

Abstract
Photosystem II uses light to drive water oxidation and plastoquinone (PQ) reduction. PQ reduction involves two PQ cofactors, QA and QB, working in series. QA is a one-electron carrier, whereas QB undergoes sequential reduction and protonation to form QBH2. QBH2 exchanges with PQ from the pool in the membrane. Based on the atomic coordinates of the Photosystem II crystal structure, we analyzed the proton transfer (PT) energetics adopting a quantum mechanical/molecular mechanical approach. The potential-energy profile suggests that the initial PT to QB•– occurs from the protonated, D1-His252 to QB•– via D1-Ser264. The second PT is likely to occur from D1-His215 to QBH− via an H-bond with an energy profile with a single well, resulting in the formation of QBH2 and the D1-His215 anion. The pathway for reprotonation of D1-His215– may involve bicarbonate, D1-Tyr246 and water in the QB site. Formate ligation to Fe2+ did not significantly affect the protonation of reduced QB, suggesting that formate inhibits QBH2 release rather than its formation. The presence of carbonate rather than bicarbonate seems unlikely because the calculations showed that this greatly perturbed the potential of the nonheme iron, stabilizing the Fe3+ state in the presence of QB•–, a situation not encountered experimentally. H-bonding from D1-Tyr246 and D2-Tyr244 to the bicarbonate ligand of the nonheme iron contributes to the stability of the semiquinones. A detailed mechanistic model for QB reduction is presented.
Keywords: electron transfer gating, purple bacterial reaction center, low-barrier hydrogen bond, photoinhibition, tyrosine peroxide
The core of the Photosystem II (PSII) reaction center is composed of D1/D2, a heterodimer of protein subunits containing the cofactors involved in photochemical charge separation, quinone reduction, and water oxidation. These reactions are driven by light absorption by pigments absorbing around 680 nm (P680). P680 is composed of four chlorophyll a (Chla) molecules, PD1/PD2, ChlD1/ChlD2, and two pheophytin a molecules (PheoD1/PheoD2). Excitation of P680 initially leads to the formation of a range of charge separated states, with the ChlD1•+ PheoD1•− state dominating. After a short time the secondary radical pair, [PD1/PD2]•+ PheoD1•−, is formed in nearly all centers. This state is stabilized by electron transfer to the first quinone, QA, and by electron donation from a tyrosine residue, D1-Tyr160 (TyrZ), to PD1•+. TyrZ• then oxidizes the Mn4CaO5 cluster, which catalyzes the subsequent water splitting reaction. QA/QA•− acts as a one-electron redox couple, accepting electrons from PheoD1•− and donating to the second quinone, QB, without undergoing protonation itself. In contrast, QB reduction involves two consecutive one-electron reduction reactions with a series of associated proton uptake reactions (reviewed in 1–6).
QB is located near the nonheme Fe2+ and the ligand to the Fe2+, D1-His215, donates an H-bond to the QB carbonyl O atom that is nearer to the Fe complex (OQB(proximal)). The QB carbonyl O atom distal to the Fe complex (OQB(distal)) accepts an H-bond from D1-Ser264, which itself accepts an H-bond from D1-His252 (Fig. 1), which is located on the protein surface in contact with the aqueous medium (5–9). It is known that QB•− formation is linked to proton uptake (10, 11) and comparisons with the structure of the bacterial reaction center led to the first suggestion that the D1-His252 was the group undergoing protonation in response to QB•− formation (12). In theoretical studies, it has been proposed that proton uptake by D1-His252 causes reorientation of the hydroxyl group of D1-Ser264 toward the distal QB carbonyl group and stabilizes QB•−, facilitating the initial electron transfer (ET) from QA to QB (8).
Fig. 1.

Changes in the H-bond network geometry of QB in response to changes in the protonation/redox state. O and N atoms are depicted as red and blue spheres, respectively. Only the H atoms involved in H-bonds or protonation sites are depicted as cyan spheres.
Similar quinone reduction reactions occur in photosynthetic reaction centers from purple bacteria, which are thought to share a common ancestor with PSII (4, 13, 14). Notably, D1-Ser264 and D1-His252 in PSII are equivalent to Ser-L223 and Asp-L213, respectively, in the reaction center from Rhodobacter sphaeroides. Purple bacterial reaction centers contain an additional globular subunit, the H-subunit, that covers the quinone/Fe surface. Consequently, longer proton pathways exist to allow protons from the medium to reach the QB binding site. The residues involved in this proton transfer (PT) pathway include Glu-L212, Asp-L213, Ser-L223, Asp-M17, and several residues of the H-subunit (15, 16). Glu-L212 is protonated in response to the formation of QB•− on the first turnover. When Asp-L213 is protonated (via the same proton pathway), reorientation of the Ser-L223 hydroxyl group occurs and an H-bond forms between Ser-L223 and OQB(distal) (17–20). Protonation of QB•− to form QBH• occurs at OQB(distal) via Ser-L223 and Asp-L213 upon formation of the QA•− QB•− state. Electron transfer then takes place leading to formation of QBH−, which then undergoes protonation at OQB(proximal) forming QBH2, with the proton from Glu-L212 (15). For PSII the current state of knowledge lacks these details, but several of the reactions are considered to be similar (reviewed in 4–6, 21). There are, however, several obvious structural differences between the two systems that must result in mechanistic differences. Glu-L212, for example, which appears to be a prerequisite for the second protonation step in purple bacterial reaction centers (RCs) (15, 16), is replaced with Ala in PSII. Another potentially important difference is the presence of bicarbonate in PSII instead of the Glu-M234 in purple bacterial RCs. This exchangeable carboxylic acid has been linked to several phenomena specific to PSII, notably (i) slowed electron transfer when bicarbonate is replaced with other carboxylic acids, (ii) a specific EPR signal from the semiquinone–iron complex, and (iii) redox activity of the nonheme ion (4–6, 21).
Here, we investigated formation of QBH• and QBH2 in the PSII protein environment, by adopting a large-scale quantum mechanical/molecular mechanical (QM/MM) approach based on the crystal structure with resolution at 1.9 Å (9).
Results and Discussion
First Protonation Step: Conversion of QB•– to QBH• via D1-His215 and D1-Ser264.
To elucidate how the conversion of QB•– to QBH• occurs, we analyzed the potential-energy profiles of the two H-bonds of QB•–, OQB(distal)…H–OD1-Ser264 and OQB(proximal)…H–NδD1-His215. Because in previous electrostatic calculations, D1-His252 became protonated upon formation of the QB•– state (8), we also assumed the presence of protonated D1-His252 and the QB•– state in the present QM/MM calculation. In general, serine is unlikely to deprotonate. However, in the QB•– state, the potential-energy profile indicates that a PT from D1-Ser264 to OQB(distal) occurred very easily in an energetically downhill process (Fig. 2). This reaction was accompanied by a concerted PT from protonated D1-His252 to D1-Ser264, resulting in the formation of QB(distal)H•, deprotonated (neutral) D1-His252, and reoriented D1-Ser264 (Fig. 1, first and second panels). The QM/MM-optimized geometry indicates that the two H-bonds of D1-Ser264, OQB(distal)…H–OD1-Ser264 (2.48 Å) and OD1-Ser264–H…NδD1-His252. (2.51 Å), are unusually short, especially in the QB•– state (Table 1). The two short H-bonds were only present before the initial PT occurred, but they lengthened (to 2.73 and 2.67 Å, respectively) immediately after PT had occurred. Therefore, the presence of an unusually short H-bond indicates that PT between the donor and acceptor moieties is about to occur.
Fig. 2.

Potential-energy profiles of the H-bond donor–acceptor pairs: (Right) H-bond between D1-Ser264 and the distal QB carbonyl (purple, in the neutral QA state, and blue, in the reduced QA state, curves); (Left) H-bond between D1-His215 and the proximal QB carbonyl (red curves). At each point, all of the atomic coordinates in the QM region were fully relaxed (i.e., not fixed). Arrows indicate the directions of PT.
Table 1.
H-bond distances in optimized geometries in the PSII protein environment near QB systems (in Å)
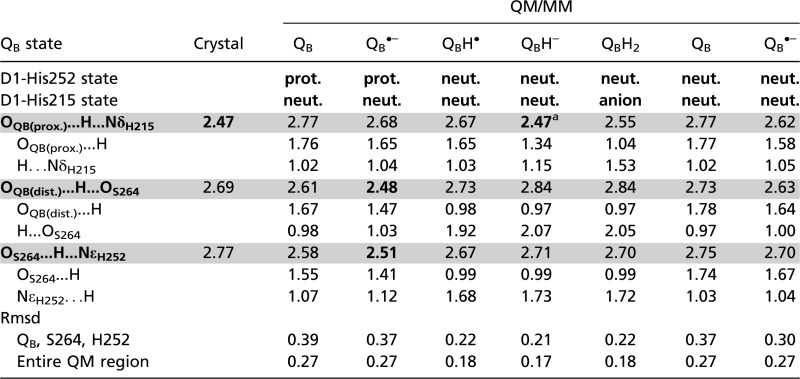 |
H-bond donor-acceptor distances of <∼2.5 Å are indicated in bold. H-bond donor-acceptor distances are shaded in gray. Crystal = 1.9-Å structure (9) (PDB entry 3ARC). anion, doubly deprotonated (negatively charged) His; prot., doubly protonated (positively charged) His; neut., singly protonated (either at Nδ or Nɛ) His.
a2.54 Å when the bicarbonate (HCO3–) ligand was replaced with carbonate (CO32–).
In contrast, the potential-energy profile of the OQB(proximal)…H–NδD1-His215 (Fig. 2) resembles that of a standard asymmetric double-well H-bond (22) (Fig. S1), suggesting that the first PT from D1-His215 to OQB(proximal) is an energetically uphill process. This is primarily because proton release from the singly protonated (neutral) His (pKa ∼14 for imidazole) (23) is unfavorable, unlike the doubly protonated (positively charged) His, for which the pKa ∼7. Although the pKa for neutral His is expected to be lowered to some extent by the positive charge and environment around the iron (see below), it is still likely to be relatively high and thus unfavorable on this step.
The potential-energy profiles in Fig. 2 indicate that the initial PT occurs more favorably when QA•– is present compared with the neutral QA state. This is similar to the situation occurring in the purple bacterial RC, where QB•– is unprotonated on the first flash and the QB•– to QBH• step only occurs when the [QA•– QB•–] state is formed, before the second electron transfer step (15, 16).
Overall, the results suggest the following model for the first protonation: Protonation of QB•– to QBH• primarily occurs at OQB(distal), this occurs as a result of concerted PT from D1-His252(H+) to OQB(distal) via D1-Ser264, and this induces reorientation of the D1-Ser264 hydroxyl group so that it can act as a H-bond acceptor from the QBH•. The preferential occurrence of the first protonation at OQB(distal) over OQB(proximal) is consistent with the QB protonation mechanism in purple bacterial RCs (15, 16, 24).
Second PT and an Unusually Short H-Bond Distance Between QBH– and D1-His215.
Because OQB(distal) is protonated upon QBH• formation (Fig. 2), the second protonation—i.e., the conversion of QBH− to QBH2—must occur at OQB(proximal), which is H-bonded by Nδ of D1-His215 (Fig. 1). The QM/MM optimized H-bond distance between OQB(proximal) of QBH– and Nδ of D1-His215 was found to be unusually short (2.47 Å) in the QBH– state (Table 1).
Intriguingly, this distance is identical to that in the 1.9 Å structural model, PSII monomer unit “A” of the PSII complexes (9). The corresponding OQB(proximal)–NδD1-His215 distances were found to be 2.77 Å in the QB state and 2.68 Å in the QB•– state (Table 1), both being significantly longer compared with that in the QBH– state. In addition, the corresponding QA-side distance (OQA(proximal)–NδD2-His214) was 2.78 Å in the 1.9 Å structure (9) and ∼2.8–2.9 Å in the purple bacterial RC (16). Given that the QB geometry was less well defined than QA geometry in the 1.9 Å structure (9), the significance of the short OQB(proximal)–NδD1-His215 distance (2.47 Å) distance should be treated with caution. Furthermore, the QBH– state is expected to be a short-lived intermediate and not a state that would be present and in PSII under normal circumstances. Nevertheless, more discussion of the possible redox states of QB in the crystal structure is given in the SI Text.
In a typical H-bond with an O–O distance longer than ∼2.6 Å, an H-atom is located near the donor moiety owing to the larger pKa value of the donor moiety relative to the acceptor moiety (having an asymmetric double-well potential H-bond) (22) (Fig. S1). On the other hand, according to the classification of H-bonds by Jeffrey (25) or Frey (26), short H-bonds with O–O distances of 2.4–2.5 Å can be classified as single-well (ionic) H-bonds (22) (Fig. S1). Because O–N distances are generally greater than O–O distances, the OQB(proximal)–NδD1-His215 of 2.47 Å is an unusually short H-bond and may possess a single-well potential. Remarkably, the calculated potential-energy profile for the OQB(proximal)–NδD1-His215 in the QBH– state resembled that of a barrierless single-well (ionic) H-bond, suggesting that the second PT can occur isoenergetically at OQB(proximal) (Fig. 2). The significantly elongated H–N bond of D1-His215 (1.15 Å) in the QBH– state implies that further migration of an H atom toward the acceptor OQB(proximal) moiety (i.e., PT) can occur easily. Indeed, the single-well potential obtained for OQB(proximal)–NδD1-His215 is symmetric (Fig. 2), implying that the pKa difference (27) between D1-His215 deprotonation and QBH– protonation is close to zero.
The pKa for the QH– to QH2 protonation for plastoquinone (PQ) is expected to be similar to that measured for ubiquinone in aqueous solution—i.e., 10.7, significantly higher than the pKa of 4.9 for the protonation of the semiquinone, Q•– to QH• (18, 28). The pKa for deprotonation of a neutral His is expected to be similar to that for imidazole—i.e., ∼14 (23). In PSII, however, the ligation of D1-His215 to the positively charged Fe2+ should lower the pKa of neutral D1-His215. The pKa of the neutral His ligand to Fe2+ in the Rieske (2Fe-2S) cluster has been measured to be ∼12.5 rather than ∼14 (29, 30). The ligand environment of the Fe2+ in PSII is more positively charged than that in the Rieske cluster; thus, the pKa of neutral D1-His215 deprotonation is expected to be lower than ∼12.5. In agreement with this, FTIR studies have indicated that deprotonation of D1-His215 occurs in response to pH changes (31). Overall then the literature suggests that the pKa value of D1-His215 is likely to be close to that for the QBH– to QBH2 protonation, in accordance with the single-well potential obtained here (Fig. 2). For further details, see Table S1.
It has been proposed that Glu-L212 in purple bacterial RC from R. sphaeroides provides a proton to QBH– (15, 32, 33). This residue is 5.7 Å away from OQB(proximal) (34), and it is not clear how this last protonation reaction occurs. One possibility is that the QBH– state is also protonated by the Fe-ligated imidazole (His-L190) and that Glu-L212 provides a proton to the deprotonated His residue (His-L190) facilitating the release of QBH2 from the site. Such a scenario has been discussed (and disfavored) previously (15). Ionizable residues corresponding to Glu-L212 are absent near QB in PSII. PSII may not require the corresponding residue, as the quinones are more exposed to the aqueous phase. Doubly deprotonated D1-His215 may be reprotonated via the bicarbonate ligand; indeed, a role for the bicarbonate in protonation of QB has been considered for many years (21). The distance between OQB(proximal) and Obicarbonate is 4.8 Å (9). Despite this long distance, rapid PT may be possible if water intermediate(s) were involved. In purple bacteria, water intermediates may also mediate PT from Glu-L212 to QBH–, perhaps via the His-L190 anion.
H-Bond Pattern of the Tyrosine Residue Pair near the Bicarbonate Ligand.
FTIR studies by Takahashi et al. have suggested that only one of the two tyrosine residues, either D1-Tyr246 or D2-Tyr244, provides an H-bond to bicarbonate (35). We investigated the influence of the H-bond pattern of these tyrosine residues on the stability of QA•– and QB•–. To do this, we redefined the QM region such that it included QA, the nonheme Fe and its ligands, QB, D1-Tyr246 and D2-Tyr244. Note that in the calculations, in the absence of the PSII protein environment (i.e., in vacuum), electrons were almost evenly distributed over QA and QB owing to the structural symmetry (Table S2).
When the tyrosine hydroxyl groups were oriented toward QA in the presence of deprotonated (neutral) D1-His252, QA•– formation (QA•– favored orientation) was more pronounced ([QA]–0.50 [Fe(His)4(HCO3)–]+0.77 [QB]–0.22) with values of QA/QB = 50/22 for electron distribution and 69/33 for spin distribution (Table S2; Fig. 3). On the other hand, when the tyrosine hydroxyl groups were oriented toward QB in the presence of protonated D1-His252 (QB•– favored orientation), QB•– formation was high ([QA]+0.04 [Fe(His)4(HCO3)–]+0.80 [QB]–0.84; QA/QB = 0/84 for electron distribution and 8/98 for spin distribution) (Table S2; Fig. 3). QB•– formation was predominantly influenced by the protonation state of D1-His252 and orientation of D1-Ser264, as suggested in previous theoretical studies (8). On the other hand, donation of an H-bond from D1-Tyr246 to bicarbonate (i.e., QA•– favored orientation) appears to play an important role particularly in the stability of QA•–, because D1-His252/D1-Ser264-like residues are absent near QA. The orientation of the tyrosines may well be related to communication between the quinone sites via the H-bonding network of bicarbonate, gating, and/or protonation reactions during electron transfer (Fig. 3).
Fig. 3.

H-bond arrangements of D1-Tyr246, D2-Tyr244, and the bicarbonate ligand of the nonheme Fe complex upon formation of QA•– (H atoms in cyan and C atoms in blue) or QB•– (H atoms in pink and C atoms in yellow). For clarity, D1-His272 and D2-His268, which were also included in the QM region, are not shown in the figure. Atomic coordinates are provided in Dataset S1.
The two calculated conformations shown in Fig. 3 differ not only in terms of the orientations of D1-Tyr246 and D2-Tyr244 but also in terms of the orientation of bicarbonate, implying a link between bicarbonate and the quinone redox states as suggested by FTIR studies (31, 35). The two tyrosine residues are located in the D-de loop region (D1-225–250 and D2-224–248), which is believed to be crucial to the stability of the QA•– state (36). The corresponding loop region is absent in the purple bacterial RC. Thus, the semiquinone stabilization mechanism, involving D1-Tyr246, D2-Tyr244, bicarbonate, and the rearrangement of the H-bond network, as suggested here, is only relevant to PSII.
Recently it was suggested that the ligation of the Fe2+ by bicarbonate changes during electron transfer, going from bidentate to monodentate upon formation of QA•– and returning to the bidentate form upon electron transfer to QB (37). In ref. 37, a monodentate ligand yielded two significantly different Fe–Obicarbonate distances of 2.3 and 3.2 Å for QB•– formation. Because the atomic coordinates were unavailable, we could not evaluate the specific models discussed in ref. 37, however we addressed the same question in our calculations using the most recent structure (9). We could not observe such a dramatic (∼1 Å) change of the Fe–Obicarbonate bond. Our calculations showed that bicarbonate was clearly a bidentate ligand in both conformers irrespective of the reduction state of the two quinones: the two Fe–Obicarbonate distances were 2.21 and 2.29 Å in the presence of QA•– and 2.27 and 2.32 Å in the presence of QB•– (Fig. 3). We conclude that the formation of stable QA•– or QB•– states do not result in dramatic differences in the bicarbonate ligation such as those proposed by Chernev et al. (37). It seems possible that the changes in the environment of the nonheme Fe2+ reported by Chernev et al. using X-ray absorption spectroscopy could have resulted from the changes in the H-bond network associated with D1-Tyr246, D2-Tyr244, and the bicarbonate (Fig. 3). These results appear to fit better with FTIR studies by Takahashi et al. (35). A further argument against electron transfer-induced changes in the ligation of the nonheme Fe2+ comes from EPR studies. The EPR spectra from QA•– Fe2+ formed at room and cryogenic temperature and QB•– Fe2+ are all essentially the same (38, 39). A difference in the number of ligands to the Fe would be expected to result in more marked differences in these spectra.
Replacement of the Bicarbonate Ligand.
Formate.
The depletion of (bi)carbonate or its substitution by formate results in a slowing of quinone reduction (6, 21). Recent EPR studies have suggested that formate inhibits release of QBH2 (39). The PT mechanisms reported here suggest that the efficient release of QBH2 requires the reprotonation of the anionic D1-His215. This process may involve bicarbonate, forming carbonate and releasing a proton to anionic D1-His215, as suggested here and earlier (21, 31). When bicarbonate is replaced with formate, the potential-energy profile indicates that the second protonation remains essentially unchanged and can occur isoenergetically at OQB(proximal) (Fig. S2). Note that the calculated OQB(proximal)–NδD1-His215 distance was 2.49 Å in the QBH– state with the formate ligand, which is essentially the same as that with bicarbonate ligand (2.47 Å, Table 1). In the EPR work, a new EPR signal was reported from the formate-inhibited enzyme when reduced by three electrons, attributed to QA•– Fe2+ in the presence of a two-electron reduced form of QB (39). In light of the present work, we suggest that this could correspond to QA•– Fe2+ QBH2 with the anionic D1-His215.
Carbonate.
Recently, the characteristic g-value of ∼1.9 for the semiquinone and nonheme Fe complex in EPR spectroscopy (40) was reinvestigated in theoretical simulations. On the basis of the simulations, it was proposed that the native ligand to the nonheme Fe was carbonate (CO32–) rather than bicarbonate (HCO3–) (41). In contrast, FTIR studies suggested that the bicarbonate ligand does not deprotonate even upon oxidation of the nonheme Fe (42). When the bicarbonate was substituted with fully ionized carbonate in our calculations, the QBH– to QBH2 protonation process became slightly energetically uphill and the OQB(proximal)–NδD1-His215 distance lengthened to 2.54 Å in the QBH– state (Table 1 and Fig. S2). These results suggest that the pKa for D1-His215 deprotonation was upshifted upon replacement of bicarbonate with carbonate and that the ligand plays a role in affecting the pKa of D1-His215, as proposed previously (31, 41).
We also calculated the charge distribution in the PSII protein environment with carbonate (QM region: QA, the nonheme Fe, the His and carbonate ligands, QB, D1-Tyr246, and D2-Tyr244). Starting with carbonate-ligated Fe2+ and two neutral quinones, the QM/MM calculations resulted in the oxidized Fe3+, a reduced quinone, and a neutral quinone (Table S3). QM/MM calculations for the carbonate-ligated Fe2+, one reduced quinone, and one neutral quinone resulted in oxidized Fe3+ and two reduced quinones. These results suggest that the fully ionized carbonate ligand forces Fe2+ to release an electron to one of the quinones. This does not reflect experimental findings, so these results argue against carbonate being a stable ligand to the iron and indicate another explanation must be found for the characteristic EPR spectra of the semiquinone-iron signals seen in PSII. More discussion of the possible carbonate ligand is given in the SI Text.
Tyrosine Peroxide at D1-Tyr246, Possible Link with Photoinhibition, and a Water-Mediated H-Bond Network.
In both of the PSII monomer units of the 1.9-Å structure, an elongation of density is clearly seen near the hydroxyl O atom of D1-Tyr246. In QM/MM calculations, atomic coordinates of a tyrosine peroxide (Tyr-OOH) form (rather than a tyrosine with a water molecule) fitted to the density (Fig. 4). Root-mean-square deviation of the optimized coordinates for Tyr-OOH with respect to those in the 1.9-Å structure is small (0.123 Å), even slightly smaller than that for Tyr-OH (0.149 Å). A Tyr-OOH at this position may be relevant to the function of PSII and is particularly intriguing in light of the literature: (i) Under strong light, the nonheme Fe2+ is thought to be involved in the generation of hydroxyl radicals (OH•) via iron-peroxo intermediates (43, 44) and Tyr-OOH can be generated in the presence of OH• (45). (ii) The D-de loop (D1-238–249 including D1-Tyr246) was proposed to be the first target for cleavage during photodegradation of D1 protein (46) and OH• may trigger this process (44). (iii) Spectroscopic studies suggest that the nonheme Fe undergoes an increase in its redox potential and a minor modification of the bicarbonate binding site during photodamage (47). These and other observations in the literature could be linked to Tyr-OOH formation.
Fig. 4.

(Upper) The electron density map in the neighborhood of D1-Tyr246. QM/MM optimized geometry of D1-Tyr246 is shown as (Left) Tyr-OOH or (Right) Tyr-OH and H2O in the presence of QB. (Lower) H-bond network (red dotted lines) linking bicarbonate to D1-His215. D1-Tyr246 was modeled as Tyr-OH. (Left) A single water molecule (red cross) is required to establish the H-bond network with His215 in the presence of QBH2, (Right) whereas two water molecules are required in the absence of QBH2.
Although it is possible that D1-Tyr246 may become a peroxide under photoinhibitory conditions, its presence in the crystal structure could represent OH• generated by the X-ray beam (48). The absence of the peroxide-like density for its counterpart D2-Tyr244 and the presence in both monomers in the crystal structure suggests that peroxide generation specifically occurs at D1-Tyr246. The long distances (4.8/6.5 Å) between bicarbonate and D1-His215/QBH– imply that a direct PT would be kinetically unfavorable. QM/MM optimized geometries indicate that inclusion of water completes an H-bond network from bicarbonate to D1-His215 (Fig. 4). There is, however, no resolved water molecule in this region in the 1.9 Å structure (9). We speculate that the proposed Tyr-OOH at D1-Tyr246 could have arisen from Tyr-OH and a bridging water molecule. Photolytic-generation of Tyr-OOH at D1-Tyr246 is expected to remove the water molecule and disconnect the PT path between bicarbonate and D1-His215. The presence of Tyr-OOH specifically at D1-Tyr246 but its absence at its counterpart D2-Tyr244 on the QA side fits with the expectation that protons are excluded from the QA side, as it functions as a one-electron couple that does not show pH dependence (49).
Working Model.
Based on the findings reported here and the literature discussed above, we are able to propose a mechanism for quinone reduction in PSII: (i) Protonation of D1-His252 occurs upon electron transfer from QA•– to QB forming QB•–. (ii) The presence of QA•–, formed on the second turnover, triggers protonation of the QB•– from D1-Ser264 with the concerted arrival on the serine of a proton from the protonated D1-His252, leading to neutral histidine formation. (iii) As in the purple bacterial reaction center, QBH• may be transiently formed before the second electron arrives from QA•–, forming QBH–. (iv) QBH– has a single-well H-bond to the D1-His215 (ligand to the nonheme iron) and this favors the PT forming QBH2, which remains H-bonded to the D1-His215 anion. (v) The release of QBH2 is facilitated by reprotonation of D1-His215 anion and this may occur through water in the QB-site, which is part of an H-bonded network with the bicarbonate (a ligand to the Fe2+) and groups exposed to the aqueous medium. (vi) The orientation of the tyrosine hydroxyl groups (e.g., D1-Tyr246) contributes to stabilizing reduced states of quinones. D1-Tyr246 may be involved in the H-bond network involving bicarbonate, water, D1-His215, and QBH2. This working model should allow specific features to be tested by future experimentation.
Computational Procedures
Coordinates and Atomic Partial Charges.
The atomic coordinates of PSII were taken from the X-ray structure of PSII monomer unit “A” of the PSII complexes from Thermosynechococcus vulcanus at a 1.9-Å resolution (PDB code, 3ARC) (9). Hydrogen atoms were generated and energetically optimized with CHARMM (50), whereas the positions of all nonhydrogen atoms were fixed, and all titratable groups were kept in their standard protonation states (i.e., acidic groups were ionized and basic groups were protonated). For the QM/MM calculations, we added additional counter-ions to neutralize the entire system. Atomic partial charges of the amino acids were adopted from the all-atom CHARMM22 (51) parameter set. The atomic charges of Chla, Pheoa, and quinones were taken from our previous studies on PSII (52). We considered the Mn4CaO5 cluster as the (O4)2−(O5)H− model (53) in the S1 state (see ref. 53 for the atomic coordinates and charges).
QM/MM Calculations.
We used the electrostatic embedding QM/MM scheme, in which electrostatic and steric effects created by a protein environment were explicitly considered, and we used the Qsite (54) program code as used in previous studies (52). Owing to the large system size of PSII, we considered residues and cofactors in only the D1 and D2 subunits as the protein environment. We used the unrestricted density functional theory method with the B3LYP functional and LACVP**+ basis sets. To analyze the effects of the H-bond pattern near the bicarbonate-tyrosine moiety, the QM region was defined as (QA, QB, bicarbonate, Fe, D1-His215, D1-His272, D2-His214, D2-His268, D1-Tyr246, and D2-Tyr244), whereas other protein units and all cofactors were approximated by the MM force field. To analyze the H-bond potential energy profiles, the QM region was redefined as (QB, D1-His252, D1-Ser264, bicarbonate, Fe, D1-His215, D1-His272, D2-His214, and D2-His268). The tail of PQ was replaced with a methyl group at C11. As in a previous study (37), we assumed a high-spin state (S = 2) of Fe2+ and set the spin multiplicity of the system to S = 2 in calculations for QB, QBH–, and QBH2, and S = 5/2 for QB•–, QBH•, and [QAQB] •–. The geometries were refined by constrained QM/MM optimization. Specifically, the coordinates of the heavy atoms in the surrounding MM region were fixed to the original X-ray coordinates, whereas those of the H atoms in the MM region were optimized using the OPLS2005 force field. All of the atomic coordinates (except for the C11 atom of PQs) in the QM region were fully relaxed (i.e., not fixed) in the QM/MM calculation.
The potential-energy profile of the H-bond was obtained as follows: First, we prepared for the QM/MM optimized geometry without constraints, and we took the resulting geometry as the initial geometry. The H atom was then moved from the H-bond donor atom (Odonor) to the acceptor atom (Oacceptor) by 0.05 Å, after which the geometry was optimized by constraining the Odonor–H and H–Oacceptor distances, and the energy of the resulting geometry was calculated. This procedure was repeated until the H atom reached the Oacceptor atom.
Supplementary Material
Acknowledgments
We thank James Murray of Imperial College for useful discussion. This research was supported by the Japan Science and Technology Agency, Precursory Research for Embryonic Science and Technology program (K.S. and H.I.); Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan [22740276 (K.S.)]; Special Coordination Fund for Promoting Science and Technology of MEXT (H.I.); Takeda Science Foundation (H.I.); Kyoto University Step-Up Grant-in-Aid for young scientists (H.I.); and a Grant for Basic Science Research Projects from the Sumitomo Foundation (H.I.). A.W.R. is the recipient of the Wolfson Merit Award of the Royal Society.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1212957110/-/DCSupplemental
References
- 1.Diner BA, Rappaport F. Structure, dynamics, and energetics of the primary photochemistry of photosystem II of oxygenic photosynthesis. Annu Rev Plant Biol. 2002;53:551–580. doi: 10.1146/annurev.arplant.53.100301.135238. [DOI] [PubMed] [Google Scholar]
- 2.Renger G, Renger T. Photosystem II: The machinery of photosynthetic water splitting. Photosynth Res. 2008;98(1-3):53–80. doi: 10.1007/s11120-008-9345-7. [DOI] [PubMed] [Google Scholar]
- 3.Holzwarth AR. Ultrafast primary reactions in the photosystems of oxygen evolving organisms. In: Braun M, Gilch P, Zinth W, editors. Ultrashort Laser Pulses in Biology and Medicine, Biological and Medical Physics, Biomedical Engineering. Dordrecht, The Netherlands: Springer; 2008. pp. 141–164. [Google Scholar]
- 4.Cardona T, Sedoud A, Cox N, Rutherford AW. Charge separation in photosystem II: A comparative and evolutionary overview. Biochim Biophys Acta. 2012;1817(1):26–43. doi: 10.1016/j.bbabio.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 5.Müh F, Glöckner C, Hellmich J, Zouni A. Light-induced quinone reduction in photosystem II. Biochim Biophys Acta. 2012;1817(1):44–65. doi: 10.1016/j.bbabio.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 6.Petrouleas V, Crofts AR. The iron-quinone acceptor complex. In: Wydrzynski T, Satoh K, editors. Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase. Dordrecht, The Netherlands: Springer; 2005. pp. 177–206. [Google Scholar]
- 7.Kawakami K, Umena Y, Kamiya N, Shen J-R. Structure of the catalytic, inorganic core of oxygen-evolving photosystem II at 1.9 Å resolution. J Photochem Photobiol B. 2011;104(1-2):9–18. doi: 10.1016/j.jphotobiol.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 8.Ishikita H, Knapp E-W. Control of quinone redox potentials in photosystem II: Electron transfer and photoprotection. J Am Chem Soc. 2005;127(42):14714–14720. doi: 10.1021/ja052567r. [DOI] [PubMed] [Google Scholar]
- 9.Umena Y, Kawakami K, Shen J-R, Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature. 2011;473(7345):55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 10.Robinson HH, Crofts AR. Kinetics of proton uptake and the oxidation-reduction reactions of the quinone acceptor complex of photosystem_II from pea chloroplasts. In: Sybesma C, editor. Advances in Photosynthesis Research. Vol 1. The Hague, The Netherlands: Martinus Nijhoff/Dr. W. Junk Publishers; 1984. pp. 477–480. [Google Scholar]
- 11.Rutherford AW, Renger G, Koike H, Inoue Y. Thermoluminescence as a probe of photosystem II. The redox and protonation states of the secondary acceptor quinone and the O2-evolving enzyme. Biochim Biophys Acta. 1984;767(3):548–556. [Google Scholar]
- 12.Crofts AR, et al. Catalytic sites for reduction and oxidation of quinones. In: Papa S, Chance B, Ernster L, editors. Cytochrome Systems: Molecular Biology and Bioenergetics. New York: Plenum; 1987. pp. 617–624. [Google Scholar]
- 13.Michel H, Deisenhofer J. Relevance of the photosynthetic reaction center from purple bacteria to the structure of photosystem II. Biochemistry. 1988;27:1–7. [Google Scholar]
- 14.Rutherford AW, Faller P. Photosystem II: Evolutionary perspectives. Philos Trans R Soc Lond B Biol Sci. 2003;358(1429):245–253. doi: 10.1098/rstb.2002.1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wraight CA. Proton and electron transfer in the acceptor quinone complex of photosynthetic reaction centers from Rhodobacter sphaeroides. Front Biosci. 2004;9:309–337. doi: 10.2741/1236. [DOI] [PubMed] [Google Scholar]
- 16.Okamura MY, Paddock ML, Graige MS, Feher G. Proton and electron transfer in bacterial reaction centers. Biochim Biophys Acta. 2000;1458(1):148–163. doi: 10.1016/s0005-2728(00)00065-7. [DOI] [PubMed] [Google Scholar]
- 17.Ishikita H, Knapp E-W. Variation of Ser-L223 hydrogen bonding with the QB redox state in reaction centers from Rhodobacter sphaeroides. J Am Chem Soc. 2004;126(25):8059–8064. doi: 10.1021/ja038092q. [DOI] [PubMed] [Google Scholar]
- 18.Zhu Z, Gunner MR. Energetics of quinone-dependent electron and proton transfers in Rhodobacter sphaeroides photosynthetic reaction centers. Biochemistry. 2005;44(1):82–96. doi: 10.1021/bi048348k. [DOI] [PubMed] [Google Scholar]
- 19.Paddock ML, et al. ENDOR spectroscopy reveals light induced movement of the H-bond from Ser-L223 upon forming the semiquinone (QB–•) in reaction centers from Rhodobacter sphaeroides. Biochemistry. 2007;46(28):8234–8243. doi: 10.1021/bi7005256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin E, et al. Hydrogen bonding and spin density distribution in the QB semiquinone of bacterial reaction centers and comparison with the QA site. J Am Chem Soc. 2011;133(14):5525–5537. doi: 10.1021/ja2001538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shevela D, Eaton-Rye JJ, Shen J-R. Govindjee (2012) Photosystem II and the unique role of bicarbonate: A historical perspective. Biochim Biophys Acta. 1817(8):1134–1151. doi: 10.1016/j.bbabio.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 22.Perrin CL, Nielson JB. “Strong” hydrogen bonds in chemistry and biology. Annu Rev Phys Chem. 1997;48:511–544. doi: 10.1146/annurev.physchem.48.1.511. [DOI] [PubMed] [Google Scholar]
- 23.Bruice TC, Schmir GL. Imidazole catalysis. II. The reaction of substituted imidazoles with phenyl acetates in aqueous solution. J Am Chem Soc. 1958;80(1):148–156. [Google Scholar]
- 24.Rabenstein B, Ullmann GM, Knapp E-W. Energetics of electron-transfer and protonation reactions of the quinones in the photosynthetic reaction center of Rhodopseudomonas viridis. Biochemistry. 1998;37(8):2488–2495. doi: 10.1021/bi971921y. [DOI] [PubMed] [Google Scholar]
- 25.Jeffrey GA. An Introduction to Hydrogen Bonding. Oxford, UK: Oxford Univ Press; 1997. [Google Scholar]
- 26.Frey PA. In: Isotope Effects in Chemistry and Biology. Kohen A, Limbach H-H, editors. Boca Raton, FL: CRC Press; 2006. pp. 975–993. [Google Scholar]
- 27.Schutz CN, Warshel A. The low barrier hydrogen bond (LBHB) proposal revisited: The case of the Asp... His pair in serine proteases. Proteins. 2004;55(3):711–723. doi: 10.1002/prot.20096. [DOI] [PubMed] [Google Scholar]
- 28.Swallow AJ. In: Function of Quinones in Energy Conserving Systems. Trumpower BL, editor. New York: Academic Press; 1982. pp. 59–72. [Google Scholar]
- 29.Zu Y, et al. Reduction potentials of Rieske clusters: Importance of the coupling between oxidation state and histidine protonation state. Biochemistry. 2003;42(42):12400–12408. doi: 10.1021/bi0350957. [DOI] [PubMed] [Google Scholar]
- 30.Hsueh KL, Westler WM, Markley JL. NMR investigations of the Rieske protein from Thermus thermophilus support a coupled proton and electron transfer mechanism. J Am Chem Soc. 2010;132(23):7908–7918. doi: 10.1021/ja1026387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berthomieu C, Hienerwadel R. Iron coordination in photosystem II: Interaction between bicarbonate and the QB pocket studied by Fourier transform infrared spectroscopy. Biochemistry. 2001;40(13):4044–4052. doi: 10.1021/bi002236l. [DOI] [PubMed] [Google Scholar]
- 32.Paddock ML, McPherson PH, Feher G, Okamura MY. Pathway of proton transfer in bacterial reaction centers: Replacement of serine-L223 by alanine inhibits electron and proton transfers associated with reduction of quinone to dihydroquinone. Proc Natl Acad Sci USA. 1990;87(17):6803–6807. doi: 10.1073/pnas.87.17.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kálmán L, Maróti P. Stabilization of reduced primary quinone by proton uptake in reaction centers of Rhodobacter sphaeroides. Biochemistry. 1994;33(31):9237–9244. doi: 10.1021/bi00197a027. [DOI] [PubMed] [Google Scholar]
- 34.Stowell MHB, et al. Light-induced structural changes in photosynthetic reaction center: Implications for mechanism of electron-proton transfer. Science. 1997;276(5313):812–816. doi: 10.1126/science.276.5313.812. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi R, Boussac A, Sugiura M, Noguchi T. Structural coupling of a tyrosine side chain with the non-heme iron center in photosystem II as revealed by light-induced Fourier transform infrared difference spectroscopy. Biochemistry. 2009;48(38):8994–9001. doi: 10.1021/bi901195e. [DOI] [PubMed] [Google Scholar]
- 36.Maenpaa P, et al. A mutation in the D-de Loop of D1 modifies the stability of the S2QA- and S2QB- states in Photosystem II. Plant Physiol. 1995;107(1):187–197. doi: 10.1104/pp.107.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chernev P, Zaharieva I, Dau H, Haumann M. Carboxylate shifts steer interquinone electron transfer in photosynthesis. J Biol Chem. 2011;286(7):5368–5374. doi: 10.1074/jbc.M110.202879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fufezan C, Zhang C, Krieger-Liszkay A, Rutherford AW. Secondary quinone in photosystem II of Thermosynechococcus elongatus: Semiquinone-iron EPR signals and temperature dependence of electron transfer. Biochemistry. 2005;44(38):12780–12789. doi: 10.1021/bi051000k. [DOI] [PubMed] [Google Scholar]
- 39.Sedoud A, et al. Effects of formate binding on the quinone-iron electron acceptor complex of photosystem II. Biochim Biophys Acta. 2011;1807(2):216–226. doi: 10.1016/j.bbabio.2010.10.019. [DOI] [PubMed] [Google Scholar]
- 40.Rutherford AW, Zimmermann J-L. A new EPR signal attributed to the primary plastosemiquinone acceptor in Photosystem II. Biochim Biophys Acta. 1984;767(1):168–175. [Google Scholar]
- 41.Cox N, et al. The semiquinone-iron complex of photosystem II: Structural insights from ESR and theoretical simulation; evidence that the native ligand to the non-heme iron is carbonate. Biophys J. 2009;97(7):2024–2033. doi: 10.1016/j.bpj.2009.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hienerwadel R, Berthomieu C. Bicarbonate binding to the non-heme iron of photosystem II investigated by Fourier transform infrared difference spectroscopy and 13C-labeled bicarbonate. Biochemistry. 1995;34(50):16288–16297. doi: 10.1021/bi00050a008. [DOI] [PubMed] [Google Scholar]
- 43.Pospísil P, Arató A, Krieger-Liszkay A, Rutherford AW. Hydroxyl radical generation by photosystem II. Biochemistry. 2004;43(21):6783–6792. doi: 10.1021/bi036219i. [DOI] [PubMed] [Google Scholar]
- 44.Miyao M, Ikeuchi M, Yamamoto N, Ono T. Specific degradation of the D1 protein of photosystem II by treatment with hydrogen peroxide in darkness: Implications for the mechanism of degradation of the D1 protein under illumination. Biochemistry. 1995;34(31):10019–10026. doi: 10.1021/bi00031a025. [DOI] [PubMed] [Google Scholar]
- 45.Calabrese V, Boyd-Kimball D, Scapagnini G, Butterfield DA. Nitric oxide and cellular stress response in brain aging and neurodegenerative disorders: The role of vitagenes. In Vivo. 2004;18(3):245–267. [PubMed] [Google Scholar]
- 46.Greenberg BM, Gaba V, Mattoo AK, Edelman M. Identification of a primary in vivo degradation product of the rapidly-turning-over 32 kd protein of photosystem II. EMBO J. 1987;6(10):2865–2869. doi: 10.1002/j.1460-2075.1987.tb02588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vass I, Sanakis Y, Spetea C, Petrouleas V. Effects of photoinhibition on the QA-Fe2+ complex of photosystem II studied by EPR and Mössbauer spectroscopy. Biochemistry. 1995;34(13):4434–4440. doi: 10.1021/bi00013a036. [DOI] [PubMed] [Google Scholar]
- 48.Berglund GI, et al. The catalytic pathway of horseradish peroxidase at high resolution. Nature. 2002;417(6887):463–468. doi: 10.1038/417463a. [DOI] [PubMed] [Google Scholar]
- 49.Krieger A, Rutherford AW, Johnson GN. On the determination of redox midpoint potential of the primary quinone electron transfer acceptor, QA, in photosystem II. Biochim Biophys Acta. 1995;1229(2):193–201. [Google Scholar]
- 50.Brooks BR, et al. CHARMM: A program for macromolecular energy minimization and dynamics calculations. J Comput Chem. 1983;4(2):187–217. [Google Scholar]
- 51.MacKerell AD, Jr, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 52.Saito K, et al. Distribution of the cationic state over the chlorophyll pair of the photosystem II reaction center. J Am Chem Soc. 2011;133(36):14379–14388. doi: 10.1021/ja203947k. [DOI] [PubMed] [Google Scholar]
- 53.Saito K, Shen J-R, Ishida T, Ishikita H. Short hydrogen bond between redox-active tyrosine YZ and D1-His190 in the photosystem II crystal structure. Biochemistry. 2011;50(45):9836–9844. doi: 10.1021/bi201366j. [DOI] [PubMed] [Google Scholar]
- 54. (2012) QSite, version 5.8 (Schrödinger, LLC, New York, NY)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.