

Abstract
Tumor suppressor PAR-4 acts in part by modulating sensitivity to apoptosis but the basis for its activity is not fully understood. In this study, we describe a novel mechanism of anti-apoptosis by NF-kappaB, revealing that it can block PAR-4-mediated apoptosis by downregulating trafficking of the PAR-4 receptor GRP78 from the endoplasmic reticulum (ER) to the cell surface. Mechanistic investigations revealed that NF-kappaB mediated this anti-apoptotic mechanism by upregulating expression of UACA, a pro-inflammatory protein in certain disease settings. In clinical specimens of cancer, a strong correlation existed between NF-kappaB activity and UACA expression, relative to normal tissues. UACA bound to intracellular PAR-4 in diverse cancer cells, where it prevented translocation of GRP78 from the ER to the cell surface. This pathway of anti-apoptosis could be inhibited by suppressing levels of NF-kappaB or UACA expression, which enhanced ER stress and restored GRP78 trafficking to the cell surface, thereby sensitizing cancer cells to apoptosis by extracellular PAR-4 or GRP78 agonistic antibody. In summary, our results identify a novel intracellular pathway of apoptosis mediated by NF-kappaB through UACA elevation, which by attenuating ER stress and GRP78 translocation to the cell surface can blunt the sensitivity of cancer cells to apoptosis.
Keywords: Par-4, UACA, Apoptosis
Introduction
The tumor suppressor Par-4 is expressed ubiquitously across various tissue types and is localized at various cell compartments, including the ER, cytosol, and nucleus (1). Endogenous Par-4 is essential for the apoptotic function of diverse cytotoxic agents (2). Nuclear translocation and the apoptotic function of Par-4 is activated by chemotherapeutic agents, such as vincristine, that cause phosphorylation of Par-4 (2). Activated Par-4 is capable of nuclear entry, NF-κB inhibition, caspase activation and induction of apoptosis (2).
Par-4 protein is secreted in response to ER stress, and extracellular Par-4 protein produces autocrine and paracrine effects by binding to its cell surface receptor GRP78 (1). Apoptosis by extracellular Par-4 acting via GRP78 receptor requires intracellular Par-4 function for trafficking GRP78 to the membrane, and ER stress for activation of the caspase 8/caspase 3 pathway (1). Furthermore, the ability to systemically express Par-4 can be transferred from cancer-resistant mice to cancer sensitive mice by bone marrow transplantation, implying a role for systemic Par-4 in suppression of tumor growth and metastasis (3).
As susceptibility to extracellular Par-4 is dependent on cell surface GRP78 (1), we sought to identify cell survival factors that may broadly regulate GRP78 trafficking to the cell surface. Our studies indicated that cell survival NF-κB activity (4) suppressed GRP78 translocation to the cell surface and rendered cancer cells resistant to apoptosis by extracellular Par-4.
Materials and Methods
Cell culture
All the cancer cell lines and HEL cells were obtained from American Type Culture Collection in the past two years and authenticated (on 2-29-12 and 8-16-12) at Genetica DNA Laboratories (Cincinnati, OH) using short tandem repeats profiling of DNA.
Co-immunoprecipitation and Western blot analysis
Precleared cell lysates were subjected to immunoprecipitation with 4 μg of antibody conjugated to 50 μl of protein G-Sepharose beads. The immunoprecipitates were washed with lysis buffer and subjected to Western blot analysis as described (1).
IHC, ICC, proliferation, and apoptosis
These standard procedures have been previously described (1,2), and additional details are presented in Supplemental section.
NF-κB reporter assays and FACS analysis
NF-κB transcription activity in the cells were determined using transfection with NF-κB-luc construct as reporter and β-galactosidase expression construct to normalize luc activity, as previously described (1,2). Cell surface GRP78 analysis was performed (without fixing the cells to allow the detection of GRP78 at their surface) by FACS analysis using either GRP78 primary antibody (N-20, SantaCruz Biotechnology, Inc.) or no primary antibody as control, and R-phycoerythrin-conjugated secondary antibody (R&D Systems) (1).
Quantitative Real-Time-PCR Analysis
To determine the expression of UACA, total RNA was prepared from cells using Trizol reagent (Invitrogen Corp.) and subjected to qRT-PCR (see Supplemental section).
Statistical analysis
All experiments were performed in triplicate to verify the reproducibility of the findings. Statistical analyses were carried out with Statistical Analysis System software (SAS Institute, Cary, NC) and P values were calculated using the Student t test. The effect of interaction between two different treatments was analyzed using a two-way ANOVA model with data normality and equality of variance assumptions. Mean of three experiments +Standard Deviation bars are shown.
All other cell lines, reagents and procedural details are presented in the Supplemental Section.
Results
NF-κB activity regulates apoptosis by extracellular Par-4
Constitutively activated NF-κB pathway is one of the most commonly activated mechanisms that promotes anti-apoptosis and therapeutic resistance in diverse human cancers (5). To determine whether NF-κB activity regulates apoptosis by extracellular Par-4, we used two different approaches to block NF-κB activity: (a) the IκB-super repressor (IκB-SR) S32A/S36A mutant of IκB, and (b) treatment with PS-1145, an inhibitor of IKK activation. Various cancer cells were infected with IκB-SR or control GFP adenovirus and then treated with recombinant control protein thioredoxin (TRX) or TRX-Par-4. TRX-Par-4, but not TRX, produced apoptosis in a dose-dependent manner, and reporter assays confirmed inhibition of NF-κB activity by IκB-SR (Figure S1A,B,C). The apoptotic sensitivity of PC-3 and H460 cells, which are intrinsically sensitive to TRX-Par-4, was further enhanced by IκB-SR (Figure 1A). Moreover, A549 cells, which are resistant to the action of TRX-Par-4, were rendered highly susceptible to TRX-Par-4 by IκB-SR (Figure 1A). These findings suggest that elevated NF-κB activity in cancer cells mitigates susceptibility to extracellular Par-4.
Figure 1. Apoptosis by extracellular Par-4 is regulated by NF-κB.
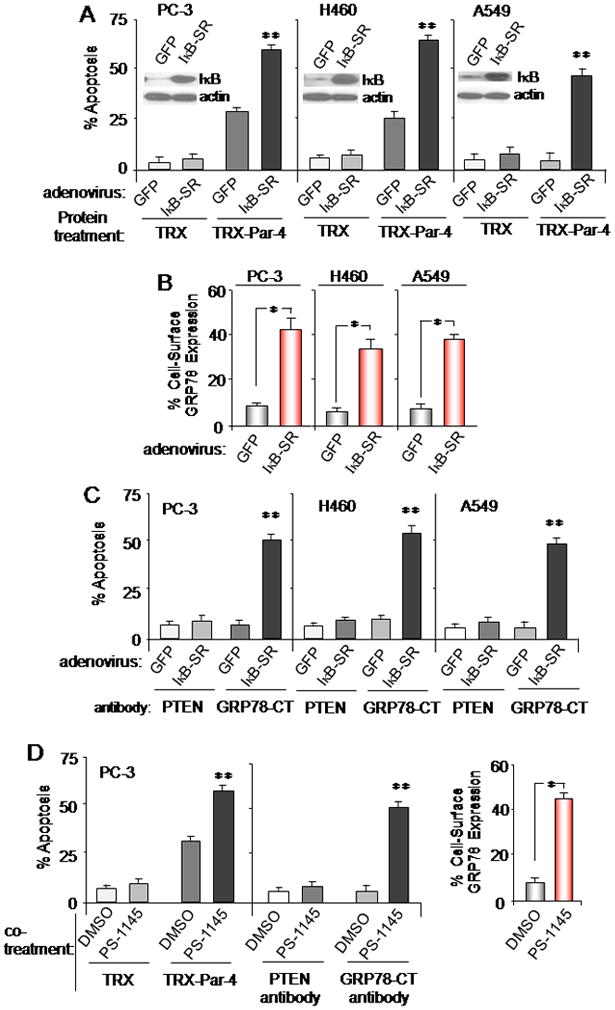
(A,B,C) IκB-SR up-regulates cell surface GRP78 and sensitizes cancer cells to apoptosis. Cells were transduced with IκB-SR or control GFP adenovirus; and then (B) unfixed cells were subjected to FACS analysis for cell surface GRP78, or (A, B) the cells were treated with (A) TRX-Par-4 or TRX (100 nM of each protein) or (B) GRP78 C-terminal agonistic antibody or PTEN control antibody (1 μg/ml) for 24 h. The cells were subjected to ICC for active caspase-3 and apoptotic cells were scored under a fluorescent microscope. Expression of IκB-SR was confirmed by Western blot analysis (A, inset).
(D) PS-1145 up-regulates cell surface GRP78 and sensitizes cells to apoptosis. Cells were co-treated with PS-1145 (10 μM) and TRX or TRX-Par-4 protein (100 nM), or GRP78 agonistic antibody or PTEN antibody (1 μg/ml) for 24 h, and subjected to FACS analysis for cell surface GRP78 (right panel) or to apoptotic assays (left panel).
Asterisk (*) indicates statistically significant (P < 0.001) difference by the Student t test, and (**) indicates that the effect is significantly (P < 0.001) higher based on two-way ANOVA with data normality and equality of variance assumptions.
We next examined whether NF-κB activity also regulated GRP78 levels at the cell surface. Increased expression of GRP78 at the cell surface was noted in response to IκB-SR adenoviral expression (Figure 1B). These findings were corroborated by cell surface biotinylation studies (Figure S1D).
To further confirm the up-regulation of cell surface GRP78, we tested the effect of IκB-SR on cellular response to the GRP78 (carboxyl-terminal) agonistic antibody (6). Cells infected with IκB-SR- but not GFP-producing adenovirus showed enhanced apoptosis of cancer cells with GRP78 agonistic antibody (Figure 1C). Moreover, PS-1145 significantly elevated GRP78 expression at the cell surface, and increased the susceptibility of the cells to apoptosis by TRX-Par-4 or the GRP78 antibody (Figure 1D and Figure S1E,F). TRX-Par-4 protein by itself did not inhibit NF-κB activity (Figure S1G). Collectively, these findings indicate that NF-κB activity negatively regulates the expression of cell surface GRP78 and apoptosis by extracellular Par-4 in cancer cells.
Par-4 interacts with UACA
Since intracellular Par-4 function is essential for translocation of GRP78 to the cell surface (1), we hypothesized that an NF-κB-regulated protein may bind to Par-4 and thereby prevent Par-4 from translocating GRP78 to the cell surface. We screened for Par-4 binding proteins: (a) that were up-regulated by NF-κB, and (b) that inhibited GRP78 translocation to the cell surface. We performed a yeast-two hybrid (Y2H) screen using a human lung cDNA library and full length Par-4 as bait. The interactions were further confirmed by a one-on-one Y2H analysis. UACA (7) emerged as a strong binding partner of Par-4 in the presence of highly selective growth medium (Figure 2A).
Figure 2. UACA binds to Par-4 and is regulated by NF-κB.

(A) UACA binds to Par-4 in one-on-one Y2H assay. Yeast cells were co-transformed with various constructs. 1: Positive Control (pB27-TCF4 and pP7-βcatenin); 2: Negative Control (pB27empty, which contains only the DBD; and pP7empty, which contains only the TA domain and is identical to pP6); 3: Negative Control (Par-4 + pP7empty); 4 : Negative Control (pB27empty + Sfrs11); 5 : Par-4 + Sfrs11; 6 : Negative Control (pB27empty + UACA); 7: Par-4 + UACA. The cells were then cultured in a 96-well plate; and the indicated serial dilutions were spotted on solid medium lacking tryptophan, leucine and histidine, and containing 0, 5, or 10 mM 3-amino-1,2,4-triazole (3AT). Sfrs11 is another prospective, albeit weaker, binding partner of Par-4. Interaction for two independent yeast clones is shown.
(B) UACA binds to Par-4 in human cell lines. Whole-cell lysates from the indicated cell lines were subjected to co-immunoprecipitation with Par-4 or p65/NF-κB (control) antibody. The immunoprecipitates were subjected to Western blot analysis. Lysates from each cell line were included as Input samples. UACA, ~160 kDa; p65, 65 kDa; Par-4, ~40 kDa; GRP78, ~78 kDa.
(C) UACA co-localizes with Par-4. H460 cells were subjected to ICC analysis for UACA (green fluorescence) and Par-4 (red fluorescence), or for calnexin (red fluorescence) and UACA (green fluorescence), and nuclei were stained with DAPI. The images were overlayed to determine co-localization (yellow fluorescence). Over 70 percent cells showed co-localization of Par-4 and UACA.
(D) NF-κB activity is essential for UACA expression. Western blot (WB) analysis for the indicated proteins was performed on whole-cell lysates from: MEFs from IKKβ−/− or IKKβ+/+ mice (left panel); NIH 3T3/Ras G12V or PC-3 cells infected for 18 h with IκB-SR adenovirus or GFP adenovirus (middle panels); or various cell lines treated with PS-1145 or DMSO for 24 h (right panels).
The interaction between Par-4 and UACA was validated by co-immunoprecipitation (co-IP) studies using various human cell lines. The Par-4 antibody immunoprecipitated Par-4 and co-immunoprecipitated UACA in all the cell lines (Figure 2B). The Par-4 antibody also co-immunoprecipitated UACA from whole-tissue extracts from the prostate and lung of C57/BL6 mice (Figure S2A). The UACA antibody immunoprecipitated UACA, and co-immunoprecipitated Par-4 protein (Figure S2B). Neither UACA nor Par-4 was co-immunoprecipitated with the p65/NF-κB control antibody. As expected from our previous studies (1), the Par-4 antibody also co-immunoprecipitated GRP78 (Figure 2B). These findings indicate that Par-4 binds to UACA in mammalian cells and tissues.
The interaction between Par-4 and UACA was further confirmed by immuno-cytochemical (ICC) analysis in cancer cells. UACA co-localizes with Par-4, as well as with calnexin, an ER resident protein (Figure 2C). Par-4 also co-localizes with calnexin (1). These findings indicate that Par-4 and UACA co-localize with each other. Consistently, Par-4 and UACA showed co-expression within the cytoplasmic compartment of cancer tissues (Figure S2C). Importantly, cell fractionation studies indicated that Par-4 and UACA were present in both the ER- and non-ER sub-cellular fraction of cells, and the Par-4 antibody co-immunoprecipitated UACA from the ER-fraction but not from the non-ER fraction (Figure S2D). Together, these studies provide evidence that Par-4 and UACA bind to each other.
To test whether the basal level of UACA expression is regulated by NF-κB in unstimulated mouse and human cells, IKKβ+/+ and IKKβ−/− MEFs were tested for UACA expression. IKKβ+/+ MEFs showed UACA expression, but IKKβ−/− MEFs showed severely diminished expression of UACA (Figure 2D). Lack of NF-κB activity in IKKβ−/− MEFs was confirmed by NF-κB reporter assays (Figure S2E). IκB-SR inhibited the expression of UACA in NIH 3T3/RasG12V cells or PC-3 cells (Figure 2D). Similarly, PS-1145 and Bay-11-7082 inhibited UACA expression in diverse cell lines (Figure 2D, Figure S2F). UACA expression was regulated by NF-κB at the RNA level (Figure S2G). Collectively, these findings indicate that UACA is up-regulated by NF-κB activity and binds to Par-4 in mouse and human cells.
Expression of UACA in normal and tumor tissues
We next examined UACA levels and p65/NF-κB nuclear expression in human tissues. UACA expression showed 3-fold or more elevation in prostate cancer specimens compared to normal counterpart tissues, as judged by IHC (Figures 3A and S3A). Similarly, UACA expression was markedly higher in lung adenocarcinoma and in squamous cell carcinoma relative to normal lung specimens (Figure 3B). Up-regulation of UACA was seen in all the grades of cancer and there was no further increase with tumor grade (Figure 3A). We consistently noted a correlation between nuclear expression of p65/NF-κB and high levels (≥3+) of UACA in the tissues (Figures 3C, left panel; and S3C). These data suggest that UACA expression and nuclear expression of p65/NFκB are particularly elevated in diverse cancers. Moreover, GRP78 levels were overall higher in cancer tissues than in normal tissues (Figure S3D). A majority of the cancer tissues that co-expressed ‘nuclear p65 and high levels of UACA’ showed ‘2+’ (rather than ≥3+) levels of GRP78 (compare Figure 3C, left and right panels).
Figure 3. UACA expression is elevated in cancer tissues.

(A,B,C) Expression of UACA, p65/NF-κB, or GRP78 in normal and cancer tissues. Sections of normal prostate (n=20) or grade 1 prostate adenocarcinoma (n=24) (A,C); and normal lung (n=10), lung adenocarcinoma (n=38) or squamous cell lung carcinoma (n=39) (B,C); were subjected to IHC with either a control antibody or UACA antibody (A,B,C), p65/NF-κB antibody (C, left panel), or GRP78 antibody (C, right panel), using DAB (brown), and nuclei were counterstained blue with hematoxylin. As expected, no staining was observed with the control antibody (Figure S3A), and detection of UACA with the UACA antibody was neutralized by corresponding UACA peptide (Figure S3B). Representative sections are shown at 100X (A and B). UACA or GRP78 expression in the normal and cancer tissues was scored as ‘≤ 1+’, ‘2+’, or ‘≥ 3+’. Percentage of tissues co-expressing ‘nuclear p65 expression and high (‘≥ 3+’) UACA’ (C, left panel), or ‘nuclear p65 expression/high (‘≥ 3+’) UACA’ and ‘≤ 1+’, ‘2+’, or ‘≥ 3+’ levels of GRP78 (C, right panel) is shown.
(D) UACA up-regulation correlates with NF-κB activation. Western blot (WB) analysis was performed on lysates prepared from freshly frozen prostate cancer or lung cancer tissues and corresponding matched normal tissue controls.
Western blot analysis indicated that UACA and XIAP (a surrogate marker for NF-κB activity) was up-regulated in prostate and lung cancer tissues relative to the corresponding normal tissues (Figure 3D). On the other hand, Par-4 levels were similar in the normal and cancer tissue pairs. These findings suggested that an increase in NF-κB activity correlates with an increase in UACA expression in cancer tissues. Similarly, we noted an increase in NF-κB activity and UACA expression in prostate cancer or lung cancer cell lines relative to corresponding normal cells (Figure S3E), and in NIH 3T3/RasG12V transformed cells relative to genetically matched control cells (Figure S3F).
UACA blocks apoptosis by Par-4
We investigated whether the interaction of intracellular Par-4 with UACA regulated the apoptotic effects of extracellular Par-4. To address this question, we knocked down the expression of UACA with siRNA in PC-3, H460, and A549 cell lines, and assessed the apoptotic effects of extracellular TRX-Par-4 protein added to the medium. UACA knock-down rendered A549 cells sensitive to extracellular TRX-Par-4 relative to TRX control (Figure 4A). PC-3 and H460 cells were rendered further sensitive to TRX-Par-4 protein following UACA knock-down with multiple RNAi approaches (Figure 4A and S4, data not shown). These findings indicate that UACA inhibits apoptosis by extracellular Par-4.
Figure 4. UACA knock-down induces apoptosis by Par-4.
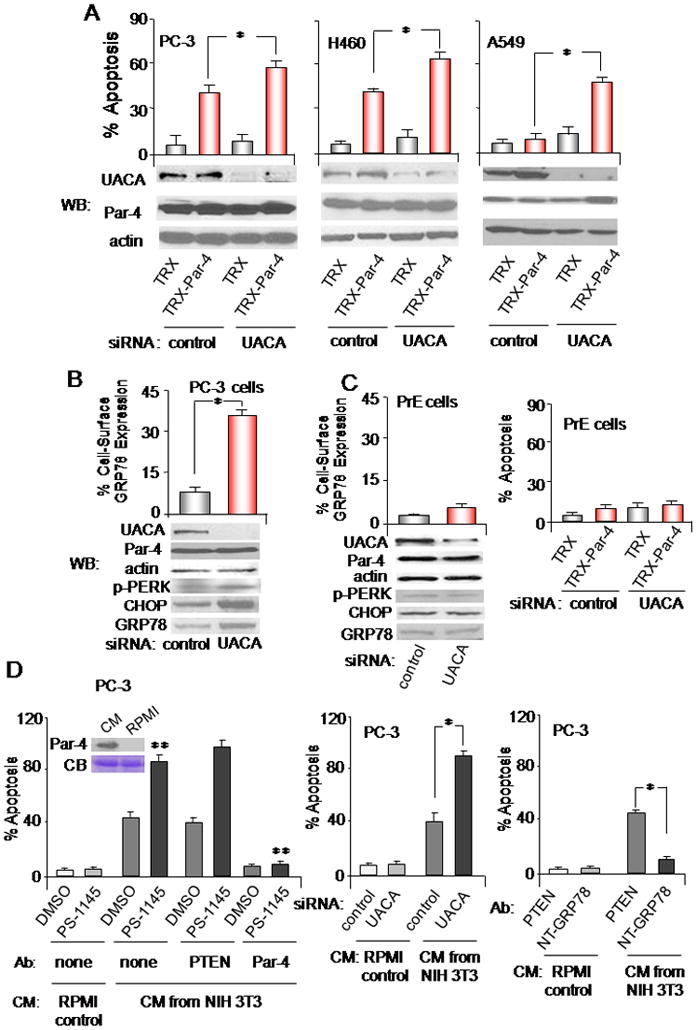
(A,B) UACA knock down affects cancer cells. The indicated cell lines were transfected with UACA siRNA or control siRNA duplexes. As indicated in Figure S4, transfection with two different siRNA duplexes and one shRNA was used to confirm the findings. (B) Up-regulation of GRP78 at the cell surface was determined by FACS analysis of unfixed cells. (A) The cells were treated with TRX or TRX-Par-4 (100 nM) for 24 h, and subjected to ICC for active caspase 3 and scored for apoptosis. Knock-down of UACA was confirmed by Western blot (WB) analysis.
(C) UACA knock-down does not affect normal cells. PrE cells were transfected with siRNA duplexes, and up-regulation of GRP78 at the cell surface was determined by FACS analysis of unfixed cells (left panel), knock-down of UACA was confirmed by Western blot (WB) analysis (left panel), or the cells were treated with TRX or TRX-Par-4 (100 nM) for 24 h, and scored for apoptosis (right panel).
(D) Effects of Par-4 protein secreted by mammalian cells. PC-3 cells were treated with CM from NIH 3T3 fibroblasts or with RPMI control medium, in the presence of PS-1145 or DMSO, and no antibody, PTEN antibody or Par-4 antibody (left panel), or GRP78 N-terminal antibody or control antibody (right panel). Moreover, the cells were transfected with UACA siRNA or control siRNA duplexes, then treated with CM from NIH 3T3 fibroblasts or with RPMI control medium for 24 h, and scored for apoptosis (middle panel). Par-4 in the CM was confirmed by Western blot analysis, using albumin in the Coomassie blue (CB) stained gel to normalize loading (left panel, inset). Each treatment used 5 nM secreted Par-4 protein, as judged by quantitative Western blot analysis (not shown).
Asterisk (*) indicates statistically significant (P < 0.001) difference by the Student t test; and (**) indicates that the effect is significant (P < 0.001) based on two-way ANOVA with data normality and equality of variance assumptions.
As UACA knock-down resulted in sensitization to apoptosis by extracellular Par-4, we investigated whether UACA prevents intracellular Par-4 from translocating GRP78 to the cell surface. UACA knock-down in PC-3 cells resulted in a significant (P < 0.001) increase in the number of GRP78 receptors at the cell surface (Figure 4B). These results imply that UACA modulates sensitivity to extracellular Par-4 preventing translocation of GRP78 to the cancer cell surface.
Primary prostate epithelial cells PrE, on the other hand, were resistant to cell surface GRP78 translocation and apoptosis by TRX-Par-4 after knock down of UACA (Figure 4C). Moreover, consistent with our previous observation that GRP78 cell surface trafficking is associated with an ER-stress response (1), we noted that expression of the ER-stress response proteins, GRP78, phospho-PERK and CHOP/GADD153 was elevated after UACA knock down in the cancer cells but not in the normal cells (Figure 4B and C). These findings indicate that UACA regulates ER-stress and GRP78 cell surface translocation in cancer cells but not in normal cells.
Our previous studies (1) have indicated that normal fibroblasts secrete Par-4 in their conditioned medium (CM). To confirm that Par-4 secreted by mammalian cells shows results similar to those seen with recombinant TRX-Par-4, we tested the CM from NIH 3T3 cells for apoptosis. The CM from NIH 3T3 cells but not RPMI 1640 growth medium, produced apoptosis of PC-3 cells (Figure 4D, left panel). The action of the CM was neutralized by the Par-4 antibody, indicating that the apoptotic activity of the CM was induced by Par-4. PS-1145 sensitized the cells to Par-4 in the CM (Figure 4D, left panel). The action of Par-4 in the CM was also enhanced by siRNA-mediated knock down of UACA (Figure 4D, middle panel). The action of Par-4 in the CM was abrogated by an amino-terminal GRP78 antibody, which blocks binding of extracellular Par-4 to the amino-terminus of cell surface GRP78 (1), but not with control PTEN antibody (Figure 4D, right panel). Collectively, these findings indicate that NF-κB activity and UACA negatively regulate apoptosis by Par-4 secreted naturally by mammalian cells.
We then interrogated whether UACA suppression following NF-κB inhibition was essential for enhanced apoptosis. A549 cells were transfected with RFP-UACA or RFP expression constructs driven by the CMV-promoter, then infected with IκB-SR or GFP adenovirus and treated with TRX-Par-4 or TRX protein. RFP-UACA but not RFP control vector rescued the cells from the apoptotic effect of IκB-SR plus TRX-Par-4 (Figure 5A) or IκB-SR plus the GRP78 carboxyl-terminal antibody (Figure 5B). RFP-UACA inhibited cell surface GRP78 levels that were induced by IκB-SR (Figure 5C). Together, these results indicate that suppression of UACA following inhibition of NF-κB activity is necessary for enhancing cell surface expression of GRP78 and apoptosis by Par-4.
Figure 5. UACA up-regulation by NF-κB is essential for apoptosis by Par-4.
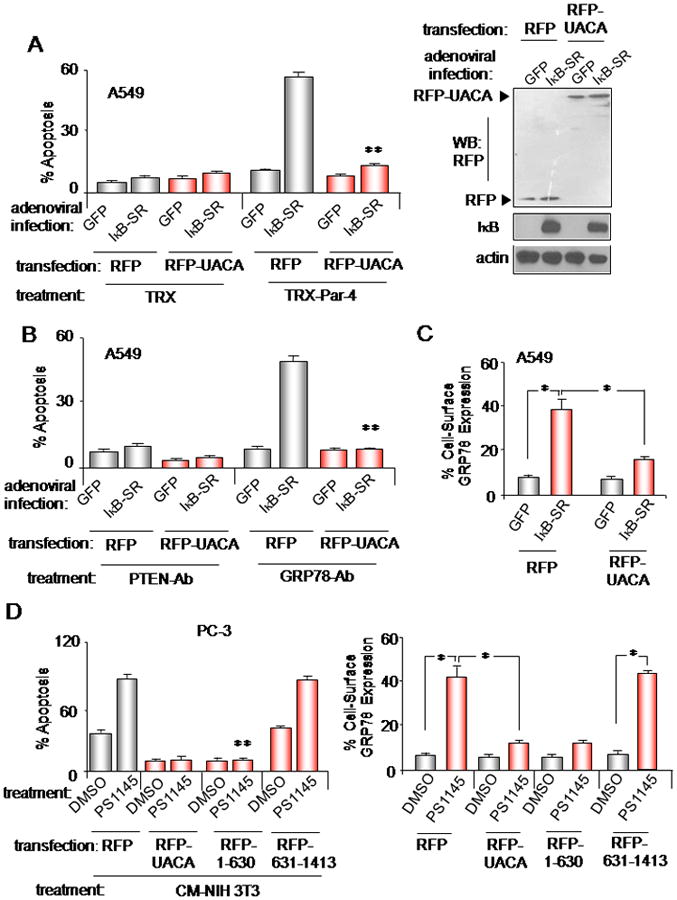
(A,B,C) UACA restoration inhibits GRP78 cell surface translocation and apoptosis. A549 cells were transfected with RFP-UACA- or RFP-expression constructs driven by the CMV-promoter and infected with IκB-SR or GFP adenovirus. Western blot analysis confirmed expression of the constructs (A, right panel). The cells were treated with TRX-Par-4 or TRX (A), or GRP78 agonistic antibody or PTEN antibody (B), and RFP+ cells were scored for apoptosis (A, B). Up-regulation of cell surface GRP78 was determined by FACS analysis of unfixed cells (C).
(D) Binding of UACA to intracellular Par-4 is essential for inhibition of cell surface GRP78 expression and apoptosis. Cells were transfected with expression constructs for RFP control, RFP-UACA, RFP-1-630 mutant UACA, which binds to endogenous Par-4, or RFP-mutant UACA, which failed to bind to Par-4 (see Figure S5), then treated with PS-1145 or DMSO control. Up-regulation of GRP78 at the cell surface was determined by FACS analysis of unfixed cells (D, right panel). The cells were treated with CM from NIH 3T3 (containing 5 nM secreted Par-4) or RPMI control medium, and RFP+ cells were scored for apoptosis (D, left panel).
Asterisk (*) indicates statistically significant (P < 0.001) difference by the Student t test and (**) indicates significance (P < 0.001), based on two-way ANOVA with data normality and equality of variance assumptions.
We next tested whether binding of UACA to intracellular Par-4 was necessary to rescue the cells from apoptosis by extracellular Par-4. These experiments used full-length UACA and a deletion mutant of UACA (1-630aa), which could bind to endogenous Par-4, or another deletion mutant of UACA (631-1413aa) that failed to bind to endogenous Par-4 (Figure S5). Consistently, full-length UACA and 1-630aa mutant, but not 631-1413aa mutant, prevented PS-1145-inducible GRP78 translocation to the cell surface (Figure 5D, right panel), and rescued the cells from the apoptotic action of naturally secreted Par-4 (±PS1145) in the CM from NIH 3T3 fibroblasts (Figure 5D, left panel). Collectively, these findings indicate that UACA binding to intracellular Par-4 is essential for inhibition of GRP78 translocation to the cell surface and apoptosis by extracellular Par-4.
Discussion
GRP78 is an ER-resident protein involved in cell survival and autophagy (8,9,10). GRP78 is selectively expressed at the plasma membrane of cancer cells owing to increased ER stress caused by cumulative genetic aberrations and altered metabolic dependency (8). Par-4, localized at the ER, binds and promotes the translocation of GRP78 to the cancer cell surface (1). The present study uncovered UACA as a Par-4 binding protein, which sequesters Par-4 and prevents it from translocating GRP78 to the cell surface. Accordingly, suppression of UACA expression by RNAi led to GRP78 translocation to the cell surface and sensitization of the cells to apoptosis by extracellular Par-4 protein or GRP78 agonistic antibody. Thus, UACA is a functional regulator of sensitivity to extracellular inducers of apoptosis in cancer cells. Importantly, UACA expression is regulated by NF-κB activity, which is elevated in cancer cells, and NF-κB-specific inhibitors cause suppression of UACA expression to promote GRP78 translocation to the cell surface. The endogenous NF-κB-UACA-Par-4-GRP78 pathway was recapitulated by using vincristine, which activates endogenous Par-4 by PKA-mediated phosphorylation and thereby inhibits NF-κB activity (Figure S6). Because cell surface GRP78 also responds to apoptosis by the GRP78 agonistic antibody and several other growth inhibitory proteins and small molecules (6,11), our findings imply that inhibition of the NF-κB pathway or UACA may be an effective strategy for sensitization and selective targeting of cancer cells. The NF-κB pathway is known to promote cell survival by induction of proteins (Bcl2, BclXL, A1/BFL1, c-FLIP, and IAPs) that inhibit progression of apoptotic pathways at the downstream steps involving intracellular changes in mitochondrial membrane potential or caspase activation. This study indicates that NF-κB activity inhibits apoptosis by naturally secreted/extracellular Par-4 at the proximal step of receptor-ligand interaction by down-regulating the translocation of GRP78 to the cell surface (Figure S7).
NF-κB activation plays a pivotal role in anti-apoptosis, tumor promotion and progression, and therapeutic resistance in diverse cancers, and NF-κB inhibitors are considered promising candidates in cancer therapy (4,5). We noted that UACA protein levels were elevated in cancer tissues and cell lines relative to counterpart normal tissues and cells, and elevated UACA expression correlated with p65/NF-κB nuclear localization/activity. These observations are in striking contrast with the reduced expression of UACA reported recently in lung tumors, in which NF-κB activity was not revealed (12). Furthermore, a majority of cancer tissues that co-expressed ‘nuclear p65 and high levels of UACA’ showed relatively lower (i.e., 2+ rather than ≥3+) levels of GRP78. Inhibition of UACA expression by RNAi led to an ER-stress response, elevated total and cell surface levels of GRP78 and sensitization to apoptosis by extracellular Par-4 in cancer cells (Figure 4B). Consistently, all the treatments that cause UACA down-regulation result in increased levels of total GRP78 in cancer cells (Figure S6E). GRP78 is a regulator of the unfolded protein response (UPR) pathway and is also induced by UPR signaling. An overall increase in total GRP78 may contribute to elevated cell surface expression of GRP78. However, intracellular Par-4 function is essential for translocation of GRP78 to the cell surface (Figure S6C and reference 1). Increased levels of CHOP (a downstream pro-apoptotic UPR signaling component) in cancer cells in response to UACA inhibition (Figure 4B) may contribute to the pro-apoptotic effect of extracellular Par-4. The precise molecular link between UACA inhibition and increased phosphorylation of PERK (Figure 4B), a proximal sensor of UPR, is currently being investigated.
The levels of GRP78 were overall lower in normal cells and tissues relative to cancer cells and tissues, and although the NF-κB pathway may regulate UACA in normal cells, UACA knock-down in normal cells did not result in GRP78 translocation to the cell surface or sensitization to apoptosis by extracellular Par-4. This implies that the NF-κB-UACA-ER-stress-GRP78 translocation pathway is truncated in normal cells but intact and fully functional in cancer cells. Together with the finding that a knock out mouse for UACA lacks a clear phenotype (13), these data indicate that in normal cells either UACA is dispensable or other redundant gene(s) compensate for loss of UACA function. UACA suppression combined with extracellular molecules that function via cell surface GRP78 signaling may therefore be an effective strategy for selective elimination of cancer cells.
In summary, our findings indicated that inhibition of GRP78 translocation to the cell membrane is a downstream effect of NF-κB activation. The action of NF-κB is mediated through UACA, which sequesters Par-4 and prevents it from translocating GRP78 to the cell surface. Importantly, this pathway can be targeted to overcome apoptosis-resistance in cancer cells. Thus, this study suggests a novel link between NF-κB activity, UACA levels, and resistance to extracellular inducers of apoptosis that act via cell surface GRP78 in cancer cells.
Supplementary Material
Acknowledgments
This study was supported by KLCR grant and NIH/NCI grant CA060872 (to VMR).
Footnotes
The authors declare no conflicts
References
- 1.Burikhanov R, Zhao Y, Goswami A, Qiu S, Schwarze SR, Rangnekar VM. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell. 2009;138:377–88. doi: 10.1016/j.cell.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gurumurthy S, Goswami A, Vasudevan KM, Rangnekar VM. Phosphorylation of Par-4 by protein kinase A is critical for apoptosis. Mol Cell Biol. 2005;25:1146–61. doi: 10.1128/MCB.25.3.1146-1161.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao Y, Burikhanov R, Brandon J, Qiu S, Shelton BJ, Spear B, et al. Systemic Par-4 inhibits non-autochthonous tumor growth. Cancer Biol Ther. 2011;12:152–57. doi: 10.4161/cbt.12.2.15734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilmore TD, Garbati MR. Inhibition of NF-kappaB signaling as a strategy in disease therapy. Curr Top Microbiol Immunol. 2011;349:245–63. doi: 10.1007/82_2010_105. [DOI] [PubMed] [Google Scholar]
- 5.Kim HJ, Hawke N, Baldwin AS. NF-kappaB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006;13:738–47. doi: 10.1038/sj.cdd.4401877. [DOI] [PubMed] [Google Scholar]
- 6.Misra UK, Mowery Y, Kaczowka S, Pizzo SV. Ligation of cancer cell surface GRP78 with antibodies directed against its COOH-terminal domain up-regulates p53 activity and promotes apoptosis. Mol Cancer Ther. 2009;8:1350–62. doi: 10.1158/1535-7163.MCT-08-0990. [DOI] [PubMed] [Google Scholar]
- 7.Yamada K, Senju S, Nakatsura T, Murata Y, Ishihara M, Nakamura S, et al. Identification of a novel autoantigen UACA in patients with panuveitis. Biochem Biophys Res Commun. 2001;280:1169–76. doi: 10.1006/bbrc.2001.4189. [DOI] [PubMed] [Google Scholar]
- 8.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434:181–88. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cook KL, Shajahan AN, Wärri A, Jin L, Hilakivi-Clarke LA, Clarke R. Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer Res. 2012;72:3337–49. doi: 10.1158/0008-5472.CAN-12-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grkovic S, O’Reilly VC, Han S, Hong M, Baxter RC, Firth SM. IGFBP-3 binds GRP78, stimulates autophagy and promotes the survival of breast cancer cells exposed to adverse microenvironments. Oncogene. 2012 doi: 10.1038/onc.2012.264. [DOI] [PubMed] [Google Scholar]
- 11.Sato M, Yao VJ, Arap W, Pasqualini R. GRP78 signaling hub a receptor for targeted tumor therapy. Adv Genet. 2010;69:97–114. doi: 10.1016/S0065-2660(10)69006-2. [DOI] [PubMed] [Google Scholar]
- 12.Moravcikova E, Krepela E, Prochazka J, Rousalova I, Cermak J, Kenkova K. Down-regulated expression of apoptosis-associated genes APIP and UACA in non-small cell lung carcinoma. Int J Oncol. 2012;4:2111–21. doi: 10.3892/ijo.2012.1397. [DOI] [PubMed] [Google Scholar]
- 13.Sakai T, Liu L, Teng X, Ishimaru N, Mukai-Sakai R, Tran NH, et al. Inflammatory disease and cancer with a decrease in Kupffer cell numbers in Nucling-knockout mice. Int J Cancer. 2010;126:1079–94. doi: 10.1002/ijc.24789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.