10.1016/j.biochi.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 19.D’Ambrosio K, Limauro D, Pedone E, Galdi I, Pedone C, Bartolucci S, De Simone G. Insights into the catalytic mechanism of the Bcp family: Functional and structural analysis of Bcp1 from Sulfolobus solfataricus. Proteins: Struct, Funct, and Bioinf. 2009;76:995–1006. doi: 10.1002/prot.22408. [DOI] [PubMed] [Google Scholar]
- 20.Choi J, Choi S, Chon JK, Choi J, Cha MK, Kim IH, Shin W. Crystal structure of the C107S/C112S mutant of yeast nuclear 2-Cys peroxiredoxin. Proteins: Struct, Funct, and Bioinf. 2005;61:1146–1149. doi: 10.1002/prot.20704. [DOI] [PubMed] [Google Scholar]
- 21.Kabsch W. XDS. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karplus PA, Diederichs K. Linking Crystallographic Model and Data Quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans P. Scaling and assessment of data quality. Acta Crystallogr, Sect D: Biol Crystallogr. 2005;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 24.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr, Sect D: Biol Crystallogr. 2012;68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Afonine PV, Grosse-Kunstleve RW, Adams PD, Lunin VY, Urzhumtsev A. On macromolecular refinement at subatomic resolution with interatomic scatterers. Acta Crystallogr, Sect D: Biol Crystallogr. 2007;63:1194–1197. doi: 10.1107/S0907444907046148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Davis IW, Murray LW, Richardson JS, Richardson DC. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liao SJ, Yang CY, Chin KH, Wang AHJ, Chou SH. Insights into the Alkyl Peroxide Reduction Pathway of Xanthomonas campestris Bacterioferritin Comigratory Protein from the Trapped Intermediate–Ligand Complex Structures. J Mol Biol. 2009;390:951–966. doi: 10.1016/j.jmb.2009.05.030. [DOI] [PubMed] [Google Scholar]
- 31.Limauro D, Pedone E, Galdi I, Bartolucci S. Peroxiredoxins as cellular guardians in Sulfolobus solfataricus– characterization of Bcp1, Bcp3 and Bcp4. FEBS Journal. 2008;275:2067–2077. doi: 10.1111/j.1742-4658.2008.06361.x. [DOI] [PubMed] [Google Scholar]
- 32.Wakita M, Masuda S, Motohashi K, Hisabori T, Ohto H, Takamiya K. The Significance of Type II and PrxQ Peroxiredoxins for Antioxidative Stress Response in the Purple Bacterium Rhodobacter sphaeroides. J Biol Chem. 2007;282:27792–27801. doi: 10.1074/jbc.M702855200. [DOI] [PubMed] [Google Scholar]
- 33.Horta BB, Oliveira MAD, Discola KF, Cussiol JRR, Netto LES. Structural and Biochemical Characterization of Peroxiredoxin Qβ from Xylella fastidiosa Catalytic Mechanism and High Reactivity. J Biol Chem. 2010;285:16051–16065. doi: 10.1074/jbc.M109.094839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madern D, Ebel C, Zaccai G. Halophilic adaptation of enzymes. Extremophiles. 2000;4:91–98. doi: 10.1007/s007920050142. [DOI] [PubMed] [Google Scholar]
- 35.Sako Y, Norimichi N, Uchida A, Ishida Y, Morii H, Koga Y, Hoaki T, Maruyama T. Aeropyrum pernix, gen. nov., sp nov., a Novel Aerobic Hyperthermophilic Archaeon Growing at Temperatures up to 100°C. Int J Syst Bacteriol. 1996;46:1070–1077. doi: 10.1099/00207713-46-4-1070. [DOI] [PubMed] [Google Scholar]
- 36.Berkholz DS, Faber HR, Savvides SN, Karplus PA. Catalytic Cycle of Human Glutathione Reductase Near 1 Å Resolution. J Mol Biol. 2008;382:371–384. doi: 10.1016/j.jmb.2008.06.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ådén J, Wallgren M, Storm P, Weise CF, Christiansen A, Schroder WP, Funk C, Wolf-Watz M. Extraordinary μs–ms backbone dynamics in Arabidopsis thaliana peroxiredoxin Q. Biochim Biophys Acta, Proteins and Proteomics. 2011;1814:1880–1890. doi: 10.1016/j.bbapap.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 38.Wood ZA, Poole LB, Hantgan RR, Karplus PA. Dimers to doughnuts: redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry. 2002;41:5493–5504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- 39.Parsonage D, Youngblood DS, Sarma N, Wood ZA, Karplus PA, Poole LB. Analysis of the Link between Enzymatic Activity and Oligomeric State in AhpC, a Bacterial Peroxiredoxin. Biochemistry. 2005;44:10583–10592. doi: 10.1021/bi050448i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chandonia JM, Brenner SE. The Impact of Structural Genomics: Expectations and Outcomes. Science. 2006;311:347–351. doi: 10.1126/science.1121018. [DOI] [PubMed] [Google Scholar]
- 41.Knaus T, Eger E, Koop J, Stipsits S, Kinsland CL, Eali SE, Macheroux P. Reverse Structural Genomics An Unusual Flavin-Binding Site in a Putative Protease from Bacteroides thetaiotaomicron. J Biol Chem. 2012;287:27490–27498. doi: 10.1074/jbc.M112.355388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soito L, Williamson C, Knutson ST, Fetrow JS, Poole LB, Nelson KJ. PREX: PeroxiRedoxin classification indEX, a database of subfamily assignments across the diverse peroxiredoxin family. Nucleic Acids Res. 2010;39:D332–D337. doi: 10.1093/nar/gkq1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crooks GE, Hon G, Chandonia JM, Brenner SE. Weblogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.