

Abstract
Rationale
Nkx2.5 is a transcription factor that regulates cardiomyogenesis in vivo and in embryonic stem cells. It is also a common target in congenital heart disease. Although Nkx2.5 has been implicated in the regulation of many cellular processes that ultimately contribute to cardiomyogenesis and morphogenesis of the mature heart, relatively little is known about how it is regulated at a functional level.
Objective
We have undertaken a proteomic screen to identify novel binding partners of Nkx2.5 during cardiomyogenic differentiation in an effort to better understand the regulation of its transcriptional activity.
Methods and Results
Purification of Nkx2.5 from differentiating cells identified the myosin phosphatase subunits PP1β and Mypt1 as novel binding partners. The interaction with PP1β/Mypt1 resulted in exclusion of Nkx2.5 from the nucleus and consequently, inhibition of its transcriptional activity. Exclusion of Nkx2.5 was inhibited by treatment with LeptomycinB and was dependent on a Mypt1 nuclear export signal. Furthermore, in transient transfection experiments, Nkx2.5 co-localized outside the nucleus with phosphorylated Mypt1 in a manner dependent on Wnt signalling and Rho-associated protein kinase. Treatment of differentiating mouse embryonic stem cells with Wnt3a resulted in enhanced phosphorylation of endogenous Mypt1, increased nuclear exclusion of endogenous Nkx2.5 and a failure to undergo terminal cardiomyogenesis. Finally, knockdown of Mypt1 resulted in rescue of Wnt3a-mediated inhibition of cardiomyogenesis, indicating that Mypt1 is required for this process.
Conclusions
We have identified a novel interaction between Nkx2.5 and myosin phosphatase. Promoting this interaction represents a novel mechanism whereby Wnt3a regulates Nkx2.5 and inhibits cardiomyogenesis.
Keywords: myogenesis, stem cell, signal transduction, gene regulation, cardiac differentiation, cardiac transcription factors, differentiation, embryonic stem cells
INTRODUCTION
During development, differentiation of myocardial cells and morphogenesis of the heart are controlled by transcription factors that are in turn regulated by the convergence of several extracellular signalling cascades. Cardiac progenitor cells originate from the splanchic mesoderm and are segregated into two distinct populations, termed the first and second heart fields (SHF), that give rise to distinct structures within the mature heart.1–3 The differentiation of these progenitors into cardiomyocytes is regulated by transcription factors including GATA4, Nkx2.5, MEF2C and members of the Tbx family. These same factors regulate the cardiomyogenesis in embryonic stem cells (ESCs).4
Nkx2.5 is among the earliest markers of cardiomyogenic differentiation.5 Mutations in Nkx2.5 are a common cause of congenital heart disease, with more than 40 known mutations associated with various phenotypes.6 In the mouse, loss of Nkx2.5 results in failure of cardiac looping, and embryonic lethality.7, 8 Although cardiomyocytes are specified normally, the expression of downstream genes, including the ventricular isoform of myosin light chain 2 (Mlc2v) and Connexin40 (Cnx40) is disrupted. Studies in mouse ESCs (mESCs) have supported the notion that Nkx2.5 is not required for cardiomyocyte specification, although, as in the null mouse, downstream genes were aberrantly expressed.9 Expression of a dominant negative form of Nkx2.5 fused to the repressor domain of engrailed resulted in inhibition of cardiomyogenesis in P19 cells, indicating that some Nkx activity is required for cardiac differentiation.10 It has also been shown that overexpression of Nkx2.5 induced cardiomyogenesis in this system, suggesting that its activity is sufficient to drive differentiation under certain conditions.11
The role of Nkx factors during heart development has been examined on a finer scale. Analysis of the role of Nkx2.5 in the SHF identified a negative feedback loop in which Nkx2.5 inhibits Bmp2/Smad1 signalling, ultimately controlling the specification and proliferation of progenitors.12 In zebrafish, morpholino mediated inhibition of nkx2.5 and nkx2.7 expression resulted in the formation of an excess number of atrial cardiomyocytes paralleled by a decrease in the number of ventricular cardiomyocytes, suggesting that Nkx genes differentially regulate distinct progenitor populations within the developing heart. 13
Nkx2.5 regulates many targets via direct interactions with other cardiomyogenic transcription factors including GATA4, Tbx5 and MEF2C.14–16 These interactions confer specificity and modulate Nkx2.5 activity. Although Nkx2.5 usually acts as a transcriptional activator, it can repress target gene expression in the context of particular regulatory complexes. Specifically, cooperation of Nkx2.5 with Tbx5 promotes expression of ANF while interaction with Tbx2 is repressive.15, 17, 18 This differential partnership results in repression of ANF expression and the formation of chamber myocardium in Tbx2 expressing regions of the developing heart.18 These findings highlight the importance of cofactor interactions for the regulation of Nkx2.5 activity.
Cardiomyogenesis is also regulated by the action of signalling molecules derived from surrounding tissue structures. In particular, the Wnt signalling cascade plays opposing roles at different stages of cardiomyogenesis.19 Wnt signalling mediated by Wnt3a is important for the induction of mesoderm both in vivo and in mESCs 20, 21. Indeed, a lack of canonical Wnt signalling resulted in the loss of Mesp1 expression, which is perhaps the earliest marker of cardiogenic mesoderm21, 22 Conversely, specification of cardiac progenitor cells from Mesp1+ve cells is potentiated by the Wnt inhibitor Dkk1, indicating that Wnt signalling is inhibitory at this later stage.23, 24 Consistent with this, activation of Wnt/β-catenin signalling in differentiating mESCs at later stages resulted in reduced cardiomyogenesis.25,26 Although the multiphasic and multifunctional nature of Wnt signalling has been studied extensively19, the molecular mechanisms whereby Wnts exert these differential effects on cardiomyogenesis remain largely undefined.
In order to gain insight into the complex function of Nkx2.5, we have undertaken a proteomic screen to identify novel cofactors in the context of cardiomyogenesis. The β-isoform of protein phosphatase 1 (PP1β, also known as PP1δ) and its regulatory subunit Myosin Phosphatase Targeting Subunit 1 (Mypt1), two subunits of the myosin phosphatase (MP) enzyme complex, were identified as putative cofactors of Nkx2.5 and validated by co-immunoprecipitation. Together with Myosin light chain kinase (MLCK), MP modulates the balance of phosphorylation of the regulatory light chain of myosinII, thereby regulating muscle contraction.27 We show that MP induces exclusion of Nkx2.5 from the nucleus and inhibits its transcriptional activity. This occurs downstream of Wnt signalling and is modulated by ROCK-mediated phosphorylation of Mypt1. Inhibition of cardiomyogenesis by Wnt3a26 is accompanied by increased nuclear exclusion of endogenous Nkx2.5 and is rescued my knockdown of Mypt1. Our findings suggest a novel mechanism whereby Wnt3a inhibits the terminal stages of cardiomyogenesis in stem cells.
METHODS
Protocols are described in detail in the Online Data Supplement. mESCs were maintained as previously described.28 Briefly, differentiation was induced by embryoid body (EB) formation for 5 days followed by two days of adherent culture. All transfections were performed in HEK293 cells or P19 cells using Fugene (Roche) or Lipofectamine2000 (Life Technologies). Luciferase assays were performed using the Dual-Luciferase reporter assay system (Promega). Stable cell lines were generated in mESCs using Expression Arrest GIPZ lentiviral shRNAmir particles targeted to Mypt1, along with a non-silencing control (Thermo Fisher Scientific). Images were captured using a Zeiss LSM 510 Meta microscope or a Zeiss LSM 710 Multiphoton microscope equipped with Zen Software (Zeiss) and subsequently edited in Canvas11. Gene expression analysis was performed by QPCR as described previously.29 Expression data were normalized to β-actin, and averages +/− standard error of the mean are shown expressed as fold-change relative to vehicle treatment.
RESULTS
PP1β and Mypt1 interact with Nkx2.5 and inhibit its transcriptional activity
In order to identify novel interacting partners of Nkx2.5 during cardiomyogenesis in stem cells, we performed a proteomic screen using affinity purification of Nkx2.5. P19 stable cell lines were generated to overexpress PGK-Nkx2.5TAP or PGK-TAP as a control. These cell lines, termed P19[Nkx2.5TAP] and P19[TAP] respectively, were differentiated into cardiomyocytes and protein was harvested on Day 6. Lysates were subjected to immunoprecipitation and subsequently analyzed by mass spectrometry. Using this proteomic screen technique, we identified PP1β and Mypt1 as putative binding partners of Nkx2.5 (Supplementary Table I). Both PP1β and Mypt1 are subunits of the MP enzyme complex. Sequence analysis of the three unique peptides identified for Mypt1 revealed that only one of these contained sequences that were also found in Mypt2, indicating that Mypt1 is the major isoform interacting with Nkx2.5.
We validated this interaction by co-immunoprecipitation experiments, in which Nkx2.5-Flag was transiently co-expressed with HA-PP1β and myc-Mypt1 in HEK293 cells. We immunoprecipitated each subunit with antibodies specific to Flag, HA or myc epitopes and detected the presence of interacting proteins by western blot. Both HA-PP1β and myc-Mypt1 co-immunoprecipitated with Flag-Nkx2.5, using an anti-Flag antibody (Fig. 1A). Reciprocal immunoprecipitations of HA-PP1β or myc-Mypt1 also co-immunoprecipitated Flag-Nkx2.5. Therefore, we have identified PP1β and Mypt1 as novel interacting partners of Nkx2.5.
Figure 1. PP1β and Mypt1 interact with Nkx2.5 and inhibit its transcriptional activity.

A) HEK293 cells were transiently transfected with Flag-Nkx2.5, HA-PP1β and myc-Mypt1. Total protein was harvested and immunoprecipitated with antibodies specific for Flag, HA and myc or with mouse IgG as a control. Immunoprecipitated lysates and input (30μg/0.02% of input) fractions were analyzed by western blots with the antibodies indicated. B) P19 cells were transiently transfected with the plasmids indicated or with empty vector, along with the ANF-luciferase reporter and SV40-Renilla as an internal control. Firefly luciferase activity was normalized to Renilla luciferase and reporter activation was quantified as a percentage of the treatment resulting in maximum activation of the reporter. Statistical significance was calculated using ANOVA with a Dunnett’s post-hoc test (n=4–7,**p<0.01, ***p<0.001).
In order to understand the implications of the interaction between Nkx2.5 and MP, we sought to examine its effect on Nkx2.5 transcriptional activity using an ANF-luciferase reporter.30 While Nkx2.5 was able to activate this reporter when transfected alone, co-transfection with HA-PP1β and myc-Mypt1 resulted in significant inhibition of reporter activation, indicating that this interaction is inhibitory to Nkx2.5-dependent transcription (Fig. 1B). Catalytic PP1 subunits can interact with many different regulatory subunits. Therefore, we sought to ascertain whether the inhibition of Nkx2.5 was specific to MP or whether other PP1 regulatory subunits could regulate Nkx2.5. We substituted NIPP1, the nuclear targeting subunit of PP131 for Mypt1, which resulted in a loss of inhibition of Nkx2.5 activity, indicating that the Mypt1 subunit is specifically required (Fig. 2). Interestingly, co-transfection of Nkx2.5 with HA-PP1β and both myc-Mypt1 and myc-NIPP1 resulted in a loss of inhibition, suggesting that competition from other targeting subunits may block the association of MP subunits and its inhibitory effect on Nkx2.5.
Figure 2. Coexpression with MP results in exclusion of Nkx2.5 from the nucleus.

P19 cells were transiently transfected with the plasmids indicated and fixed one day later for analysis of subcellular localization. A) Fixed cells were labelled with antibodies specific to Nkx2.5 and myc-tag as well as with Hoechst dye to detect nuclei. Scale bar represents 10μm. B) Subcellular localization of Nkx2.5 was quantified by imaging 10 fields in three independent experiments. An average of 142 cells were quantified per treatment (n=3, *p<0.05).
Interaction with MP results in exclusion of Nkx2.5 from the nucleus
MP dephosphorylates myosin and as such, the enzyme complex is localized primarily in the cytoplasm. Given that Nkx2.5 is a nuclear transcription factor, we sought to determine the subcellular location of the Nkx2.5/MP interaction. Flag-Nkx2.5 was transfected with and without the MP subunits and its subcellular localization was visualized by immunofluorescence. When transfected alone, Flag-Nkx2.5 was localized almost exclusively to the nucleus (Fig. 2A&B). However, when co-expressed with MP, Nkx2.5 was redistributed to the cytoplasm in a perinuclear pattern (Fig. 2A). Quantification confirmed that more than 60% of co-expressing cells exhibited a perinuclear distribution of Nkx2.5 (Fig. 2B). In agreement with our reporter assays, the redistribution of Nkx2.5 did not occur in the presence of NIPP1 (Fig. 2A&B). Interestingly, treatment with LeptomycinB, an inhibitor of CRM1-dependent nuclear export, prevented nuclear exclusion of Nkx2.5 in the presence of MP, indicating that Nkx2.5 is exported from the nucleus via this system. Finally, co-expression with MP did not result in an altered subcellular distribution of GATA4, suggesting that this is not a regulatory mechanism affecting all cardiomyogenic transcription factors (Fig. 2A).
MP-induced nuclear exclusion of Nkx2.5 is modulated by phosphorylation of Mypt1
Although MP is primarily localized to the cytoplasm, phosphorylation of Mypt1 at Thr853 by Rho-associated kinase (ROCK) causes a redistribution of the MP complex to the perinuclear region and the nucleus.32 In addition, this phosphorylation event inhibits PP1 activity >100 fold.33, 34 The perinuclear localization of Nkx2.5 indicated that it might interact with this phosphorylated form of MP. Indeed, co-labelling with antibodies against Nkx2.5 and pMypt1T853 identified co-localization of Nkx2.5 with pMypt1T853 in the perinuclear region of co-transfected cells (Fig. 3A). To determine whether modulation of Mypt1 phosphorylation could affect the subcellular localization of Nkx2.5, cells co-transfected with Flag-Nkx2.5 and the MP subunits were treated with the ROCK inhibitor y-27632. Consistent with previous reports, this resulted in a significant decrease (≈30%) in the levels of pMypt1 relative to total Mypt1 (Fig. 3B&C).33 Redistribution of Nkx2.5 to the perinuclear region in response to co-expression with MP was significantly attenuated in the presence of y-27632 (≈40%) relative to untreated control cultures (Fig. 3D,E&F). However, treatment with y-27632 did not completely abrogate phosphorylation of Mypt1 and there remained some pMypt1T853-positive cells in which Nkx2.5 accumulated in the perinuclear region (Fig. 3F, arrow). Interestingly, we observed a perinuclear accumulation of the myc-labelled Mypt1 in Figure 3E, indicating that in at least some cells, most of the Mypt1 is phosphorylated.
Figure 3. MP-induced nuclear exclusion of Nkx2.5 is modulated by phosphorylation of Mypt1.

P19 cells were transiently transfected with Flag-Nkx2.5, HA-PP1β and myc-Mypt1. A) Cells were labelled by immunofluorescence with antibodies specific to Nkx2.5 and pMypt1Thr853. Scale bar represents 10μm. B) Total protein was harvested from y-27632-treated and control cultures and analyzed by western blot using antibodies specific to the factors indicated. C) The relative levels of pMypt1 were quantified by densitometry and are expressed relative to total Mypt1 (n=3, *p<0.05). D, E&F) Cultures were treated with y-27632 and labelled by immunofluorescence with antibodies specific to Nkx2.5 and myc or pMypt1T853 as well as with Hoechst dye to detect nuclei. Scale bar represents 10μm. The subcellular localization of Nkx2.5 was quantified in control and y-27632-treated cultures and expressed relative to the level of nuclear exclusion of Nkx2.5 in untreated controls (n=3, *p<0.05).
Thr853 and a nuclear export signal in Mypt1 are required for nuclear exclusion of Nkx2.5
To determine whether phosphorylation of Mypt1 at T853 is critical for Nkx2.5 nuclear export, we mutated T853 to alanine (Mypt1T853A) to generate a phospho-deficient form of MP and examined its ability to support Nkx2.5 exclusion. Coexpression with HA-PP1 and myc-Mypt1T853A resulted in a significant loss (>60%) of nuclear exclusion of Flag-Nkx2.5 relative to coexpression with wild-type MP, supporting the requirement of Mypt1 phosphorylation for this process (Fig. 4A&C). We also generated a phosphomimetic Mypt1 mutant (Mypt1T853E) although this mutant did not exclude Nkx2.5 more efficiently than wild-type Mypt1 (data not shown).
Figure 4. Thr853 and a nuclear export signal in Mypt1 are required for nuclear exclusion of Nkx2.5.

P19 cells were transiently transfected with Flag-Nkx2.5, HA-PP1β and myc-Mypt1, myc-Mypt1Δ853A (A, C) or myc-Mypt1 NES (B, D, E). A &B) Cultures were labelled by immunofluorescence with antibodies specific to Nkx2.5 and myc as well as with Hoechst dye to detect nuclei. Scale bar represents 10μm. C & E) The subcellular localization of Nkx2.5 was quantified in wild-type, Mypt1T853A, and myc-Mypt1ΔNES expressing cultures (n=3, *p<0.05). D) The subcellular localization of Mypt1 and Mypt1ΔNES was quantified (n=3, *p<0.05).
The ability of LeptomycinB to inhibit MP-induced nuclear exclusion of Nkx2.5 (Fig 2A) indicated that this process is mediated by Crm1. We used NetNES1.1 to search for leucine-rich nuclear export signal (NES) sequences in the MP subunits and Nkx2.5. While neither Nkx2.5, nor PP1β contained putative NES signals, two leucine residues near the C-terminus of Mypt1 (962 and 964 in the human sequence) were predicted to form an NES.35 We mutated these residues to alanine (Mypt1ΔNES) and examined the subcellular localization of Mypt1 and Nkx2.5. Quantification of myc+ve nuclei revealed that while cultures expressing wild-type Mypt1 exhibited nuclear myc staining in approximately 40% of cells, almost 80% of cells expressing Mypt1ΔNES were positive for nuclear myc (Fig. 4B&D). Strikingly, co-expression with HA-PP1 and myc-Mypt1ΔNES resulted in a drastic (>80%) loss of MP-induced nuclear exclusion of Nkx2.5 (Fig. 4B&E), indicating that Crm1-mediated nuclear export of Nkx2.5 occurs in a Mypt1-NES-dependent fashion. These data suggest a mechanism whereby phosphorylation of Mypt1 by ROCK results in translocation of MP to the nucleus, where it interacts with Nkx2.5. The complex is then exported to the perinuclear region via the Mypt1 NES.
The Wnt inhibitor Dkk1 blocks MP-dependent redistribution of Nkx2.5
ROCK-mediated phosphorylation of Mypt1, and subsequent nuclear exclusion of Nkx2.5, might be regulated via a signalling cascade that is inhibitory toward cardiomyogenesis. Wnt3a is a good candidate in this instance as it is known to inhibit terminal differentiation of cardiac progenitors and it also modulates ROCK activity and expression.26, 36, 37 The inhibition of late stages of cardiomyogenesis by Wnt3a has primarily been attributed to the canonical/β-catenin pathway, while non-canonical Wnt signalling is usually considered supportive for terminal differentiation.19, 25, 26 Although the activation of ROCK is generally associated with the non-canonical/planar cell polarity (PCP) pathway, the Rho/ROCK pathway is activated by Wnt3a 37, 38 and is required for subsequent β-catenin-dependent gene activation.36 We tested whether the Wnt inhibitor Dkk1 could modulate ROCK-mediated phosphorylation of Mypt1 at T853, which, in addition to being relevant to our study, is a reliable measure of ROCK activity.39 Although a previous study reported that Dkk1 does not modulate Rho activation in myeloma cells,40 we found that in our system, Dkk1 treatment resulted in a reproducible decrease (≈20%) in the level of phosphorylated Mypt1 relative to total Mypt1 (Fig. 5A&B). Therefore, blocking Wnt signalling results in decreased ROCK activity and Mypt1 phosphorylation.
Figure 5. The redistribution of Nkx2.5 in response to coexpression with MP is attenuated by treatment with Dkk1.

P19 cells were transiently cotransfected with Flag-Nkx2.5, HA-PP1β and myc-Mypt1. A) Total protein was harvested from Dkk1- and vehicle-treated cultures and analyzed by western blots using antibodies specific to the factors indicated. B) The relative levels of pMypt1 were quantified by densitometry and are expressed relative to total Mypt1 (n=3, *p<0.05). C&D) Cultures were treated with 150ng/ml Dkk1 or vehicle (PBS+0.1% BSA) and labelled with antibodies specific to Nkx2.5 and myc as well as with Hoechst dye to detect nuclei. Scale bar represents 10μm. The subcellular localization of Nkx2.5 was quantified in vehicle- and Dkk1-treated cultures and expressed relative to the level of nuclear exclusion of Nkx2.5 in vehicle-treated controls (n=4, *p<0.05).
We next sought to establish a link between Wnt signalling and nuclear exclusion of Nkx2.5. We co-transfected Flag-Nkx2.5 with MP and analyzed the subcellular distribution of Nkx2.5 in the presence and absence of Dkk1. By immunofluorescence analysis, treatment with Dkk1 significantly inhibited the MP-dependent nuclear exclusion of Nkx2.5 when compared to vehicle (Fig. 5C&D). These data identify a correlation between the Wnt-regulated level of Mypt1 phosphorylation and the MP-dependent nuclear exclusion of Nkx2.5.
Wnt3a treatment of differentiating mESCs excludes Nkx2.5 from the nucleus and inhibits cardiomyogenesis
As activation of Wnt signalling at late stages of mESC cardiomyogenesis is known to be inhibitory,25, 26 we sought to determine whether this involves nuclear exclusion of Nkx2.5. Differentiating mESCs were treated with Wnt3a or vehicle control from days 4–7 and the effects on cardiomyogenesis were examined by gene expression analysis and immunofluorescence. Treatment of differentiating mESCs with Wnt3a resulted in a reduced number of Mlc2v+ve cardiomyocytes (Fig. 6A&B). This was accompanied by a significant reduction in transcript levels of cardiomyogenic markers including Nkx2.5, Mlc2v, Cnx40, ANF and myosin heavy chain 6 (MyHC6) (Fig. 6C). Notably, the expression of GATA4 was not significantly affected by Wnt3a treatment (Fig. 6C). The transcript levels of Nkx2.5 were strongly downregulated in response to Wnt3a treatment, which is consistent with recent evidence that Wnt signalling modulates Nkx2.5 expression.41
Figure 6. Treatment of differentiating mESCs with Wnt3a results in reduced cardiomyogenesis associated with increased exclusion of endogenous Nkx2.5 from the nucleus.

Differentiating D3 mESCs were treated with Wnt3a or vehicle (PBS + 0.1% BSA) control from days 4 to 7. A) Wnt3a- and vehicle- treated cells were fixed on day 7 and labelled with antibodies specific to Mlc2v and stained with Hoechst dye to detect nuclei. Scale bar represents 10μm. B) The number of Mlc2v+ cells was quantified in vehicle- and Wnt3a-treated cultures (n=3, *p<0.05). C) The expression of genes associated with cardiomyogenesis was analyzed by QPCR in vehicle- and Wnt3a-treated cultures. Data are expressed relative to expression levels in vehicle treated cultures (n=3, *p<0.05). D) Total protein was harvested from Wnt3a- and vehicle- treated mESCs on day 7 and analyzed by western blot with antibodies specific to the factors indicated. E) The exclusion of endogenous Nkx2.5 from the nucleus was quantified in vehicle- and Wnt3a-treated mESC cultures (n=3, *p<0.05). F) Differentiated mESCs from vehicle- and Wnt3a-treated cultures were fixed on day 7 and labelled with antibodies specific to Nkx2.5 and Mlc2v. Scale bar represents 10μm. G) Differentiated mESCs from Wnt3a-treated cultures were fixed on day 7 and labelled with antibodies specific to Nkx2.5 and pMypt1. Scale bar represents 10μm.
To determine if reduced cardiomyogenesis correlated with changes in MP phosphorylation, we examined levels of pMypt1T853 in Wnt3a and vehicle-treated cultures. Consistent with the results of Dkk1 treatment, Wnt3a treatment resulted in increased levels of pMypt1T853 (Fig. 6D). We next analyzed the subcellular distribution of endogenous Nkx2.5 in Wnt3a and vehicle-treated cultures. In vehicle-treated mESCs, Nkx2.5 was primarily localized to the nucleus, with only a small number (approximately 2%) of cells displaying a perinuclear pattern (Fig. 6E&F). In Wnt3a-treated cells however, the fraction of cells in which endogenous Nkx2.5 was excluded from the nucleus increased significantly to approximately 8% (Fig. 6E&F). In order to determine the differentiation status of these cells, we co-labelled them with antibodies against Mlc2v (Fig. 6F). Whereas cells which exhibited a nuclear pattern of Nkx2.5 expression differentiated into Mlc2v+ve cardiomyocytes, we did not observe any Mlc2v+ve cardiomyocytes in which Nkx2.5 was excluded from the nucleus, suggesting that this phenomenon is associated with a failure to undergo terminal differentiation. As in our transient transfection experiments, excluded Nkx2.5 co-localized with pMypt1T853 (Fig. 6G). Therefore, endogenous Nkx2.5 is excluded from the nucleus in response to Wnt3a treatment of differentiating mESCs and this exclusion correlates with a failure to differentiate.
Knockdown of Mypt1 results in enhanced cardiac gene expression and rescue of Wnt3a-mediated inhibition of cardiomyogenesis
We next sought to determine whether the endogenous Nkx2.5 and MP proteins interacted during mESC differentiation. Nkx2.5 was immunoprecipitated from differentiating mESCs using anti-Nkx2.5 antibodies and non-specific IgG; Nkx2.5 and Mypt1 proteins were visualized by western blotting. Consistent with our previous analyses, Mypt1 was detected in Nkx2.5-immunoprecipitated fractions, indicating that the endogenous proteins interact during mESC differentiation (Figure 7A). Interestingly, the major Nkx2.5 band identified in the immunoprecipitation migrated with the 50kDA marker, which is significantly larger than the predicted molecular weight of 34kDa. This has been observed in other systems42, 43 and likely represents post-translational modification of Nkx2.5. Given that the proteins form a complex during mESC differentiation, we next sought to determine if the loss of MP would enhance mESC differentiation into cardiomyocytes. We infected mESCs with lentiviruses expressing 3 shRNAs targeted to Mypt1, two of which (sh152 and sh346) produced stable lines exhibiting significant knockdown of Mypt1 expression (>60%) with a concomitant decrease in pMypt1T853 levels (~50%) (Fig. 7B&C). The two knockdown lines (termed shMypt1) were differentiated and the level of cardiac gene expression was compared to a stable cell line expressing a non-target (NT) shRNAmir control. Consistent with an inhibitory role for Mypt1, shMypt1 cells expressed significantly higher levels of Nkx2.5, Mlc2v and MyHC6 relative to control (Fig. 7D).
Figure 7. Knockdown of Mypt1 results in enhanced cardiac gene expression and rescue of Wnt3a-mediated inhibition of cardiomyogenesis.

A) Protein from differentiated mESCs was subjected to immunoprecipitation with Nkx2.5 antibodies or IgG. Immunoprecipitated fractions were analyzed by Western Blot with antibodies against Nkx2.5 and Mypt1. B) mESCs were infected with lentiviral particles carrying shRNAmir targeting sequences for Mypt1 or a non-targeting control. The levels of pMypt1T853 and Mypt1 were assessed by western blot and compared to α-tubulin. C) The levels of Mypt1 and pMypt1T853 knockdown were quantified by densitometry (n=3). D) shMypt1 cells and control cells were differentiated and the expression of the genes indicated was analyzed by QPCR (n=4, *p<0.05). E) Control and shMypt1 cells were differentiated and treated with Wnt3a or vehicle. Gene expression was assessed on day 7 (n=4, *p<0.05). F) The number of MyHC+ve cells was quantified in control and shMypt1 cells treated with Wnt3a or vehicle (n=6, *p<0.05). G) Knockdown and control cell lines were labelled with antibodies against MyHC and stained with Hoechst dye to detect nuclei.
To examine whether Mypt1 is required for inhibition of cardiomyogenesis by Wnt3a, we treated differentiating shMypt1 and control cells with Wnt3a and assessed the extent of cardiomyogenic differentiation by gene expression analysis and immunofluorescence. As in our previous experiments, the expression levels of Nkx2.5, MyHC6 and Cnx40 in control cells (Fig. 7E) were reduced upon treatment with Wnt3a. In shMypt1 cells however, there was no significant difference between vehicle and Wnt3a treatments, indicating that these cells are refractory to Wnt3a-mediated inhibition (Fig. 7E). We also observed an alleviation of Wnt3a inhibition by quantitative analysis of the number of MyHC+ve cells in control and shMypt1 cultures (Fig. 7F&G). These data are consistent with our model and support a mechanism in which Mypt1 is required for Wnt3a-mediated inhibition of cardiomyogenesis in mESCs.
DISCUSSION
Nkx2.5 is among the earliest genes expressed during cardiomyogenesis and regulates many targets genes that are essential for heart development.7, 8 Despite this, little is known about how Nkx2.5 function is regulated. We have shown that PP1β and Mypt1, subunits of the MP complex, are novel interacting partners of Nkx2.5 (S.Table 1 and Fig. 1A). Nkx2.5 interacted specifically with the phosphorylated form of MP, resulting in nuclear exclusion of Nkx2.5 and a decrease in its transcriptional activity, in a mechanism dependent on a newly identified Mypt1 NES (Fig. 1–4). This interaction was modulated by the Wnt inhibitor Dkk1, which reduced Mypt1 phosphorylation (Fig. 5). Consistent with this, treatment of differentiating mESCs with Wnt3a resulted in enhanced nuclear exclusion of endogenous Nkx2.5, associated with downregulation of cardiac gene expression and inhibition of cardiomyogenesis (Fig. 6). Finally, knockdown of Mypt1 enhanced overall cardiomyogenesis and rescued Wnt3a-mediated inhibition (Fig. 7). Taken together, these findings suggest a model whereby Wnt3a activates ROCK, which in turn phosphorylates MP, stimulating its translocation to the nucleus. MP then engages Nkx2.5 and the complex is exported to the perinuclear region via the Crm1 pathway. These findings are summarized in a model, shown in Figure 8.
Figure 8. MP interacts with Nkx2.5 and controls its subcellular localization downstream of ROCK/Wnt3a.
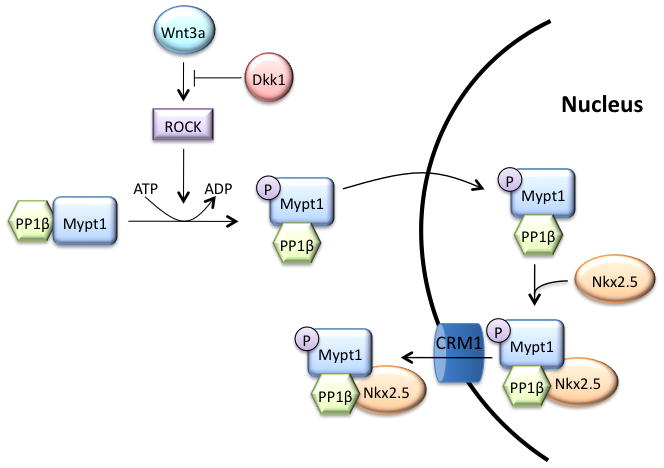
Under cardiomyogenic conditions, Nkx2.5 resides in the nucleus and activates cardiac gene expression. In response to Wnt3a signalling, Mypt1 is phosphorylated at Thr853 by ROCK, causing MP to translocate into the nucleus, where it can engage Nkx2.5 and form a complex, which is then exported from the nucleus in a Crm1-dependent pathway.
It has previously been reported that MP interacts with, and dephosphorylates HDAC7, causing it to translocate to the nucleus.44 We have shown that Nkx2.5 interacts with a form of the MP complex containing phosphorylated Mypt1. This phosphorylation event has profound inhibitory effects (>100-fold decrease in activity) on the phosphatase activity of the PP1β subunit, which result from docking of the phosphorylated T853 residue within its active site.33, 34 Therefore, it is unlikely that the regulation of the subcellular localization of Nkx2.5 is dependent on dephosphorylation by MP, although our data cannot rule out this possibility. We have also not ruled out that Nkx2.5 phosphorylation via other mechanisms may play a role in its subcellular distribution. However, we have shown that modulating ROCK activity can alter the subcellular redistribution of Nkx2.5 in response to coexpression with MP by regulating the levels of pMypt1T853. Notably, inhibition of ROCK activity significantly enhances survival of hESC-derived cardiomyocytes after dissociation of EBs.45 Given that Nkx2.5 protects cardiomyocytes from stress-induced cell death, it is tempting to speculate that changes in the subcellular distribution of Nkx2.5 may contribute to the protective effects conferred by ROCK inhibition.46
Wnt3a is known to inhibit terminal cardiac differentiation of mESCs, although mechanistic insight into this regulation is not fully developed.26 We have shown in this study that treatment of differentiating mESCs with Wnt3a results in exclusion of Nkx2.5 from the nucleus, which is accompanied by reduced cardiomyogenic differentiation. Consistent with this, knockdown of Mypt1 in mESCs results in rescue of Wnt3a-mediated inhibition of cardiomyogenesis. This represents a novel mechanism whereby Wnt3a signalling inhibits differentiation of cardiac progenitors. Although ROCK has traditionally been associated with the Wnt/PCP pathway, it is activated by Wnt3a 37, 38 and is required for β-catenin function.36 We have shown that Dkk1 can partially block ROCK-mediated phosphorylation of Mypt1, supporting a possible link between ROCK activation and signalling downstream of the Lrp5/6 receptor. While our data are consistent with recent work indicating that ROCK is involved in Wnt/β-catenin signalling36, others have shown that Dkk1 cannot inhibit Wnt activation of rho in myeloma cells.40 This difference may be representative of both the complexity of Wnt signalling and dissimilarity between the biological systems. Alternatively, it has been shown that Wnt3a treatment of mesenchymal stem cells results in increased expression of the ROCK2 isoform.36 Therefore, it is possible that the reduction in ROCK activity upon treatment with Dkk1 is reflective of reduced gene expression levels rather than direct inhibition of ROCK activation by rhoA. The extent of inhibition of Nkx2.5 exclusion by Dkk1 (≈40%) was stronger than expected based on the changes in Mypt1 phosphorylation levels (Figure 5). This may be indicative of a threshold effect whereby pMypt1T853 must accumulate within the nucleus at high levels in order for exclusion of MP/Nkx2.5 to be initiated or of additional roles for Dkk1 in this process.
Previous studies have shown that Nkx2.5 activity is not essential for cardiac differentiation of mESCs.9 Therefore, while promoting nuclear exclusion of Nkx2.5 may represent at least part of the anti-cardiomyogenic effects exerted by Wnt3a, additional mechanisms are likely at play, such as transcriptional regulation of Nkx2.5 and other important cardiomyogenic factors.41 It is also possible that nuclear exclusion of Nkx2.5 may contribute to enhanced protein turnover. Furthermore, given that knockdown of Mypt1 was able to fully rescue Wnt3a-mediated inhibition, it is possible that MP is involved in additional inhibitory pathways mediated by Wnt. Whether nuclear exclusion can also inhibit the repressive function of Nkx2.5 remains to be determined.12
A recent study47 showed that treatment of EBs with Wnt3a results in activation of a Wnt-responsive promoter throughout the EB and that this was delayed by treatment with Dkk1. Visualization of endogenous Wnt signalling using a reporter in EBs revealed that it is initially localized to distinct regions of the EB and is self-perpetuating until it encompasses the whole EB.47 We consistently observed exclusion of Nkx2.5 in localized ‘patches’ (Figure 7F). Perhaps the regions that fail to become Wnt-responsive are those that express endogenous antagonistic signalling molecules, such as Dkk1, that override Wnt3a and block exclusion of Nkx2.5, resulting in ‘patches’ of excluded and nuclear Nkx2.5. While we have shown that Wnt3a inhibition requires Mypt1, given that most cells retain Nkx2.5 in the nucleus even after treatment with Wnt3a, it is also likely that other mechanisms contribute to this pathway.
We have recently shown that MLCK enhances skeletal myogenesis by phosphorylating MEF2C.48 When considered together with the model described here, as well as work done by others, it appears that modulating the balance of myosin phosphorylation represents only one aspect of a more diverse role played by MLCK/MP. 44 In summary, we have found that interaction with MP regulates the subcellular distribution of Nkx2.5 downstream of Wnt3a and consequently modulates cardiomyogenesis in stem cells. These findings provide novel mechanistic insight into Wnt-mediated modulation of the cardiac cell fate in embryonic stem cells.
Supplementary Material
Novelty and Significance.
What Is Known?
The transcriptional activity of Nkx2.5 is important for proper heart formation.
In embryonic stem cells, Wnt3a signaling inhibits the formation of cardiac myocytes from cardioblast precursors.
Wnt3a signaling results in the phosphorylation and inhibition of the myosin phosphatase subunit Mypt1.
What New Information Does This Article Contribute?
Identification of myosin phosphatase as a novel interacting partner of Nkx2.5 protein.
Myosin phosphatase inhibits Nkx2.5-dependent transcription by mediating its export out of the nucleus.
Increased nuclear sequestration of NKx2.5 by Mypt1 phosphorylation dependent increase in the association between myosin phosphatase and NKx2.5 represents a new mechanism by which Wnt3a inhibits cardiomyogenic differentiation.
The transcription factor Nkx2.5 is critical for proper heart development; however, the mechanisms regulating its activity remain poorly understood.. We show here that myosin phosphatase binds Nkx2.5 and exports it from the nucleus, attenuating its nuclear transcriptional activity. This process is regulated by phosphorylation of the Mypt1 subunit by ROCK, which is in turn regulated by Wnt signaling. Knockdown of Mypt1 in embryonic stem cells rescues the inhibitory effects of Wnt3a on cardiomyogenesis, indicating that Wnt3a requires Mypt1 for inhibition. This is the first evidence that Nkx2.5 activity is regulated by signaling pathways that induce changes in sub-cellular localization. These findings also provide novel insight into the mechanisms by which Wnt inhibits the terminal stages of cardiomyogenesis in stem cells and may have implications for designing future cell therapy approaches.
Acknowledgments
The authors would like to acknowledge Laura Trinkle-Mulcahy, Masumi Eto and Anthony Firulli for the generous gift of plasmids.
SOURCES OF FUNDING
T.R. was supported by awards from the Heart and Stroke Foundation of Ontario and the Government of Ontario. J.P.L. was supported by awards from the Government of Ontario and the Natural Sciences and Engineering Research Council of Canada. D.F. is a Canada Research Chair (CRC) Tier I in Proteomics and Systems Biology. This work was supported by a grant to I.S.S. and M.R. from the Canadian Institutes of Health Research, MOP-53277. PLP is supported by R01AR056712 and R01AR052779 from the National Institute of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) as well as by the Muscular Dystrophy Association (186668).
Non-standard Abbreviations
- mESCs
mouse embryonic stem cells
- ANF
atrial natriuretic factor
- Cnx40
connexin40
- Mlc2v
Myosin light chain 2v
- PPI
protein phosphatase 1
- MP
myosin phosphatase
- MLCK
myosin light chain kinase
- ROCK
Rho-associated protein kinase
- TAP
Tandem affinity purification
- EB
embryoid body
- PLA
proximity ligation assay
- MyHC
myosin heavy chain
- SHF
second heart field
Footnotes
DISCLOSURES
The authors disclose no potential conflicts of interest.
References
- 1.Kelly RG, Brown NA, Buckingham ME. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev Cell. 2001;1:435–440. doi: 10.1016/s1534-5807(01)00040-5. [DOI] [PubMed] [Google Scholar]
- 2.Cai CL, Liang X, Shi Y, Chu PH, Pfaff SL, Chen J, Evans S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell. 2003;5:877–889. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. 2005;6:826–835. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- 4.Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 5.Akazawa H, Komuro I. Cardiac transcription factor Csx/Nkx2-5: Its role in cardiac development and diseases. Pharmacol Ther. 2005;107:252–268. doi: 10.1016/j.pharmthera.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 6.Reamon-Buettner SM, Borlak J. NKX2-5: an update on this hypermutable homeodomain protein and its role in human congenital heart disease (CHD) Hum Mutat. 2010;31:1185–1194. doi: 10.1002/humu.21345. [DOI] [PubMed] [Google Scholar]
- 7.Lyons I, Parsons LM, Hartley L, Li R, Andrews JE, Robb L, Harvey RP. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 1995;9:1654–1666. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka M, Chen Z, Bartunkova S, Yamasaki N, Izumo S. The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development. 1999;126:1269–1280. doi: 10.1242/dev.126.6.1269. [DOI] [PubMed] [Google Scholar]
- 9.Nakashima Y, Ono K, Yoshida Y, Kojima Y, Kita T, Tanaka M, Kimura T. The search for Nkx2-5-regulated genes using purified embryonic stem cell-derived cardiomyocytes with Nkx2-5 gene targeting. Biochem Biophys Res Commun. 2009;390:821–826. doi: 10.1016/j.bbrc.2009.10.056. [DOI] [PubMed] [Google Scholar]
- 10.Jamali M, Rogerson PJ, Wilton S, Skerjanc IS. Nkx2-5 activity is essential for cardiomyogenesis. J Biol Chem. 2001;276:42252–42258. doi: 10.1074/jbc.M107814200. [DOI] [PubMed] [Google Scholar]
- 11.Skerjanc IS, Petropoulos H, Ridgeway AG, Wilton S. Myocyte enhancer factor 2C and Nkx2-5 up-regulate each other’s expression and initiate cardiomyogenesis in P19 cells. J Biol Chem. 1998;273:34904–34910. doi: 10.1074/jbc.273.52.34904. [DOI] [PubMed] [Google Scholar]
- 12.Prall OW, Menon MK, Solloway MJ, Watanabe Y, Zaffran S, Bajolle F, Biben C, McBride JJ, Robertson BR, Chaulet H, Stennard FA, Wise N, Schaft D, Wolstein O, Furtado MB, Shiratori H, Chien KR, Hamada H, Black BL, Saga Y, Robertson EJ, Buckingham ME, Harvey RP. An Nkx2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell. 2007;128:947–959. doi: 10.1016/j.cell.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Targoff KL, Schell T, Yelon D. Nkx genes regulate heart tube extension and exert differential effects on ventricular and atrial cell number. Dev Biol. 2008;322:314–321. doi: 10.1016/j.ydbio.2008.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Durocher D, Charron F, Warren R, Schwartz RJ, Nemer M. The cardiac transcription factors Nkx2-5 and GATA-4 are mutual cofactors. EMBO J. 1997;16:5687–5696. doi: 10.1093/emboj/16.18.5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hiroi Y, Kudoh S, Monzen K, Ikeda Y, Yazaki Y, Nagai R, Komuro I. Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat Genet. 2001;28:276–280. doi: 10.1038/90123. [DOI] [PubMed] [Google Scholar]
- 16.Vincentz JW, Barnes RM, Firulli BA, Conway SJ, Firulli AB. Cooperative interaction of Nkx2.5 and Mef2c transcription factors during heart development. Dev Dyn. 2008;237:3809–3819. doi: 10.1002/dvdy.21803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, Seidman JG. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001;106:709–721. doi: 10.1016/s0092-8674(01)00493-7. [DOI] [PubMed] [Google Scholar]
- 18.Habets PE, Moorman AF, Clout DE, van Roon MA, Lingbeek M, van Lohuizen M, Campione M, Christoffels VM. Cooperative action of Tbx2 and Nkx2.5 inhibits ANF expression in the atrioventricular canal: implications for cardiac chamber formation. Genes Dev. 2002;16:1234–1246. doi: 10.1101/gad.222902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gessert S, Kuhl M. The multiple phases and faces of wnt signaling during cardiac differentiation and development. Circ Res. 2010;107:186–199. doi: 10.1161/CIRCRESAHA.110.221531. [DOI] [PubMed] [Google Scholar]
- 20.Liu P, Wakamiya M, Shea MJ, Albrecht U, Behringer RR, Bradley A. Requirement for Wnt3 in vertebrate axis formation. Nat Genet. 1999;22:361–365. doi: 10.1038/11932. [DOI] [PubMed] [Google Scholar]
- 21.Lindsley RC, Gill JG, Kyba M, Murphy TL, Murphy KM. Canonical Wnt signaling is required for development of embryonic stem cell-derived mesoderm. Development. 2006;133:3787–3796. doi: 10.1242/dev.02551. [DOI] [PubMed] [Google Scholar]
- 22.Bondue A, Lapouge G, Paulissen C, Semeraro C, Iacovino M, Kyba M, Blanpain C. Mesp1 acts as a master regulator of multipotent cardiovascular progenitor specification. Cell Stem Cell. 2008;3:69–84. doi: 10.1016/j.stem.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 23.Lindsley RC, Gill JG, Murphy TL, Langer EM, Cai M, Mashayekhi M, Wang W, Niwa N, Nerbonne JM, Kyba M, Murphy KM. Mesp1 coordinately regulates cardiovascular fate restriction and epithelial-mesenchymal transition in differentiating ESCs. Cell Stem Cell. 2008;3:55–68. doi: 10.1016/j.stem.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.David R, Brenner C, Stieber J, Schwarz F, Brunner S, Vollmer M, Mentele E, Muller-Hocker J, Kitajima S, Lickert H, Rupp R, Franz WM. MesP1 drives vertebrate cardiovascular differentiation through Dkk-1-mediated blockade of Wnt-signalling. Nat Cell Biol. 2008;10:338–345. doi: 10.1038/ncb1696. [DOI] [PubMed] [Google Scholar]
- 25.Ueno S, Weidinger G, Osugi T, Kohn AD, Golob JL, Pabon L, Reinecke H, Moon RT, Murry CE. Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc Natl Acad Sci U S A. 2007;104:9685–9690. doi: 10.1073/pnas.0702859104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naito AT, Shiojima I, Akazawa H, Hidaka K, Morisaki T, Kikuchi A, Komuro I. Developmental stage-specific biphasic roles of Wnt/beta-catenin signaling in cardiomyogenesis and hematopoiesis. Proc Natl Acad Sci U S A. 2006;103:19812–19817. doi: 10.1073/pnas.0605768103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsumura F, Hartshorne DJ. Myosin phosphatase target subunit: Many roles in cell function. Biochem Biophys Res Commun. 2008;369:149–156. doi: 10.1016/j.bbrc.2007.12.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kennedy KA, Porter T, Mehta V, Ryan SD, Price F, Peshdary V, Karamboulas C, Savage J, Drysdale TA, Li SC, Bennett SA, Skerjanc IS. Retinoic acid enhances skeletal muscle progenitor formation and bypasses inhibition by bone morphogenetic protein 4 but not dominant negative beta-catenin. BMC Biol. 2009;7:67. doi: 10.1186/1741-7007-7-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Savage J, Conley AJ, Blais A, Skerjanc IS. SOX15 and SOX7 differentially regulate the myogenic program in P19 cells. Stem Cells. 2009;27:1231–1243. doi: 10.1002/stem.57. [DOI] [PubMed] [Google Scholar]
- 30.Fan C, Liu M, Wang Q. Functional analysis of TBX5 missense mutations associated with Holt-Oram syndrome. J Biol Chem. 2003;278:8780–8785. doi: 10.1074/jbc.M208120200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SE, Shima H, Nakamura K, Kikuchi K. Broad specificity in binding of NIPP-1, nuclear inhibitor of protein phosphatase-1, to PP1 isoforms in vivo. Tohoku J Exp Med. 2000;191:39–45. doi: 10.1620/tjem.191.39. [DOI] [PubMed] [Google Scholar]
- 32.Lontay B, Kiss A, Gergely P, Hartshorne DJ, Erdodi F. Okadaic acid induces phosphorylation and translocation of myosin phosphatase target subunit 1 influencing myosin phosphorylation, stress fiber assembly and cell migration in HepG2 cells. Cell Signal. 2005;17:1265–1275. doi: 10.1016/j.cellsig.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 33.Muranyi A, Derkach D, Erdodi F, Kiss A, Ito M, Hartshorne DJ. Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: inhibitory effects and occurrence in A7r5 cells. FEBS Lett. 2005;579:6611–6615. doi: 10.1016/j.febslet.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 34.Khromov A, Choudhury N, Stevenson AS, Somlyo AV, Eto M. Phosphorylation-dependent autoinhibition of myosin light chain phosphatase accounts for Ca2+ sensitization force of smooth muscle contraction. J Biol Chem. 2009;284:21569–21579. doi: 10.1074/jbc.M109.019729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.la Cour T, Kiemer L, Molgaard A, Gupta R, Skriver K, Brunak S. Analysis and prediction of leucine-rich nuclear export signals. Protein Eng Des Sel. 2004;17:527–536. doi: 10.1093/protein/gzh062. [DOI] [PubMed] [Google Scholar]
- 36.Rossol-Allison J, Stemmle LN, Swenson-Fields KI, Kelly P, Fields PE, McCall SJ, Casey PJ, Fields TA. Rho GTPase activity modulates Wnt3a/beta-catenin signaling. Cell Signal. 2009;21:1559–1568. doi: 10.1016/j.cellsig.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kishida S, Yamamoto H, Kikuchi A. Wnt-3a and Dvl induce neurite retraction by activating Rho-associated kinase. Mol Cell Biol. 2004;24:4487–4501. doi: 10.1128/MCB.24.10.4487-4501.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Endo Y, Wolf V, Muraiso K, Kamijo K, Soon L, Uren A, Barshishat-Kupper M, Rubin JS. Wnt-3a-dependent cell motility involves RhoA activation and is specifically regulated by dishevelled-2. J Biol Chem. 2005;280:777–786. doi: 10.1074/jbc.M406391200. [DOI] [PubMed] [Google Scholar]
- 39.Garton AJ, Castaldo L, Pachter JA. Quantitative high-throughput cell-based assays for inhibitors of ROCK kinases. Methods Enzymol. 2008;439:491–500. doi: 10.1016/S0076-6879(07)00433-8. [DOI] [PubMed] [Google Scholar]
- 40.Qiang YW, Endo Y, Rubin JS, Rudikoff S. Wnt signaling in B-cell neoplasia. Oncogene. 2003;22:1536–1545. doi: 10.1038/sj.onc.1206239. [DOI] [PubMed] [Google Scholar]
- 41.Klaus A, Muller M, Schulz H, Saga Y, Martin JF, Birchmeier W. Wnt/beta-catenin and Bmp signals control distinct sets of transcription factors in cardiac progenitor cells. Proc Natl Acad Sci U S A. 2012;109:10921–10926. doi: 10.1073/pnas.1121236109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costa MW, Lee S, Furtado MB, Xin L, Sparrow DB, Martinez CG, Dunwoodie SL, Kurtenbach E, Mohun T, Rosenthal N, Harvey RP. Complex SUMO-1 regulation of cardiac transcription factor Nkx2-5. PLoS One. 2011;6:e24812. doi: 10.1371/journal.pone.0024812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang J, Zhang H, Iyer D, Feng XH, Schwartz RJ. Regulation of cardiac specific nkx2.5 gene activity by small ubiquitin-like modifier. J Biol Chem. 2008;283:23235–23243. doi: 10.1074/jbc.M709748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parra M, Mahmoudi T, Verdin E. Myosin phosphatase dephosphorylates HDAC7, controls its nucleocytoplasmic shuttling, and inhibits apoptosis in thymocytes. Genes Dev. 2007;21:638–643. doi: 10.1101/gad.1513107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Braam SR, Nauw R, Ward-van Oostwaard D, Mummery C, Passier R. Inhibition of ROCK improves survival of human embryonic stem cell-derived cardiomyocytes after dissociation. Ann N Y Acad Sci. 2010;1188:52–57. doi: 10.1111/j.1749-6632.2009.05083.x. [DOI] [PubMed] [Google Scholar]
- 46.Monzen K, Zhu W, Kasai H, Hiroi Y, Hosoda T, Akazawa H, Zou Y, Hayashi D, Yamazaki T, Nagai R, Komuro I. Dual effects of the homeobox transcription factor Csx/Nkx2-5 on cardiomyocytes. Biochem Biophys Res Commun. 2002;298:493–500. doi: 10.1016/s0006-291x(02)02497-x. [DOI] [PubMed] [Google Scholar]
- 47.ten Berge D, Koole W, Fuerer C, Fish M, Eroglu E, Nusse R. Wnt signaling mediates self-organization and axis formation in embryoid bodies. Cell Stem Cell. 2008;3:508–518. doi: 10.1016/j.stem.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Al Madhoun AS, Mehta V, Li G, Figeys D, Wiper-Bergeron N, Skerjanc IS. Skeletal myosin light chain kinase regulates skeletal myogenesis by phosphorylation of MEF2C. EMBO J. 2011 doi: 10.1038/emboj.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.