

Abstract
The molecular etiology of human progenitor reprogramming into self-renewing leukemia stem cells (LSC) has remained elusive. Although DNA sequencing has uncovered spliceosome gene mutations that promote alternative splicing and portend leukemic transformation, isoform diversity also may be generated by RNA editing mediated by adenosine deaminase acting on RNA (ADAR) enzymes that regulate stem cell maintenance. In this study, whole-transcriptome sequencing of normal, chronic phase, and serially transplantable blast crisis chronic myeloid leukemia (CML) progenitors revealed increased IFN-γ pathway gene expression in concert with BCR-ABL amplification, enhanced expression of the IFN-responsive ADAR1 p150 isoform, and a propensity for increased adenosine-to-inosine RNA editing during CML progression. Lentiviral overexpression experiments demonstrate that ADAR1 p150 promotes expression of the myeloid transcription factor PU.1 and induces malignant reprogramming of myeloid progenitors. Moreover, enforced ADAR1 p150 expression was associated with production of a misspliced form of GSK3β implicated in LSC self-renewal. Finally, functional serial transplantation and shRNA studies demonstrate that ADAR1 knockdown impaired in vivo self-renewal capacity of blast crisis CML progenitors. Together these data provide a compelling rationale for developing ADAR1-based LSC detection and eradication strategies.
Although advanced malignancies are diverse in phenotype, they often exhibit stem-cell properties including enhanced survival, differentiation, quiescence, and self-renewal potential (1, 2). Early insights into the molecular pathogenesis of cancer stemmed from the discovery of the Philadelphia chromosome (Ph+) and its constitutively active BCR-ABL1 tyrosine kinase in chronic myeloid leukemia (CML) (3–5). Tyrosine kinase inhibitor (TKI) therapy targeting BCR-ABL1 suppresses CML during the chronic phase (CP) of the disease in most patients able to tolerate long-term therapy (6). Although the CP stage of CML often can be controlled for long periods of time with standard TKI therapies, subsequent genetic and epigenetic alterations promote progenitor expansion and the generation of self-renewing leukemia stem cells (LSC) that fuel disease progression and blast crisis (BC) transformation along with TKI resistance (7, 8). Furthermore, TKI discontinuation usually results in CML resurgence, suggesting that quiescent progenitors persist despite therapy (9–12).
Mutations in spliceosome genes and alternative splicing of coding and noncoding RNAs are emerging as important drivers of transcriptomic diversity that fuel leukemic progression and therapeutic resistance (7, 8, 13). Moreover, previous studies reveal extensive RNA editing in the human transcriptome (14–17), primarily in primate-specific Alu sequences (18–20), which promotes splice isoform diversity. RNA editing activity is mediated by the adenosine deaminase acting on dsRNA (ADAR) family of editases (21), which includes ADAR1 (also known as ADAR), ADAR2 (ADARB1), and ADAR3 (ADARB2). ADAR1 and ADAR2 are active in embryonic cell types (18), and ADAR3 may play a nonenzymatic regulatory role in RNA editing activity (22). ADAR enzymes regulate fetal and adult hematopoietic stem cell (HSC) maintenance and stem cell responses to inflammation (23–26). ADAR-mediated adenosine-to-inosine (A-to-I) RNA editing in an Alu sequence containing dsRNA hairpin structures (14, 20) can generate alternative donor or acceptor splice sites (27, 28), alter RNA structure (21), modulate regulatory RNAs and gene silencing activities (29), and introduce codon sequence alterations (29). Interestingly, ADAR deregulation has been implicated in a variety of malignant cell types (30, 31). However, the functional effects of RNA editing in leukemia have not been elucidated. Here we examined the role of ADAR1-mediated RNA editing in malignant reprogramming of myeloid progenitors into LSC that drive BC transformation in CML.
Whole-transcriptome sequencing coupled with quantitative RT-PCR (qRT-PCR) analysis demonstrated increased IFN-responsive ADAR1 expression in LSC from primary BC CML patient samples as compared with CP CML and normal cord blood progenitors. This increase coincided with enhanced expression of inflammatory pathway genes during the progression from CP to BC. Moreover, ADAR1 p150 mRNA expression correlated with BCR-ABL amplification and increased A-to-I editing with differential expression of ADAR target genes in BC CML. Lentiviral ADAR1 p150 expression induced PU.1 expression that promotes expansion of myeloid progenitors and was associated with production of a misspliced form of glycogen synthase kinase (GSK)-3β that has been implicated in LSC self-renewal (7). Lentiviral ADAR1 knockdown also reduced BC LSC self-renewal capacity in RAG2−/−γc−/− mice. These data shed light on the contribution of ADAR1-mediated RNA editing to malignant progenitor reprogramming driving leukemic progression.
Results
Inflammatory Mediator-Driven RNA Editing Portends Blastic Transformation.
To investigate the mechanisms driving malignant reprogramming of myeloid progenitors into LSC, we performed whole-transcriptome sequencing (RNA-seq) on FACS-purified CD34+CD38+Lin− progenitor cells isolated from primary CML patient samples (Table S1). We identified 2,228 genes that were differentially expressed in BC and CP progenitors, and pathway analysis demonstrated overrepresentation of inflammatory IFN-related and proteoglycan-related pathways involved in hematological development (Fig. 1A) in BC as compared with CP CML. Among the significantly affected networks identified using Ingenuity Pathway Analysis (IPA), IFN-γ, cytokine, and TNF signaling pathways (Fig. S1A) along with self-renewal and reprogramming factors (KLF family and LEF1; Fig. S1B) were found to be enriched for differentially expressed genes in BC as compared with CP CML.
Fig. 1.

Inflammatory mediator-driven RNA editing portends blastic transformation. (A) Significantly over-represented pathways enriched for 2,228 differentially expressed genes in BC (n = 8) versus CP (n = 8) CML progenitors. (B) Human ADAR1 p150 and p110 were analyzed in CD34+CD38+Lin− progenitors from normal cord blood (n = 8), CML CP (n = 6), and CML BC (n = 7) by qRT-PCR. Ratios of p150 to p110 were determined (overall P = 0.0067; *P < 0.05 compared with normal cord blood; **P < 0.05 compared with CML CP by one-way ANOVA with post hoc Tukey test). (C) Pearson correlation analysis of ADAR1 isoforms and BCR-ABL mRNA levels in CML BC progenitors (n = 6). (D) RNA-seq–based analysis of A-to-G (=I) changes at putative editing sites (35) in CML BC (n = 8) versus CP progenitors (n = 8) (P < 0.05 by unpaired two-tailed Student t test). For each site, the percentage of reads that contained G versus A bases was calculated for each sample. The differences between average percentages were computed between disease stages (BC–CP). Data were reported as the number of sites showing significantly different editing ratios. (E) Volcano plot analysis showing enrichment of more highly edited sites in CML BC (n = 8) compared with CP progenitors (n = 8). (F) Differential editing at ADAR target sites in CML BC (n = 8) versus CP (n = 8). All sites shown were significantly different (P < 0.05 by Student t test, Dataset S1). (G) One hundred seventy-five putative ADAR target genes were differentially expressed in CP (n = 8) versus BC (n = 8). Unsupervised hierarchical clustering (see SI Materials and Methods) separated CP and BC samples. A select subset of genes (boxes) discriminated BC from cord blood (n = 3).
Analysis of inflammatory mediator splice isoform expression revealed up-regulation of numerous inflammation-associated receptors and signaling molecules (Fig. S2A). The most significantly up-regulated transcripts included an LSC marker, IL-3Rα (CD123), IFN receptors, and TNF receptors (TNFRSF) (Fig. S2A). Notably, transcription of the IFN-responsive isoform of ADAR1 has been shown to be stimulated by a variety of inflammatory molecules (32, 33). Because of known functional differences between ADAR1 p150 and p110 that are not evident by whole-gene expression studies, we performed a sensitive qRT-PCR analysis in CD34+CD38+Lin− progenitor cells from primary CML patient samples (Table S1) using splice isoform-specific primers (Table S2). Expression levels of the inflammation-responsive ADAR1 p150 isoform were increased approximately eightfold in CML BC progenitors in comparison with normal cord blood (Fig. S1C), but levels of the constitutively active p110 isoform did not differ significantly among groups (Fig. S1D). Analysis of the ratio of p150 to p110 showed a relative enrichment of the p150 ADAR1 isoform associated with CML progression (Fig. 1B) and BCR-ABL amplification in BC CML progenitors (Fig. 1C). We observed similar patterns of increased ADAR1 p150 expression in the granulocyte/macrophage progenitor (GMP) fraction of primary CML samples (Fig. S1E), which expands during CML progression (Fig. S1F) (34). To confirm sensitivity of HSC to inflammatory mediator-induced ADAR1 p150 expression, qRT-PCR analysis was performed on CD34+ cord blood cells treated with IFN-γ or TNF-α, and results showed a twofold up-regulation of ADAR1 p150 (Fig. S1G).
Additional analysis of ADAR family gene expression in the RNA-seq data demonstrated that although total ADAR1 (ADAR) expression was detectable in all samples, ADAR2 and ADAR3 expression was below detection thresholds (Fig. S2B). This result is consistent with previous reports showing that ADAR1 is the most highly expressed RNA-editing enzyme in tumor cells (35, 36) and suggests that ADAR1 is likely the primary functional A-to-I RNA editase in CML LSC.
To investigate the contribution of ADAR1 up-regulation to RNA editing during CML progression, RNA-seq analysis of CML CP and BC CD34+CD38+Lin− progenitors was compared with the genomic coordinates of putative A-to-I editing sites identified in a previously published dataset (35). In eight CP and eight BC CML primary patient samples, the fraction of sites that contained guanosine bases (representing inosine substitution) compared with adenosine bases was calculated for each patient sample. This comparison revealed a striking enrichment of A-to-G changes in ADAR target sites in BC compared with CP progenitors (Fig. 1D), demonstrating a shift toward increased RNA editing during CML progression. Furthermore, volcano plot analysis of editing at ADAR target sites revealed ∼16-fold more editing sites with significantly increased editing ratios during BC transformation (Fig. 1E). A total of 274 sites exhibited significantly different editing rates (Dataset S1), and these sites were located predominantly within Alu repeat sequences (Fig. 1F). These data suggest that RNA editing activity is not only cell type- and context-specific but also is disease stage-specific and therefore could be a harbinger of disease progression.
By examining the relative levels of ADAR target gene (35) transcripts, we found that 175 of 2,593 putative ADAR target genes were differentially expressed in BC and CP progenitors (Fig. 1G). Differential expression of a subset of ADAR target genes clearly distinguished BC from CP or cord blood progenitors (Fig. 1G). In addition, nonnegative matrix factorization, using 200 random start sites, recapitulated these major groupings (Fig. S1H). Finally, cophenetic correlation coefficients calculated using this human myeloid progenitor dataset, compared with a randomized dataset, also supported three major groupings in the data (Fig. S1I).
To investigate whether there was evidence of a link between aberrant RNA editing and alternative splice isoform expression in BC versus CP CML, editing ratios at ADAR target sites were calculated for transcripts that were differentially expressed (Fig. S2 C and D). Editing ratios were found to be increased for significantly differentially expressed transcripts (Fig. S2C). Moreover, differential isoform expression of ADAR target genes revealed distinct clustering of CML BC versus CP (Fig. S2D). Thus, the shift in transcriptional patterns associated with increased RNA editing may reflect malignant reprogramming of progenitors in CML.
BCR-ABL Expression Enhances Inflammatory Mediator Gene Expression.
Because qRT-PCR analyses revealed that ADAR1 p150 expression correlated with BCR-ABL levels in BC CML (Fig. 1C), we sought to investigate the potential mechanisms driving ADAR1 induction in BC CML and the relationship between ADAR1 p150 and BCR-ABL. Both qRT-PCR analyses and RNA-seq studies were performed with CD34+ cord blood transduced with lentivirus expressing BCR-ABL p210 tagged with GFP or vector control (Fig. 2A and Fig. S3A). Increased phosphorylation of the BCR-ABL substrate Crkl by nanoproteomics analysis confirmed functional BCR-ABL activation (Fig. 2B). These analyses suggested that BCR-ABL may regulate ADAR1 indirectly. In support of this possibility, RNA-seq analyses of lenti-BCR-ABL–transduced cord blood compared with vector-transduced controls identified 45 differentially expressed genes (Fig. 2C). BCR and ABL1 were up-regulated along with inflammatory mediators such as TNFRSF9 (Fig. 2C). Other differentially expressed genes included factors involved in ECM function or intercellular junctions (Fig. 2C). Because TNF pathways, receptors, and other inflammatory mediators also were up-regulated during CML progression (Fig. 1A and Figs. S1A and S2A), it is conceivable that BCR-ABL regulates ADAR1 p150 through the induction of inflammatory receptor expression.
Fig. 2.
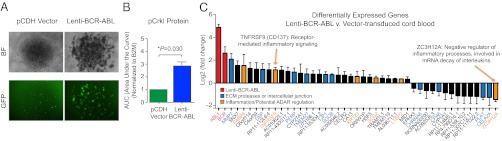
BCR-ABL expression enhances inflammatory mediator gene expression. (A) Bright-field (BF) and fluorescent (GFP) microscopy showing cord blood-derived colonies transduced with lentiviral vector backbone control (pCDH) or lentivirus expressing BCR-ABL p210 and GFP. (B) Nanoproteomics analysis of phosphorylated (p)-Crkl levels in BCR-ABL–transduced CD34+ cells from cord blood (n = 3). P < 0.05 by Student t test. (C) CD34+ cord blood cells (n = 3) were transduced with lenti-BCR-ABL or vector control and processed for RNA-seq analysis. Forty-five genes were differentially expressed in BCR-ABL–expressing cells and vector controls (P < 0.05 by DESeq).
ADAR1 Promotes Malignant Myeloid Progenitor Expansion and Alternative Splicing.
Lentiviral ADAR1 overexpression and shRNA knockdown studies were performed to examine the functional consequences of ADAR1 p150 up-regulation. Transduction efficiency of lentiviral vectors driving human ADAR1 (p150) overexpression (lenti-ADAR1) or ADAR1-targeting shRNA (lenti-shADAR1) was validated in normal cord blood (Fig. S3 B–E). Myeloid lineage skewing was observed in colony-forming assays performed with CD34+CD38+Lin− cord blood progenitors transduced with lenti-ADAR1 p150, as demonstrated by an increase in the number of macrophage colonies and a corresponding decrease in the number of erythroid burst-forming unit (BFU-E) colonies (Fig. 3A). This myeloid lineage bias coincided with up-regulation of PU.1—a myeloid transcription factor—and down-regulation of GATA1—an erythroid transcription factor—in colonies from cord blood progenitors transduced with ADAR1 p150 lentivirus (Fig. 3 B and C) and in CML BC progenitor-derived colonies (Fig. 3 D and E). RNA-seq analyses demonstrated an increase in the PU.1/GATA1 ratio in BC versus CP progenitors (Fig. 3F). Additionally, network analysis of RNA-seq data revealed down-regulation of genes involved in GATA1-dependent processes (Fig. 3G).
Fig. 3.

ADAR1 promotes malignant myeloid progenitor expansion. (A) Lentiviral overexpression of ADAR1 p150 in FACS-purified CD34+38+Lin− normal progenitors (n = 3) reduced erythroid (BFU-E) colony formation and increased macrophage (M) cfu compared with vector (ORF) controls. Mix, macrophage/erythroid colonies; GM, granulocyte/macrophage colonies; G, granulocyte colonies. *P < 0.05 and **P < 0.01 compared with vector-transduced controls or CP progenitors by Student t test. (B–E) Pearson correlation analysis of HPRT-normalized ADAR1 mRNA levels and PU.1 or GATA1 in individual colonies derived from normal progenitors (n = 3) transduced with lenti-ADAR1 p150 (B and C) or CML BC progenitor-derived colonies (D and E). r2 values were calculated using Pearson correlation analysis. (F) Ratio of PU.1 to GATA1 expression levels by RNA-seq in CP (n = 8) versus BC (n = 8) CML. (G) RNA-seq–based IPA network analysis of GATA1-associated genes in BC CML (n = 8) compared with CP (n = 8).
Short-term culture of normal progenitors transduced with lenti-ADAR1 at increasing multiplicity of infection (MOI) confirmed a positive correlation between PU.1 and ADAR1 expression, whereas the levels of GATA1 expression remained constant (Fig. S3F). The ADAR1-mediated myeloid lineage skewing recapitulates qRT-PCR data reported with aged human HSC (37) and the expansion of GMP (34) during progression of CML from CP to BC (Fig. S1F). Because we previously identified production of a misspliced form of GSK3β lacking exons 8 and 9 (7) associated with GMP expansion in CML BC, and because activation of ADAR1 might promote alternative splicing (27), we performed splice isoform-specific qRT-PCR for GSK3β variants in individual colonies derived from lenti-ADAR1–transduced cord blood or CML CP progenitors. Although GSK3β exon 8–9del was undetectable in cord blood progenitors, in CML CP samples mRNA levels of this variant were increased relative to the exon 9del form, which represents a predominant GSK3β transcript in normal hematopoietic tissues (Fig. S3G) (7). Together, these results demonstrate that ADAR1 overexpression drives hematopoietic differentiation toward the myeloid lineage, coincident with PU.1 up-regulation and alternative splicing of GSK3β in CML progenitors.
ADAR1 Knockdown Impairs Malignant Myeloid Progenitor Self-Renewal.
Previous reports demonstrate that ADAR1 mediates mouse HSC maintenance (23, 24); however the relative effects of down-modulating ADAR1 expression in human LSC versus normal HSC have not been established. In support of a favorable therapeutic index for ADAR1 inhibitory strategies in CML progenitors versus normal cord blood, shRNA knockdown and colony assay experiments showed that lenti-shADAR1 knockdown in normal cord blood progenitors did not affect hematopoietic differentiation (Fig. S3H), whereas CML CP progenitors transduced with lenti-shADAR1 produced fewer macrophage colonies coupled with increased numbers of BFU-E colonies (Fig. S3I).
To examine the role of ADAR1 in CML progenitor survival and self-renewal capacity in vivo, CD34+ human BC CML progenitors were transduced with lenti-shADAR1 or shRNA backbone (lenti-shControl) and transplanted intrahepatically into neonatal RAG2−/−γc−/− mice (Fig. 4A). In addition, qRT-PCR analysis was performed to confirm ADAR1 p150 knockdown. Similar to experiments in normal cord blood (Fig. S3C), ADAR1 p150 levels before transplantation were reduced by ∼50% in CML BC progenitors transduced with lenti-shADAR1 as compared with vector controls (Fig. 4A).
Fig. 4.

ADAR1 knockdown impairs malignant myeloid progenitor self-renewal. (A) Diagrammatic scheme of in vivo experimental design. Before and after primary transplantation, qRT-PCR was performed in human progenitors, confirming ADAR1 knockdown. BM, bone marrow. (B) Representative FACS plots showing human CD45+ engraftment in hematopoietic organs of primary RAG2−/−γc−/− transplant recipients or untransplanted control mouse bone marrow. (C and D) FACS analysis of CD45+ human cell engraftment and progenitor cells in the bone marrow of mice transplanted with BC progenitors transduced with lenti-shControl (n = 3) or lenti-shADAR1 (n = 5). (E and F) FACS analysis of human cells in mouse bone marrow (shControl n = 7 and shADAR1 n = 8) after serial transplantation of BM CD34+ cells pooled from primary transplant-recipient mice. *P < 0.05 compared with vector-transduced controls by Student t test.
By 10 wk after transplantation, robust leukemic engraftment was detectable in hematopoietic tissues by FACS analysis of human CD45+ cells (Fig. 4B). Similar levels of human hematopoietic cell engraftment were detected in bone marrow from mice transplanted with lenti-shADAR1–transduced BC progenitors and lenti-shControl (Fig. 4 C and D). qRT-PCR analysis confirmed that pooled human progenitors from primary transplant-recipient bone marrow maintained reduced levels of ADAR1 p150, consistent with continued activity of the ADAR1 shRNA after primary engraftment (Fig. 4A). We transplanted equal numbers of human CD34+ progenitors derived from the bone marrow of primary recipients of lenti-shControl or lenti-shADAR1–transduced cells into secondary recipient mice, and the bone marrow was analyzed for human cell engraftment and CD34+CD38+Lin− progenitors by FACS (Fig. 4 E and F). Notably, serial engraftment potential of shADAR1-transduced CML BC progenitors was reduced considerably (∼20%) compared with shControls (∼50%) (Fig. 4E). Although leukemic burden was not diminished significantly, the LSC self-renewal capacity was irrevocably reduced by ADAR1 knockdown, suggesting that ADAR1 plays a pivotal role in the propagation of leukemia driven by self-renewing malignant progenitors.
Discussion
Activation of IFN-responsive ADAR1 p150 in primary CML progenitors correlated with increased A-to-I RNA editing following blastic transformation. Full transcriptome RNA-seq analyses suggest that up-regulation of ADAR1 p150 in BC CML may be related to the activation of inflammatory pathways such as cytokines and TNF in advanced disease. Hematopoietic progenitor studies demonstrated that lentivirally enforced ADAR1 p150 expression promotes human myeloid differentiation fate. Lentiviral-shRNA knockdown of ADAR1 in a xenotransplantation model impaired self-renewal capacity of BC LSC. Together, these data suggest that an inflammatory mediator-driven isoform switch favoring ADAR1 p150 expression drives expansion of malignant progenitors and contributes to CML progression.
ADAR-mediated RNA editing can regulate myriad molecular processes including RNA interference (29), microRNA function (38), and RNA stability, localization, nuclear retention, degradation, and alternative splicing (27, 39–41). Moreover, high levels of ADAR-mediated RNA editing activity may reflect a reversion to a primitive transcriptional program typical of embryonic stem cells (18). A previous study demonstrated that ADAR1 was among the top 5% of genes expressed in the mutational evolution of lobular breast cancer (36), indicating that activation of ADARs may correlate with disease progression in multiple malignant cell types (31). Although previous studies have shown ADAR1 p110 up-regulation in a murine leukemia model (42) and in pediatric acute leukemias (43), it should be emphasized that human hematopoietic tissues undergo dramatic changes during aging (44, 45) that are caused in part by increased inflammation and genomic instability (46).
Our data suggest that inflammatory cues may facilitate selective up-regulation of the IFN-responsive ADAR1 p150 isoform in hematologic malignancies. Using whole-transcriptome sequencing analysis, we found that inflammatory signaling receptors were differentially expressed in BCR-ABL–expressing cord blood. Inflammatory cytokines such as IFN, TNF-α, and other interleukins have been shown to stimulate ADAR1 p150 expression (32, 33), and IFN regulates HSC quiescence (47). Together, BCR-ABL–mediated up-regulation of inflammatory pathway receptors could sensitize hematopoietic progenitors to inflammatory stimuli that drive ADAR1 expression and promote CML LSC self-renewal.
Whole transcriptome-based RNA editing analyses revealed that CML progression is accompanied by differential RNA editing. Recent in-depth transcriptomic studies have shown that gene products with predicted A-to-I editing events are significantly enriched in cancer-related pathways (35). The present study provides further evidence supporting a role for ADAR-directed RNA editing in splice isoform diversity in cancer and demonstrates the activation of inflammation-associated RNA editing during the progression of hematologic malignancies. Our results suggest that ADAR1 p150 up-regulation may contribute to blastic transformation of CML driven by the CD34+CD38+Lin− progenitor and GMP subpopulations. Because ADAR-mediated RNA editing occurs primarily in primate-specific Alu repeat sequences (14, 19, 20), the activation of RNA editases in human cells may fuel species-specific malignant reprogramming of progenitors to adopt a more primitive stem cell fate under pathological conditions.
Myeloid lineage skewing was observed in response to enforced ADAR1 p150 expression, concomitant with PU.1 activation. Although a previous study in an in vitro model implicated PU.1 in the regulation of murine ADAR1 expression (48), cell type- and niche-specific stimuli may have differential effects on RNA editing, and our studies in human cells, in which 90% of RNA editing occurs in primate-specific Alu sequences, suggest that the converse may occur also. It is conceivable that ADAR1 directly controls PU.1 through RNA editing-dependent effects or via potential transcriptional regulation related to its Z-DNA binding function (41, 49). In support of the former possibility, DARNED (50), a database compiling RNA editing sites from multiple publications, reports that transcripts of Spi1 (a gene encoding PU.1; chromosome 11, start: 47379732, end: 47400127) showed evidence of A-to-I RNA editing at 28 sites (14). Future gene-expression analyses and identification of de novo RNA editing sites through analysis of whole-genome and transcriptome DNA-RNA differences (15) in normal HSC harboring enforced ADAR1 p150 expression and LSC will be necessary to dissect further the link between ADAR1 activation PU.1 expression.
In addition to up-regulation of PU.1 in response to ADAR1 p150 overexpression, we also detected production of a misspliced GSK3β variant in colonies derived from CML CP progenitors transduced with lentiviral ADAR1 p150. We have shown previously that this particular form of GSK3β harbors reduced activity and promotes LSC self-renewal via activation of β-catenin (7). In support of a role for ADAR1 in CML LSC self-renewal, we showed in humanized CML xenograft mouse models that ADAR1 knockdown reduced the potential of CML BC LSC for serial transplantation. Given that these CML BC progenitors expressed significantly higher levels of ADAR1 p150 than did normal cord blood progenitors, we expect that LSC are relatively more dependent on IFN-responsive ADAR1 activity (24). Together these data suggest that inflammation-dependent activation of the ADAR1 p150 editase promotes CML progression. Aberrant ADAR1 activation in CML endows myeloid progenitors with self-renewal capacity leading to LSC generation. Thus, inhibition of ADAR1 activity could represent an effective strategy to prevent LSC-driven relapse in CML while sparing normal HSC populations. Furthermore, ADAR1-mediated RNA editing activation could prove to be a diagnostic and prognostic indicator of disease progression with important implications for other cancer stem cell-driven malignancies.
Materials and Methods
Primary samples from CML patients were obtained from consenting patients at the University of California San Diego, Stanford University, and the University of Toronto Health Network. FACS-purified progenitor cells from primary samples or normal cord blood were processed for RNA-Seq, qRT-PCR, and for in vitro and xenotransplantation studies. Detailed methods are available in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Wenxue Ma, Alice Shih, and Heather Leu (all of Moores Cancer Center, University of California at San Diego) for assistance with in vivo experiments; Sa Li (BC Cancer Agency) for performing SNV analysis; Jonathan Lee (Moores Cancer Center, University of California at San Diego) for assistance with figures; Guanming Wu for providing the mapping information of pathway-to-gene relationships; and the Genome Sciences Centre Library Construction, Sequencing, and Bioinformatics teams of the BC Cancer Agency. This work was supported by California Institute for Regenerative Medicine (CIRM) Grants RN2-00910-1 (to C.H.M.J.) and DR1-01430 (to C.H.M.J.), by CIRM Training Grant TG2-01154 (to Q.J.), by CIRM Scientific Excellence through Exploration and Development (SEED) Grant RS1-00228-1, by a Cancer Stem Cell Consortium with funding from the Government of Canada through Genome Canada and the Ontario Genomics Institute (OGI-047), and through the Canadian Institutes of Health Research (CSC-105367).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1213021110/-/DCSupplemental.
References
- 1.Jamieson C. The MLLgnant consequences of reverting to an embryonic transcriptional program. Cell Stem Cell. 2009;4(2):97–98. doi: 10.1016/j.stem.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 2.Eppert K, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17(9):1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 1986;233(4760):212–214. doi: 10.1126/science.3460176. [DOI] [PubMed] [Google Scholar]
- 4.Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243(5405):290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 5.Shtivelman E, Lifshitz B, Gale RP, Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature. 1985;315(6020):550–554. doi: 10.1038/315550a0. [DOI] [PubMed] [Google Scholar]
- 6.Hughes TP, et al. International Randomised Study of Interferon versus STI571 (IRIS) Study Group Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med. 2003;349(15):1423–1432. doi: 10.1056/NEJMoa030513. [DOI] [PubMed] [Google Scholar]
- 7.Abrahamsson AE, et al. GSK3β missplicing contributes to leukemia stem cell generation. Proc Natl Acad Sci USA. 2009;106(10):3925–3929. doi: 10.1073/pnas.0900189106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: Mechanisms of blastic transformation. J Clin Invest. 2010;120(7):2254–2264. doi: 10.1172/JCI41246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Copland M, et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107(11):4532–4539. doi: 10.1182/blood-2005-07-2947. [DOI] [PubMed] [Google Scholar]
- 10.Jamieson CH, Barroga CF, Vainchenker WP. Miscreant myeloproliferative disorder stem cells. Leukemia. 2008;22(11):2011–2019. doi: 10.1038/leu.2008.290. [DOI] [PubMed] [Google Scholar]
- 11.Jørgensen HG, Allan EK, Jordanides NE, Mountford JC, Holyoake TL. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood. 2007;109(9):4016–4019. doi: 10.1182/blood-2006-11-057521. [DOI] [PubMed] [Google Scholar]
- 12.Huang X, Cortes J, Kantarjian H. Estimations of the increasing prevalence and plateau prevalence of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Cancer. 2012;118(12):3123–3127. doi: 10.1002/cncr.26679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perrotti D, Neviani P. From mRNA metabolism to cancer therapy: Chronic myelogenous leukemia shows the way. Clin Cancer Res. 2007;13(6):1638–1642. doi: 10.1158/1078-0432.CCR-06-2320. [DOI] [PubMed] [Google Scholar]
- 14.Kim DD, et al. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004;14(9):1719–1725. doi: 10.1101/gr.2855504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramaswami G, et al. Accurate identification of human Alu and non-Alu RNA editing sites. Nat Methods. 2012;9(6):579–581. doi: 10.1038/nmeth.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng Z, et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol. 2012;30(3):253–260. doi: 10.1038/nbt.2122. [DOI] [PubMed] [Google Scholar]
- 17.Levanon EY, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol. 2004;22(8):1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 18.Osenberg S, et al. Alu sequences in undifferentiated human embryonic stem cells display high levels of A-to-I RNA editing. PLoS ONE. 2010;5(6):e11173. doi: 10.1371/journal.pone.0011173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eisenberg E, et al. Is abundant A-to-I RNA editing primate-specific? Trends Genet. 2005;21(2):77–81. doi: 10.1016/j.tig.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 20.Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2(12):e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bass BL, Weintraub H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell. 1988;55(6):1089–1098. doi: 10.1016/0092-8674(88)90253-x. [DOI] [PubMed] [Google Scholar]
- 22.Chen CX, et al. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA. 2000;6(5):755–767. doi: 10.1017/s1355838200000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;10(1):109–115. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.XuFeng R, et al. ADAR1 is required for hematopoietic progenitor cell survival via RNA editing. Proc Natl Acad Sci USA. 2009;106(42):17763–17768. doi: 10.1073/pnas.0903324106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Q. RNA editing catalyzed by ADAR1 and its function in mammalian cells. Biochemistry (Mosc) 2011;76(8):900–911. doi: 10.1134/S0006297911080050. [DOI] [PubMed] [Google Scholar]
- 26.Wang Q, Khillan J, Gadue P, Nishikura K. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science. 2000;290(5497):1765–1768. doi: 10.1126/science.290.5497.1765. [DOI] [PubMed] [Google Scholar]
- 27.Rueter SM, Dawson TR, Emeson RB. Regulation of alternative splicing by RNA editing. Nature. 1999;399(6731):75–80. doi: 10.1038/19992. [DOI] [PubMed] [Google Scholar]
- 28.Lev-Maor G, et al. RNA-editing-mediated exon evolution. Genome Biol. 2007;8(2):R29. doi: 10.1186/gb-2007-8-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishikura K. Editor meets silencer: Crosstalk between RNA editing and RNA interference. Nat Rev Mol Cell Biol. 2006;7(12):919–931. doi: 10.1038/nrm2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galeano F, Tomaselli S, Locatelli F, Gallo A. A-to-I RNA editing: The “ADAR” side of human cancer. Semin Cell Dev Biol. 2012;23(3):244–250. doi: 10.1016/j.semcdb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Cenci C, et al. Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J Biol Chem. 2008;283(11):7251–7260. doi: 10.1074/jbc.M708316200. [DOI] [PubMed] [Google Scholar]
- 32.Meltzer M, et al. The RNA editor gene ADAR1 is induced in myoblasts by inflammatory ligands and buffers stress response. Clin Transl Sci. 2010;3(3):73–80. doi: 10.1111/j.1752-8062.2010.00199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang JH, et al. Widespread inosine-containing mRNA in lymphocytes regulated by ADAR1 in response to inflammation. Immunology. 2003;109(1):15–23. doi: 10.1046/j.1365-2567.2003.01598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jamieson CH, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 35.Bahn JH, et al. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res. 2012;22(1):142–150. doi: 10.1101/gr.124107.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shah SP, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461(7265):809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 37.Pang WW, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci USA. 2011;108(50):20012–20017. doi: 10.1073/pnas.1116110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang W, et al. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13(1):13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heale BS, Keegan LP, O’Connell MA. ADARs have effects beyond RNA editing. Cell Cycle. 2009;8(24):4011–4012. doi: 10.4161/cc.8.24.10214. [DOI] [PubMed] [Google Scholar]
- 40.Heale BS, et al. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J. 2009;28(20):3145–3156. doi: 10.1038/emboj.2009.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herbert A, Wagner S, Nickerson JA. Induction of protein translation by ADAR1 within living cell nuclei is not dependent on RNA editing. Mol Cell. 2002;10(5):1235–1246. doi: 10.1016/s1097-2765(02)00737-2. [DOI] [PubMed] [Google Scholar]
- 42.Ma CH, et al. [Expression of ADAR1 isoforms in murine acute T-ALL leukemia model] Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2011;19(3):566–569. [PubMed] [Google Scholar]
- 43.Ma CH, et al. Abnormal expression of ADAR1 isoforms in Chinese pediatric acute leukemias. Biochem Biophys Res Commun. 2011;406(2):245–251. doi: 10.1016/j.bbrc.2011.02.025. [DOI] [PubMed] [Google Scholar]
- 44.Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nat Med. 1996;2(9):1011–1016. doi: 10.1038/nm0996-1011. [DOI] [PubMed] [Google Scholar]
- 45.Wang LD, Wagers AJ. Dynamic niches in the origination and differentiation of haematopoietic stem cells. Nat Rev Mol Cell Biol. 2011;12(10):643–655. doi: 10.1038/nrm3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chambers SM, et al. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5(8):e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Essers MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458(7240):904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 48.Wu T, Zhao Y, Hao Z, Zhao H, Wang W. Involvement of PU.1 in mouse adar-1 gene transcription induced by high-dose esiRNA. Int J Biol Macromol. 2009;45(2):157–162. doi: 10.1016/j.ijbiomac.2009.04.021. [DOI] [PubMed] [Google Scholar]
- 49.Herbert A, et al. A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase. Proc Natl Acad Sci USA. 1997;94(16):8421–8426. doi: 10.1073/pnas.94.16.8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kiran A, Baranov PV. DARNED: A DAtabase of RNa EDiting in humans. Bioinformatics. 2010;26(14):1772–1776. doi: 10.1093/bioinformatics/btq285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.