

Abstract
Leptin, the product of the obese gene, regulates energy homeostasis by acting primarily at the level of the hypothalamus. Leptin action through its receptor involves various pathways including the signal transducer and activator of transcription (STAT3), phosphatidylinositol 3-kinase (PI3K), and phosphodiesterase 3B (PDE3B)-cAMP signaling in the CNS and peripheral tissues. In the hypothalamus, leptin stimulates STAT3 activation, and induces PI3K and PDE3B activities, among others. We have previously demonstrated that PDE3B activation in the hypothalamus is critical for transducing anorectic and body weight reducing effects of leptin. Similarly, PI3K has been implicated toplay a critical role in leptin signaling in the hypothalamus. Whereas in insulin signaling pathway, PI3K is known to be an upstream regulator of PDE3B in non-neuronal tissues, it is still unknown whether this is also the case for leptin signaling in the hypothalamus. To address this possibility, the effect of wortmannin, a specific PI3K inhibitor, was examined on the leptin-induced PDE3B activity in the hypothalamus of male rats. Intracerebroventricular (icv) injection of leptin (4 μg) significantly increased PDE3B activity by 2-fold in the hypothalamus as expected. However, prior administration of wortmannin completely reversed the stimulatory effect of leptin on PDE3B activity in the hypothalamus. To demonstrate whether leptin stimulates p-Akt levels and there by a possible upstream regulator of PDE3B, we examined the effects of icv leptin on p-Akt levels in the hypothalamus and compared that with the known stimulatory effect of insulin on p-Akt. We observed that insulin increased p-Akt levels but leptin failed to do so although it increased p-STAT3 levels in the rat hypothalamus. Immunocytochemistry confirmed the biochemical finding in that leptin failed but insulin increased the number of p-Akt positive cells in various hypothalamic nuclei. Altogether these results implicate PI3K but not Akt as an upstream regulator of the PDE3B pathway of leptin signaling in the rat hypothalamus.
Keywords: leptin, insulin, PI3K, p-Akt, PDE3B, p-STAT3, hypothalamus
Introduction
Leptin signaling in the hypothalamus is obligatory for normal energy homeostasis (1–4). Leptin administration centrally or peripherally decreases food intake and body weight in a variety of animals (1). The initial discovery of the leptin receptor as a member of the class-1 cytokine receptor super-family resulted in prompt identification of the JAK2-STAT3 pathway as a major pathway of leptin signaling in various tissues including the hypothalamus (1). While the significance of the JAK2–STAT3 pathway is unequivocal in the maintenance of normal body weight (4–6), we have demonstrated that leptin’s action in the hypothalamus is also mediated through an insulin-like signaling pathway involving induction of PI3K and phosphodiesterase-3B (PDE3B) activities and a reduction of cAMP levels (7). Our studies suggest that this molecular mechanism mediates most, if not all, of the leptin’s satiety action (7). Thus, cilostamide, a specific PDE3 inhibitor, reversed the anorectic and body weight reducing effects of leptin, and reversed leptin-induced STAT3 activation in the hypothalamus (7). In addition, PDE3B pathway appears to playan important role in mediating leptin action in prooiomelanocortin (POMC) and neurotensin (NT) neurons in the hypothalamus (8). Although the findings, such as, leptin stimulates hypothalamic PI3K activity (7, 9), and PI3K inhibitors reverse the satiety action of leptin (9) suggest that PI3K is a key enzyme in leptin signaling in the hypothalamus, it is not known whether PI3K is an upstream component of the PDE3B pathway. Since PI3K is an upstream regulator of PDE3B in non-neuronal tissues (10–12), we hypothesize that activation of PI3K is a critical step in PDE3B pathway of leptin signaling in the hypothalamus.
The Akt (also known as protein kinase B) is a downstream target and effector of PI3K action (13). It is thought to mediate many metabolic, mitogenic and anti-apoptotic effects of insulin, IGF-1, and IL-3, and other growth factors and cytokines (14–16). In adipocytes, insulin stimulates MAPK, p70S6 kinase and Akt through wortmannin sensitive mechanisms, i.e., PI3K mechanism, but the PDE3B activation by insulin is due to PKB phosphorylation (17), suggesting the role of PI3K-Akt-PDE3B pathway in insulin signaling. However, leptin’s role in Akt activation is not clear. For example, in rat primary hepatocytes, activation of Akt by leptin requires the presence of a cAMP-elevating hormone (glucagon) (12). It is still unknown whether Akt is an upstream component of the PDE3B activation during leptin signaling in the hypothalamus.
Thus, to begin to understand the regulatory mechanism(s) of the PDE3B pathway of leptin signaling in the hypothalamus, the present study sought to examine if PI3K and Akt were upstream regulators of PDE3B. To this end, we examined if PI3K inhibitor wortmannin reverses the effect of leptin on PDE3B activity and if leptin activates Akt in the rat hypothalamus. Because insulin has been demonstrated to increase both PI3K and p-Akt in the hypothalamus (18), we also examined the effect of insulin on p-Akt levels as a control experiment. Here we show that PI3K but not Akt is upstream component of the PDE3B pathway of leptin signaling in the rat hypothalamus.
Materials and methods
Adult male Sprague-Dawley rats, weighing ~250 g, obtained from Taconic Farms (Germantown, NY) were housed individually in a light (lights on 0500 h to 1900 h) and temperature (22 °C)-controlled room with food (pelleted Purina rat chow) and water available ad libitum. After 7 days of acclimatization, rats were subjected to the following experiments, all of which were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Experiment 1: effect of wortmannin on PDE3B activity in the hypothalamus
Rats were implanted stereotaxically with stainless-steel cannula into the third cerebroventricle under pentobarbital anesthesia asdescribed previously (19). Following ten days of recovery, rats were fasted for 24 hours and then injected intracerebroventricularly (icv) with wortmannin (WORT, 0.01 nmol, Refs. 9, 18) or dimethyle sulfoxide (DMSO vehicle, 100%) followed 30 min later by leptin (4 μg; A. F. Parlow, National hormone and Peptide Program, Torrance, CA) or artificial cerebrospinal fluid (aCSF) resulting in four groups: DMSO + aCSF, DMSO + leptin, WORT + aCSF and WORT + leptin. Forty-five minutes later, rats were killed by decapitation; brains were removed immediately and the medial basal hypothalamus (MBH) were dissected out (20), frozen in liquid nitrogen, and kept at −80 °C until processed for protein extraction and PDE3B assay. The MBH tissue was bounded by rostrally by the posterior border of the optic chiasma, caudally by the mammillary bodies, and laterally by the lateral sulcus, and cut to a depth of approximately 2 mm.
Experiment 2: Effects of leptin and insulin on p-AKT and p-STAT3 levels in the hypothalamus at 45 minpost injection
Rats implanted with indwelling third cerebroventricular cannula were fasted for 24 hours and then injected icv with either leptin (4 μg) or insulin (10 mU; Human insulin, Eli Lilly) in 4 μl saline or saline (4 μl) alone. Forty-five minutes later, the MBH was dissected out, frozen in liquid nitrogen, and kept at −80 °C until processed for protein extraction.
To examine whether p-Akt and p-STAT3 activation was altered in specific regions of the hypothalamus, some rats from each group was anesthetized with pentobarbital and perfused intracardially with 0.9% saline followed by ice-cold 4% paraformaldehyde in 0.1 M phosphate buffer. The brains were post fixed in the same fixative for overnight and then kept in 20% sucrose solution at 4 °C until they sank. Thereafter, brains were frozen on dry ice, and coronal 25-μm free floating sections were cut in six series through the hypothalamus on a freezing microtome (Leica Sliding Microtome; Leica Microsystems GmBH, Wetzlar, Germany), and stored in cryoprotectant at −20 °C until use. One series was subjected to immunocytochemistry (ICC) for each antibody (p-Akt or p-STAT3).
Experiment 3: Effects of leptin and insulin on p-Akt and p-STAT3 levels in the hypothalamus at 10 min post injection
Because leptin failed to increase hypothalamic p-Akt levels at 45 min post injection whereas insulin significantly increased p-Akt levels, this experiment tested if leptin increased p-Akt at earlier time point. This experiment was similar to that described previously (experiment 2) except that the effects of leptin and insulin on p-Akt and p-STAT3 levels in the hypothalamus were measured at 10 min post injection.
To examine p-Akt and p-STAT3 activation by ICC, some animals were perfused with saline followed by 4% paraformaldehyde. The brains were postfixed, equilibrated with sucrose solution, frozen on dry ice, and processed for sectioning as described previously (experiment 2).
Measurement of PDE3B activity
PDE3B assay was done using a previously described protocol (11, 12), which has been used by us (7, 21). The MBHs were homogenized in 500 μl of a homogenization buffer (10 mM sodium phosphate, pH 7.2; 50 mM NaF, 150 mM NaCl; 3 mM benzamidine, 2 mM EDTA; 0.1% Triton X-100; 0.5% Lubrol; 5 μg/ml leupeptin and 20 μg/ml Pepstatin A) with a polytron homogenizer. The homogenate was centrifuged at 38,000 g for 45 min at 4°Cusing an Optima TLX ultracentrifuge. The pellet was re-suspended in 200 μl of homogenization buffer and protein was measured by Bio-Rad protein assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA) (22).
For the PDE3B assay, 100 μg of re-suspended MBH protein in 50 μl sample volume was added to a 200 μl reaction mixture [50 μl buffer A: 100 mM MOPS, pH 7.5, 4 mM EGTA, 1 mg/ml BSA; 50 μl buffer B: 100 mM MOPS, 75 mM MgAcetate, 100,000 c.p.m. 3H-cAMP (Perkin-Elmer, Boston, MA, USA); 1 μM cAMP; 100 μl H2O] containing the specific PDE3 inhibitor cilostamide (3μM) or no inhibitor. Reaction tubes were incubated in 30 °C water bath for 10 min followed by 1 min incubation at 100 °C. Tubes were then chilled on ice and 10 μl (25 mg/ml) snake venom (Sigma Chemicals, St. Louis, MO) was added, and incubated in 30 °C water bath for 10 min. To these tubes, 250 μl of low salt buffer (20 mM Tris, pH 6.8) was added. Whole reaction mixture was then added to DEAE-Sephadex A-25 column, and 3H-cAMP was eluted from the column by washing with low salt buffer. Scintillation fluid (3ml) was added to eluate and radioactivity was counted on a scintillation counter (Packard). PDE3B activity was calculated as mole cAMP hydrolyzed per min/ml using an Excel Program. PDE3B activity was adjusted to the levels of PDE3B protein in the sample and calculated as pmol/min/densitometric unit, and then the values were expressed in relation to DMSO + aCSF control. PDE3B protein levels were measured by standard Western blotting. First, protein was immunoprecipitated with a PDE3B antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and then immunoprecipitates were subjected to SDS-PAGE, followed by transfer of the resolved polypeptides to polyscreen PVDF membranes (NEN Life Science Products). Total amount of PDE3B protein was measured by incubating the blot with PDE3B antibody followed by enhanced chemoluminescence as described by the manufacturer (Perkin-Elmer). Immunoreactive bands were scanned and analyzed by NIH Image software.
Western blotting
Western blotting was done following standard procedures (23), which has been used previously by us (20, 21). Briefly, MBH was homogenized in ice-cold extraction buffer (50 mM HEPES, 150 mM NaCl, 20 mM Na-pyrophosphate, 20 mm B-glycerophosphate, 10 mM NaF, 2 mM Na3VO4, 2 mM EDTA, 1% IGEPAL, 10% glycerol, 2 mM phenylmethylsulfonyl fluoride, 1 mM MgCl2, 1mM CaCl2, 10 μg/ml leupeptin, and 10 μg/ml aprotinin). The homogenates were then rotated end-over-end at 4 °C for 60 min, and centrifuged at 10,000g for 10 min at 4 °C. Aliquots of the supernatants were used for Western blot analysis. The samples treated with Laemmli sample buffer were subjected to SDS-PAGE (8 μg for phospho-Akt with 10% gel and 50μg for phospho-STAT3 with 7.5% gel) and then transferred to PVDF membranes, blocked in 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween 20(TBST), pH 7.5. After blocking, the membranes were incubated overnight with phospho-STAT3Tyr705 (Santa Cruz Biotechnology, 1:600), phospho-AktSer473 (Cell Signaling Technology, 1:3,000), or phospho-AktThr308 (Cell Signaling Technology, 1:2,000) at 4 °C followed by incubation for 90 min with HRP-conjugated secondary antibodies at room temperature. Immunoreactive bands were visualized by Western Lightning chemiluminescence Reagent Plus as described by the manufacturer (Perkin Elmer). Immunoreactive bands were scanned and analyzed by NIH Image Software. Membranes were stripped and then blotted with STAT3 (Santa Cruz Biotechnology, 1:2000) or Akt (Cell Signaling Technology, 1:1000) antibody as appropriate to measure STAT3 and Akt levels. For calculation, p-STAT3 and p-Akt (ser473, thr308) protein levels were first normalized with STAT3 and Akt levels, respectively; and then expressed as relative to control saline group.
ICC for p-AKT and p-STAT3 localization in the hypothalamus
The ICC localization of p-STAT3 immunoreactive protein in the hypothalamus was done as described previously by Munzberg et al. (24) and recently adopted by us (25). Briefly, free floating tissue sections were pretreated with 1% NaOH and 1% H2O2 in H2O for 20 min followed by 10 min each of 0.3% glycine and 0.03% sodium dodecyl sulfate. Sections were blocked for 1 hr with blocking solution (5% normal goat serum in PBS, 1% BSA, 0.4% Triton X-100), followed by incubation with p-STAT3 antibody (1:3,000; Cell Signalling Technology, Inc., Danvers, MA) for overnight at 4 °C. On the next day, the sections were washed, incubated with biotinylated secondary goat antirabbit antibody (1:4000) for 1 hr, and then treated with avidin-biotin-complex solution for 1 h. Finally, the signal was developed by diaminobenzidine solution containing 0.6% nickel sulfate, giving a dark-blue precipitate. For ICC localization of pAktSer473 immunoreactive protein in the hypothalamus, free floating tissue sections were treated with 0.5% NaOH and 2% H2O2 in H2O for 20 min at RT followed by 2 hr with blocking solution (10% normal goat serum in KPBS, 1% BSA, 0.4% Triton X-100) at RT. Sections were then incubated with pAktSer473 antibody (1:400; Cell Signaling Technology, Inc., Danvers, MA) in antibody dilution buffer (5% normal goat serum in KPBS, 0.5% BSA, 0.4% Triton X-100) for ~38 hours at 4 °C followed by washing in KPBS and incubation with secondary antibody (goat anti rabbit, 1:1000, Vector Lab) in antibody dilution buffer for 90 min at RT. Sections were washed and incubated in Vectastain Elite ABC reagent (Vector Lab) in KPBS containing 0.4% Triton X-100 for 90 min at RT. Finally, sections were washed and signals were developed by incubating in DAB solution containing 0.6% nickel chloride, giving a brown precipitate.
Cell counting and quantification
As stated previously, brain sections were cut in 6 series through the MBH and one series was subjected to ICC with each antibody. All sections were organized systemically in a rostral-to-caudal-manner according to the rat brain atlas (26), and then counted under light microscope for p-Akt positive cells in specific areas of the MBH, including the ARC, ventromedial nuclei (VMN), and dorsomedial nuclei (DMN), lateral hypothalamic area (LHA), ventral premammilary nucleus (PMv), ventral tuberomammillary nucleus (VTM) and paraventricular nucleus (PVN). Cells were counted from both sides of the brain in each section. The data are presented as total number of p-Akt positive cells per section for each nucleus in one of six series in each animal. . Approximate bregma levels for the sections used in counting were as follows: PVN: −1.80 to −2.12 mm; LH-R: −1.80 to −2.12 mm; LH-M: −2.12 to −2.80 mm; LH-C: −2.80 to −3.40 mm; ARC-R: −2.20 to −2.80 mm; ARC-M: −2.80 to −3.45 mm; ARC-C: −3.45 to −4.16 mm; VMN: −2.30 to −3.60 mm; DMN: −2.56 to −3.40 mm; PMv: −3.80 to −4.16 mm; and VTM: −3.80 to −4.52 mm (according to the Rat Brain Atlas, Ref. 26).
Data analysis
All values are expressed as means ± standard error (SE). Statistical significance of differences was analyzed by randomized one-way ANOVA followed by Student-Newman-Keuls multiple range test. Comparisons with p < 0.05 were considered to be significant.
Results
Effect of wortmannin on leptin-induced PDE3B activity in the rat hypothalamus
Intracerebroventricular injection of leptin increased hypothalamic PDE3B activity by 2 olds (p < 0.05) as compared to control vehicle injection, as expected. Prior treatment with WORT completely reversed the stimulatory effect of leptin on PDE3B activity in the hypothalamus. WORT alone had no effect on PDE3B activity in the hypothalamus (Fig 1).
Fig. 1.
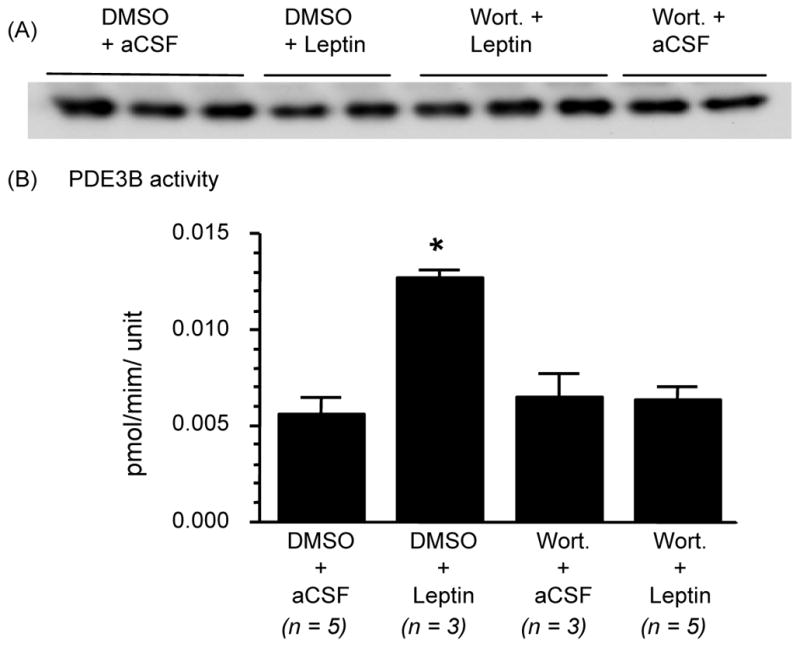
Wortmannin (Wort.), a PI3K inhibitor, reverses the effect of leptin on PD3B activity in the hypothalamus. Rats fasted for 24 hours were injected ICV with DMSO or Wort. (0.01 nmol) followed 30 min later by leptin (4 μg) or aCSF (artificial cerebrospinal fluid). (A) Immunoreactive bands for PDE3B protein in the medial basal hypothalamus extracts obtained by immunoprecipitation and western blotting. PDE3B protein levels were used to normalize PDE3B activity. (B) PDE3B activity adjusted to the level of PDE3B protein in the sample and expressed as pmol/min/densitometric unit. Values are mean ± SEM for the number of animals indicated in parentheses. *P < 0.05 versus all other groups.
Changes in p-Akt and p-STAT3 levels in the hypothalamus after 45 min of leptin or insulin injection
Because phosphorylation of Akt could occur at serine or threonine sites, we examined if leptin phosphorylates Akt either one or both sites in the hypothalamus. Icv injection of leptin did not increase Akt phosphorylation at serine or threonine sites (Fig 2). In contrast, insulin significantly increased (p < 0.05) both p-Akt levels in the hypothalamus. On the other hand, whereas leptin significantly increased (p < 0.05) p-STAT3 levels in the hypothalamus, insulin failed to do so (Fig. 2).
Fig. 2.

Changes in phosphorylation of Akt at Thr308 or Ser473 and that of STAT3 in the hypothalamus at 45 min after ICV injection of leptin (4 μg), insulin (10 mU) or saline. (A) Western blot of p-Akt (Thr308), p-Akt (Ser473), Akt, p-STAT3 and STAT3. (B) Densitometric analysis of the immunoreactive bands for p-Akt (Thr308), p-Akt (Ser473) and p-STAT3. p-Akt and p-STAT3levels were first normalized to Akt and STAT3 levels, respectively, and then expressed as relative to saline control group. Values are mean ± SEM for Values are mean ± SEM for the number of animals indicated in parentheses. *P < 0.05 versus all other groups.
To address the possibility that the effect of leptin on p-Akt levels may be confined to a specific hypothalamic sites, and p-Akt levels in whole hypothalamic extract could compromise the result, we examined if leptin induced the number of p-Akt positive cells in specific hypothalamic site(s) by ICC. Similarly, we also assessed if insulin treatment induced the number of p-STAT3 positive cells in specific hypothalamic site(s). As expected, leptin significantly increased the number of p-STAT3 positive cells in the ARC and in VMN, DMN, lateral hypothalamus (LH, data not shown), and ventral premammillary nucleus (PMv) as compared to saline control (Fig. 3). However, leptin treatment did not increase the number of p-Akt positive cells in any of these hypothalamic nuclei (Figs 4–6). There was a small number of p-Akt positive cells seen scattered through the hypothalamus in saline treated animals. In contrast, insulin significantly increased the number of p-Akt positive cells in the above mentioned hypothalamic nuclei as compared to saline treated animals (Figs. 4–6). Insulin also increased the number of p-Akt positive cells in the PVN and VTM. However, insulin failed to increase the number of p-STAT3 positive cells in the hypothalamus (Fig. 3). These results completely corroborate with the biochemical findings and suggest that while leptin increased p-STAT3 levels, it failed to increase p-Akt at 45 min post-injection. On the other hand, insulin significantly increased p-Akt but not the p-STAT3 levels in the hypothalamus.
Fig. 3.

Representative photomicrographs showing p-STAT3 positive cells in hypothalamic sections at approximately bregma -2.8(A-C) and -4.16 (D–F) from saline, leptin or insulin-treated rats that were fasted 24 hours and injected ICV with leptin (4 μg), insulin (10 mU) or saline and killed 45 min later. Scale bars, 100 μm. ARC, arcuate nucleus; VMN, ventromedial nucleus; DMN, dorsomedial nucleus; PMv, ventral premammillary nucleus; 3v, third ventricle. In this and all other figures showing immunocytochemistry, bregma are indicated in relation to the rat brain atlas (The rat brain in stereotaxic coordinates) by Paxinos and Watson (Ref. 26).
Fig. 4.

Representative photomicrographs showing p-Akt (Ser473) positive cells in the hypothalamic sections at approximately bregma −2.0 (A–C) and −2.8 (D–F) from saline, leptin or insulin-treated rats that were fasted 24 hours and injected ICV with leptin (4 μg), insulin (10 mU) or saline and killed 45 min later. Scale bars, 100 μm. PVN, paraventricular nucleus; ARC, arcuate nucleus; VMN, ventromedial nucleus; DMN, dorsomedial nucleus; 3v, third ventricle.
Fig. 6.

Changes in the number of p-Akt (Ser473) positive cells in various hypothalamic sites of saline, leptin or insulin-treated rats. One of six series from each animal was analyzed. ARC, arcuate nucleus; VMN, ventromedial nucleus; DMN, dorsomedial nucleus; LH, lateral hypothalamus; PVN, paraventricular nucleus; PMv, ventral premammillary nucleus; VTM, ventral tuberomammillary nucleus. R, M and C for respective nuclei represent rostral, middle and caudal part, respectively. Values represent the mean ± SEM for 3 animals per group.*P < 0.05 vs. saline group.
Changes in p-Akt and p-STAT3 levels in the hypothalamus after 10 min of leptin or insulin injection
As observed at 45 min post injection, whereas p-STAT3 levels were significantly increased (P <0.05; Fig. 7), the levels of p-Akt (ser473) and p-Akt (Thr308) in the hypothalamic extracts remained unchanged at 10 min after leptin injection (Fig. 7). Similarly, insulin significantly increased both p-Akt (ser473) and p-Akt (Thr308) levels (P < 0.05) but not p-STAT3 levels in the hypothalamus at 10 min post-injection (Fig. 7).
Fig. 7.

Changes in phosphorylation of Akt at Thr308 or Ser473 and that of STAT3 in the hypothalamus at 10 min after ICV injection of leptin (4 μg), insulin (10 mU) or saline. (A) Western blot of p-Akt (Thr308), p-Akt (Ser473), and Akt, p-STAT3 and STAT3. (B) Densitometric analysis of the immunoreactive bands for p-Akt (Thr308). (C) Densitometric analysis of the immunoreactive bands for p-Akt (Ser473). (D) Western blot of p-STAT3 and STAT3. (E) Densitometric analysis of the immunoreactive bands for p-STAT3. p-Akt and p-STAT3 levels were first normalized to Akt and STAT3 levels, respectively, and then expressed as relative to saline control group. Values are mean ± SEM for Values are mean ± SEM for the number of animals indicated in parentheses. *P < 0.05 versus all other groups.
In this ICC study the exact number of p-Akt or p-STAT3 positive cells were not counted, instead we only made gross comparison between the groups. There were some p-Akt positive cells in the hypothalamus of saline treated group. Leptin treatment failed to increase the number of p-Akt positive cells in various hypothalamic nuclei, but it increased the number of p-STAT3 positive cells at 10 min post-injection, as expected (Figs 8 and 9). On the other hand, insulin increased the number of p-Akt positive cells without increasing the number of p-STAT3 positive cells ((Figs 8 and 9). Importantly, as seen at 45 min post-injection, insulin also increased p-Akt positive cells in the PMv and VTM at 10 min post-injection (Fig. 9).
Fig. 8.

Representative photomicrographs of p-Akt (Ser473) positive cells (A–C) and p-STAT3 positive cells (D–F) in the hypothalamic sections at approximately bregma −2.8 from saline, leptin or insulin-treated rats. Rats fasted for 24 hours were injected ICV with leptin (4 μg), insulin (10 mU) or saline and killed 10 min later. Scale bars, 100 μm. ARC, arcuate nucleus; VMN, ventromedial nucleus; DMN, dorsomedial nucleus; 3v, third ventricle.
Fig. 9.

Representative photomicrographs of p-Akt (Ser473) positive cells (A–C, G–I) and p-STAT3 positive cells (D–F) in the hypothalamic sections at approximately bregma −3.8 (A–C), and bregma −4.16 (D–I) from saline, leptin or insulin-treated rats. Rats fasted for 24 hours were injected ICV with leptin (4 μg), insulin (10 mU) or saline and killed 10 min later. Scale bars, 100 μm. ARC, arcuate nucleus; PMv, ventral premammillary nucleus; VTM, ventral tuberomammillary nucleus; 3v, third ventricle.
Discussion
The main findings of this study were that in rat hypothalamus: 1) PI3K inhibitor wortmannin reversed the leptin-induced increase in PDE3B activities; (2) leptin failed to increase phosphorylation of Akt at Ser473 and Thr308 either at 10 or 45 min of treatment but it induced p-STAT3 activation, and 3) insulin increased phosphorylation of Akt at Ser473 and Thr308 at both 10 and 45 min post-injection without having an effect on p-STAT3 activation.
Because proper leptin signaling in the hypothalamus is required for body weight maintenance, it is essential to dissect out intracellular pathways that transduce leptin action in this brain region. In this regard besides the conventional JAK-STAT3 pathway, several non-STAT3 pathways, including the PI3K pathway, have been implicated to play critical role in energy homeostasis (4, 7, 9, 27–32). We have shown that in addition to PI3K, leptin also increases PDE3B activities in the hypothalamus, and PDE3 inhibition reverses the anorectic and bodyweight reducing effects of leptin (7). Others have shown leptin to induce PI3K activity in the hypothalamus (9), PI3K inhibition to reverse the anorectic effect of leptin (9), and PI3K to integrate leptin and insulin signaling in the hypothalamus (33). Along this line, hypothalamic PI3K pathway of leptin signaling is also defective during the development of diet-induced obesity (25). Thus it is important to characterize the downstream targets of PI3K signaling in transducing leptin action in the hypothalamus. In addition, both PI3K and PDE3B have been localized in the hypothalamic nuclei that have been implicated in energy homeostasis (2, 18, 34). Specifically, PDE3B is localized in the ARC, VMN, DMN, LH, PMv and PVN; and hypothalamic proopiomelanocortin (POMC) and neuropeptide Y (NPY) neurons express PDE3B (2, 34). Leptin also induces PI3K in POMC neurons and leptin withdrawal activates PI3K in AgRP neurons (33). PI3K is upstream of the mTOR pathway of leptin signaling in macrophages (35), and cumulative evidence suggests that PI3K pathway is upstream of several leptin-activated pathways including mTOR and Foxo1 signaling in the hypothalamus (27, 36). Importantly, PI3K is upstream of the PDE3B pathway in leptin and insulin signaling in the peripheral tissues (10, 12). Although, signaling through both PI3K and PDE3B pathways plays an important role in mediating leptin action in the hypothalamus (4, 37), it is still unknown if PI3K is upstream of PDE3B during leptin signaling in the hypothalamus. Thus, the present study tested this possibility by examining the effect of wortmannin on leptin-induced PDE3B activity in the rat hypothalamus. It has been previously demonstrated that PI3K is the only enzyme being inhibited by both wortmannin and compound LY294002 (38). In addition, wortmannin has been used in many studies defining the role of PI3K in leptin and insulin signaling in the hypothalamus in regulation of energy homeostasis (9, 18). Therefore, we used wortmannin, one of two specific PI3K inhibitors, to address if PI3K were upstream of PDE3B in the hypothalamus. Our demonstration of complete reversal of the leptin-induced PDE3B activation by a specific PI3K inhibitor wortmannin clearly identifies PI3K upstream of PDE3B signaling in the hypothalamus. Although, the mechanism by which PI3K activates PDE3B is presently unclear, there are several possibilities including a direct (10) and/or indirect effect by activating p-Akt (39).
To fully understand the mechanisms of leptin signaling in the hypothalamus in normal physiological condition and during the development of obesity, it is quite essential to establish the links between different arms of the leptin signaling network that have been demonstrated to play important role in energy homeostasis. Present finding of PI3K as upstream of the PDE3B pathway of leptin signaling together with previous demonstrations that leptin decreases cAMP levels in the hypothalamus (7) and that both PI3K and PDE3 inhibitors (7, 9) reverse the anorectic effects of leptin have provided an important link between PI3K and PDE3B-cAMP pathways in transducing leptin action in the hypothalamus in regulation of energy homeostasis. Thus, deregulation of any arm of the hypothalamic PI3K-PDE3B-cAMP pathway of leptin signaling may play a significant role in the development of central leptin resistance and obesity. Along this line, we have demonstrated impairment in the PI3K(25) and PDE3B-cAMP (A. Sahu and M. Sahu, unpublished) pathways of leptin signaling in the hypothalamus during the development of DIO.
To address if p-Akt is involved in leptin signaling and therefore could be an upstream of PDE3B, we first tested the effect of leptin on phosphorylation of Akt at Ser473 and Thr308 sites at 45 min post–injection. We initially examined the effect at this time point because our earlier studies clearly showed that leptin significantly increased both PI3K and PDE3B at 45 min (7). Nevertheless, the finding that central leptin treatment was unable to increase either p-Aktser473 or p-Aktthr308, suggests that leptin does not phosphorylates Akt in the hypothalamus. To address if phosphorylation of Akt was confined to specific region, such as the ARC, we examined the number of p-Aktser473 positive cells in the hypothalamus by ICC. The results clearly demonstrate that leptin does not increase p-Akt positive cells in any region of the hypothalamus examined. The potency of leptin was established by the demonstration of a significant increase in p-STAT3 levels in the hypothalamic extracts as well as in the number of p-STAT3 positive cells in various hypothalamic sites particularly in the ARC, VMN, DMN and PMv regions. One of the limitations in this study was that we examined changes in p-Aktthr308 levels in whole hypothalamic extracts, which could mask the effect of leptin if the changes in p-Aktthr308 were occurring in specific hypothalamic nuclei. Future study would address this issue. As previously shown (18), insulin significantly increased protein levels of both p-Aktser473 and p-Aktthr308 in the hypothalamic extracts. We also demonstrate that insulin increased the number of p-Aktser473 positive cells in various hypothalamic sites. These results together suggest that central leptin injection failed to increase hypothalamic p-Akt levels, while insulin was able to do so at 45 min post injection. Similarly, at 10 min post injection, leptin was unable to increase either p-Aktser473 or p-Aktthr308 levels in the hypothalamus or to increase the number of p-Aktser473 positive cells in any hypothalamic sites including the ARC. Leptin’s potency was again demonstrated by significant increase in p-STAT3 levels in the hypothalamus and in the number of p-STAT3 positive cells. On the other hand, whereas insulin increased phosphorylation of Akt, it failed to increase p-STAT3 levels in the hypothalamus-a finding that was confirmed by ICC.
Our finding of central leptin injection not to increase p-Aktser473 in hypothalamic extract and in the arcuate nucleus confirms previous findings (40, 41) and shows for the first time that leptin does not increase p-Aktser473 in other hypothalamic nuclei, particularly PMv and VTM (see below), that are involved in leptin signaling. In addition, we show that central leptin does not increase p-Aktthr308 levels in the hypothalamus. It appears that for full activation of Akt, its phosphorylation on both Ser473 and Thr308 is essential (42–44). It is believed that phosphatidylinositol-dependent kinase 1 (PDK1) phosphorylates Thr308 and mammalian target of rapamycin (mTOR) kinase complex 2 (mTORC2) phosphorylates Ser473 of Akt (16, 42, 44–46). However, it is still controversial if Thr308 phosphorylation is dependent on Ser473 phosphorylation (16, 45), although some studies have sown that Thr308 phosphorylation remains intact when Ser473 phosphorylation is lost or diminished (43, 46, 47). Therefore, it is essential to examine both Ser473 and Thr308 phosphorylation to address AKT activation. Thus our demonstration that central administration of leptin failed to increase both p-Aktser473 and p-Aktthr308 clearly shows that AKT is not activated by leptin, and suggests that Akt may not be upstream of the PDE3B pathway of leptin signaling in the rat hypothalamus. Because PI3K has been shown to bind and directly activate PDE3B in peripheral tissue (10, 48), it is most likely that leptin-induced PDE3B activation in the hypothalamus is mediated directly by PI3K. Inability of leptin to increase p-Akt suggests that signaling through downstream of Akt such as FoxO1, which has been shown to mediate both leptin and insulin action in NPY/AgRP and POMC neurons (27, 28), may be achieved indirectly by modifying insulin sensitivity in the hypothalamus (49). In addition despite strong evidence of leptin induced PI3K activation in the hypothalamus (7, 9), no phosphorylation of Akt at either serine or threonin suggests that Akt phosphorylation may not be used as a surrogate of PI3K activation at least in the rat hypothalamus. Although we only examined the effect of central leptin administration on p-Akt levels, it is possible that peripheral treatment of leptin may increase p-Akt levels in the hypothalamus; however it would be an indirect effect of leptin. Along this line, Koch et al. have reported a marginal increase in p-Akt positive cells after peripheral leptin administration in lean heterozygous Lep ob/+ mice (49), and we have recently observed peripheral leptin to increase p-Akt in the hypothalamus in C57BL6 mice (A. Sahu and M. Sahu, unpublished). However, whether central leptin increases p-Akt in mouse hypothalamus remains to be seen.
In a recent study Tups et al. (41) have shown intracerebroventricular insulin to increase the number of p-Aktser473 positive cells in the ARC, VMN, DMN and PVN. Our study confirms this report and further shows that insulin also increases p-Aktser473 positive cells in the PMv, lateral hypothalamus and VTM. Whereas PMv neurons are thought to link metabolic cues and reproduction (50, 51) and highly express LepRb mRNA (52), and the acute effects of leptin require PI3K signaling in these neurons (53), leptin failed to increase p-Akt positive cells in this hypothalamic area; suggesting that Akt may not be involvedin mediating leptin action in the PMv. On the other hand, increased number of p-Akt positive cells in the PMv by insulin suggests that p-Akt mediates insulin action in this hypothalamic area. Because insulin receptor is expressed in this hypothalamic nucleus (51), it remains to be seen if insulin action via p-Akt in these neurons plays any physiological role. The histaminergic VTM neurons have been implicated to be involved in mediating anorectic effect of leptin (54). Lesions in the VTM cause hyperphagia and increased body weight gain in rat (55) suggesting a role of VTM neurons in energy homeostasis. Although VTM expresses very few leptin receptors (56), VTM neurons are innervated by alpha-MSH containing neurons (57) and therefore, leptin action on these neurons is most likely mediated indirectly by activation of POMC neurons. Along this line, leptin did not induce p-STAT3 activation and had no effect on the number of p-Akt positive cells in the VTM. However, insulin significantly increased the number of p-Akt positive cells in the VTM suggesting a role of Akt in mediating insulin action in these neurons. Thus, future studies should define the role of Akt pathway of insulin signaling in VTM neurons in energy homeostasis and other physiological functions.
Finally, despite a significant increase in phosphorylation of Akt in the hypothalamic nuclei by insulin, it failed to increase p-STAT3 levels in the hypothalamus. This finding is in accord with previous report (40) showing insulin not to increase p-STAT3 in hypothalamic extracts. However, we also examined p-STAT3 positive cells in the hypothalamus by ICC to address the possibility that insulin might increase p-STAT3 in specific hypothalamic nuclei, which could be obscured by examining whole hypothalamic extracts. Here, we demonstrate that insulin failed to increase p-STAT3 positive neurons in various hypothalamic nuclei that have been implicated in energy homeostasis. Altogether, these findings suggest that insulin action in the hypothalamus is not mediated by STAT3 pathway. Thus, while leptin utilizes both STAT3 and PI3K pathways, insulin action is mediated by PI3K-Akt pathway in the hypothalamus. However, insulin can modify leptin-induced STAT3 activation in a tissue specific manner. Thus, insulin increases leptin-induced STAT3 activation in the hypothalamus (58), but it attenuates leptin-induced STAT3 activation in hepatoma cell line (59)
In summary, this study suggests that PI3K but not Akt is an upstream regulator of the PDE3B pathway of leptin signaling in the rat hypothalamus and that in addition to other hypothalamic sites insulin acts in the PMv and VTM through Akt pathway in fulfilling its role in brain function.
Fig. 5.

Representative photomicrographs of p-Akt (Ser473) ICC of hypothalamic sections at approximately bregma −4.16 (A–C) and −3.8 (D–F) from saline, leptin or insulin-treated rats that were fasted 24 hours and injected ICV with leptin (4 μg), insulin (10 mU) or saline and killed 45 min later. Scale bars, 100 μm. ARC, arcuate nucleus; PMv, ventral premammillary nucleus; VTM, ventral tuberomammillary nucleus; 3v, third ventricle.
Acknowledgments
This work was supported by NIH RO1 Grant DK61499 and DK78068 to AS. Thanks to A. F. Parlow and the NIDDK National Hormone & Pituitary Program, Torrance, CA, for supplying the recombinant murine leptin.
References
- 1.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–70. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 2.Sahu A. Leptin signaling in the hypothalamus: emphasis on energy homeostasis and leptin resistance. Front Neuroendocrinol. 2003;24:225–53. doi: 10.1016/j.yfrne.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Sahu A. Minireview: A hypothalamic role in energy balance with special emphasis on leptin. Endocrinology. 2004;145:2613–20. doi: 10.1210/en.2004-0032. [DOI] [PubMed] [Google Scholar]
- 4.Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–95. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 5.Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG., Jr STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–9. doi: 10.1038/nature01388. [DOI] [PubMed] [Google Scholar]
- 6.Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, Fu XY. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci U S A. 2004;101:4661–6. doi: 10.1073/pnas.0303992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao AZ, Huan JN, Gupta S, Pal R, Sahu A. A phosphatidylinositol 3-kinase phosphodiesterase 3B-cyclic AMP pathway in hypothalamic action of leptin on feeding. Nat Neurosci. 2002;5:727–8. doi: 10.1038/nn885. [DOI] [PubMed] [Google Scholar]
- 8.Sahu A. A role of phosphodiesterase-3B pathway in mediating leptin action on proopiomelanocortin and neurotensin neurons in the hypothalamus. Neurosci Lett. 2010;479:18–21. doi: 10.1016/j.neulet.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Jr, Schwartz MW. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature. 2001;413:794–5. doi: 10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- 10.Rahn T, Ridderstrale M, Tornqvist H, Manganiello V, Fredrikson G, Belfrage P, Degerman E. Essential role of phosphatidylinositol 3-kinase in insulin-induced activation and phosphorylation of the cGMP-inhibited cAMP phosphodiesterase in rat adipocytes. Studies using the selective inhibitor wortmannin. FEBS Lett. 1994;350:314–8. doi: 10.1016/0014-5793(94)00797-7. [DOI] [PubMed] [Google Scholar]
- 11.Zhao AZ, Bornfeldt KE, Beavo JA. Leptin inhibits insulin secretion by activation of phosphodiesterase 3B. J Clin Invest. 1998;102:869–73. doi: 10.1172/JCI3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao AZ, Shinohara MM, Huang D, Shimizu M, Eldar-Finkelman H, Krebs EG, Beavo JA, Bornfeldt KE. Leptin induces insulin-like signaling that antagonizes cAMP elevation by glucagon in hepatocytes. J Biol Chem. 2000;275:11348–54. doi: 10.1074/jbc.275.15.11348. [DOI] [PubMed] [Google Scholar]
- 13.Alessi DR, Cohen P. Mechanism of activation and function of protein kinase B. Curr Opin Genet Dev. 1998;8:55–62. doi: 10.1016/s0959-437x(98)80062-2. [DOI] [PubMed] [Google Scholar]
- 14.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88(4):435–7. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 15.Hemmings BA. Akt signaling: linking membrane events to life and death decisions. Science. 1997;275:628–30. doi: 10.1126/science.275.5300.628. [DOI] [PubMed] [Google Scholar]
- 16.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Onuma H, Makino H, Osawa H, Suzuki Y, Taira M, Kanatsuka A, Saito Y. Mitogen-activated protein kinase and p70 ribosomal protein S6 kinase are not involved in the insulin-dependent stimulation of cAMP phosphodiesterase kinase in rat adipocytes. Biochim Biophys Acta. 1998;1402:197–208. doi: 10.1016/s0167-4889(98)00003-2. [DOI] [PubMed] [Google Scholar]
- 18.Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG, Jr, Seeley RJ, Schwartz MW. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes. 2003;52:227–31. doi: 10.2337/diabetes.52.2.227. [DOI] [PubMed] [Google Scholar]
- 19.Sahu A, Kalra SP. Effects of tachykinins on luteinizing hormone release in female rats: potent inhibitory action of neuropeptide K. Endocrinology. 1992;130:1571–7. doi: 10.1210/endo.130.3.1371455. [DOI] [PubMed] [Google Scholar]
- 20.Pal R, Sahu A. Leptin signaling in the hypothalamus during chronic central leptin infusion. Endocrinology. 2003;144:3789–98. doi: 10.1210/en.2002-0148. [DOI] [PubMed] [Google Scholar]
- 21.Sahu A, Metlakunta AS. Hypothalamic phosphatidylinositol 3-kinase-phosphodiesterase 3B- cyclic AMP pathway of leptin signalling is impaired following chronic central leptin infusion. J Neuroendocrinol. 2005;17:720–6. doi: 10.1111/j.1365-2826.2005.01362.x. [DOI] [PubMed] [Google Scholar]
- 22.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 23.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols in molecular biology. New York: John Wiley, Sons; 1994. [Google Scholar]
- 24.Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–9. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- 25.Metlakunta AS, Sahu M, Sahu A. Hypothalamic phosphatidylinositol 3-kinase pathway of leptin signaling is impaired during the development of diet-induced obesity in FVB/N mice. Endocrinology. 2008;149:1121–8. doi: 10.1210/en.2007-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 4. Academic Press; San Diego: 1998. [Google Scholar]
- 27.Kim MS, Pak YK, Jang PG, Namkoong C, Choi YS, Won JC, Kim KS, Kim SW, Kim HS, Park JY, Kim YB, Lee KU. Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci. 2006;9:901–6. doi: 10.1038/nn1731. [DOI] [PubMed] [Google Scholar]
- 28.Kitamura T, Feng Y, Kitamura YI, Chua SC, Jr, Xu AW, Barsh GS, Rossetti L, Accili D. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med. 2006;12:534–40. doi: 10.1038/nm1392. [DOI] [PubMed] [Google Scholar]
- 29.Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312:927–30. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- 30.Cota D, Matter EK, Woods SC, Seeley RJ. The role of hypothalamic mammalian target of rapamycin complex 1 signaling in diet-induced obesity. J Neurosci. 2008;28:7202–8. doi: 10.1523/JNEUROSCI.1389-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–74. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 32.Martin TL, Alquier T, Asakura K, Furukawa N, Preitner F, Kahn BB. Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle. J Biol Chem. 2006;281:18933–41. doi: 10.1074/jbc.M512831200. [DOI] [PubMed] [Google Scholar]
- 33.Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. 2005;115:951–8. doi: 10.1172/JCI24301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sahu M, Litvin DG, Sahu A. Phosphodiesterase-3B is expressed in proopiomelanocortin and neuropeptide Y neurons in the mouse hypothalamus. Neurosci Lett. 2011;505:93–7. doi: 10.1016/j.neulet.2011.09.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maya-Monteiro CM, Almeida PE, D’Avila H, Martins AS, Rezende AP, Castro-Faria-Neto H, Bozza PT. Leptin induces macrophage lipid body formation by a phosphatidylinositol 3-kinase-andmammalian target of rapamycin-dependent mechanism. J Biol Chem. 2008;283:2203–10. doi: 10.1074/jbc.M706706200. [DOI] [PubMed] [Google Scholar]
- 36.Kahn BB, Myers MG., Jr mTOR tells the brain that the body is hungry. Nat Med. 2006;12:615–7. doi: 10.1038/nm0606-615. [DOI] [PubMed] [Google Scholar]
- 37.Sahu A. Intracellular leptin-signaling pathways in hypothalamic neurons: the emerging role of phosphatidylinositol-3 kinase-phosphodiesterase-3B-cAMP pathway. Neuroendocrinology. 2011;93:201–10. doi: 10.1159/000326785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351(Pt 1):95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitamura T, Kitamura Y, Kuroda S, Hino Y, Ando M, Kotani K, Konishi H, Matsuzaki H, Kikkawa U, Ogawa W, Kasuga M. Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine-threonine kinase Akt. Mol Cell Biol. 1999;19:6286–96. doi: 10.1128/mcb.19.9.6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carvalheira JB, Torsoni MA, Ueno M, Amaral ME, Araujo EP, Velloso LA, Gontijo JA, Saad MJ. Cross-talk between the insulin and leptin signaling systems in rat hypothalamus. Obes Res. 2005;13:48–57. doi: 10.1038/oby.2005.7. [DOI] [PubMed] [Google Scholar]
- 41.Tups A, Anderson GM, Rizwan M, Augustine RA, Chaussade C, Shepherd PR, Grattan DR. Both p110alpha and p110beta isoforms of phosphatidylinositol 3-OH-kinase are required for insulin signalling in the hypothalamus. J Neuroendocrinol. 2010;22:534–42. doi: 10.1111/j.1365-2826.2010.01975.x. [DOI] [PubMed] [Google Scholar]
- 42.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7(4):261–9. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 43.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15(23):6541–51. [PMC free article] [PubMed] [Google Scholar]
- 44.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 45.Bhaskar PT, Nogueira V, Patra KC, Jeon SM, Park Y, Robey RB, Hay N. mTORC1 hyperactivity inhibits serum deprivation-induced apoptosis via increased hexokinase II and GLUT1 expression, sustained Mcl-1 expression, and glycogen synthase kinase 3beta inhibition. Mol Cell Biol. 2009;29(18):5136–47. doi: 10.1128/MCB.01946-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127(1):125–37. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 47.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11(6):859–71. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 48.Rondinone CM, Carvalho E, Rahn T, Manganiello VC, Degerman E, Smith UP. Phosphorylation of PDE3B by phosphatidylinositol 3-kinase associated with the insulin receptor. J Biol Chem. 2000;275:10093–8. doi: 10.1074/jbc.275.14.10093. [DOI] [PubMed] [Google Scholar]
- 49.Koch C, Augustine RA, Steger J, Ganjam GK, Benzler J, Pracht C, Lowe C, Schwartz MW, Shepherd PR, Anderson GM, Grattan DR, Tups A. Leptin rapidly improves glucose homeostasis in obese mice by increasing hypothalamic insulin sensitivity. J Neurosci. 2010;30:16180–7. doi: 10.1523/JNEUROSCI.3202-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Donato J, Jr, Cravo RM, Frazao R, Elias CF. Hypothalamic sites of leptin action linking metabolism and reproduction. Neuroendocrinology. 2011;93:9–18. doi: 10.1159/000322472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Donato JJ, Elias CF. The ventral premammilary nucleus links metabolic cues and reproduction. Front Endcocrinol. 2011;2:57. doi: 10.3389/fendo.2011.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caron E, Sachot C, Prevot V, Bouret SG. Distribution of leptin-sensitive cells in the postnatal and adult mouse brain. J Comp Neurol. 518:459–76. doi: 10.1002/cne.22219. [DOI] [PubMed] [Google Scholar]
- 53.Williams KW, Sohn JW, Donato J, Jr, Lee CE, Zhao JJ, Elmquist JK, Elias CF. The acute effects of leptin require PI3K signaling in the hypothalamic ventral premammillary nucleus. J Neurosci. 2011;31:13147–56. doi: 10.1523/JNEUROSCI.2602-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morimoto T, Yamamoto Y, Mobarakeh JI, Yanai K, Watanabe T, Yamatodani A. Involvement of the histaminergic system in leptin-induced suppression of food intake. Physiol Behav. 1999;67:679–83. doi: 10.1016/s0031-9384(99)00123-7. [DOI] [PubMed] [Google Scholar]
- 55.Mahia J, Bernal A, Puerto A. Hyperphagia and increased body weight induced by lesions of the ventral tuberomammillary system. Behav Brain Res. 2007;181:147–52. doi: 10.1016/j.bbr.2007.03.026. [DOI] [PubMed] [Google Scholar]
- 56.Hakansson ML, Brown H, Ghilardi N, Skoda RC, Meister B. Leptin receptor immunoreactivity in chemically defined target neurons of the hypothalamus. J Neurosci. 1998;18:559–72. doi: 10.1523/JNEUROSCI.18-01-00559.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fekete C, Liposits Z. Histamine-immunoreactive neurons of the tuberomammillary nucleus are innervated by alpha-melanocyte stimulating hormone-containing axons. Generation of a new histamine antiserum for ultrastructural studies. Brain Res. 2003;969:70–7. doi: 10.1016/s0006-8993(03)02279-0. [DOI] [PubMed] [Google Scholar]
- 58.Carvalheira JB, Siloto RM, Ignacchitti I, Brenelli SL, Carvalho CR, Leite A, Velloso LA, Gontijo JA, Saad MJ. Insulin modulates leptin-induced STAT3 activation in rat hypothalamus. FEBS Lett. 2001;500:119–24. doi: 10.1016/s0014-5793(01)02591-1. [DOI] [PubMed] [Google Scholar]
- 59.Kuwahara H, Uotani S, Abe T, Degawa-Yamauchi M, Takahashi R, Kita A, Fujita N, Ohshima K, Sakamaki H, Yamasaki H, Yamaguchi Y, Eguchi K. Insulin attenuates leptin-induced STAT3 tyrosine-phosphorylation in a hepatoma cell line. Mol Cell Endocrinol. 2003;205:115–20. doi: 10.1016/s0303-7207(03)00180-1. [DOI] [PubMed] [Google Scholar]