

Abstract
Aims
The sources of cytosolic superoxide in skeletal muscle have not been defined. This study examined the subcellular sites that contribute to cytosolic superoxide in mature single muscle fibers at rest and during contractile activity. Results: Isolated fibers from mouse flexor digitorum brevis loaded with superoxide and nitric-oxide-sensitive fluorescent probes, specific pathway inhibitors and immunolocalization techniques were used to identify subcellular sites contributing to cytosolic superoxide. Treatment with the electron transport chain complex III inhibitor, antimycin A, but not the complex I inhibitor, rotenone, caused increased cytosolic superoxide through release from the mitochondrial intermembrane space via voltage-dependent anion or Bax channels, but inhibition of these channels did not affect contraction-induced increases in cytosolic superoxide. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase inhibitors decreased cytosolic superoxide at rest and following contractions. Protein and mRNA expression of NADPH oxidase subunits was demonstrated in single fibers. NOX2, NOX4, and p22phox subunits localized to the sarcolemma and transverse tubules; NOX4 was additionally expressed in mitochondria. Regulatory p40phox and p67phox proteins were found in the cytoplasm of resting fibers, but following contractions, p40phox appeared to translocate to the sarcolemma. Innovation: Superoxide and other reactive oxygen species generated by skeletal muscle are important regulators of muscle force production and adaptations to contractions. This study has defined the relative contribution of mitochondrial and cytosolic sources of superoxide within the cytosol of single muscle fibers at rest and during contractions. Conclusion: Muscle mitochondria do not modulate cytosolic superoxide in skeletal muscle but NADPH oxidase is a major contributor both at rest and during contractions. Antioxid. Redox Signal. 18, 603–621.
Introduction
Superoxide and nitric oxide (NO) are the primary free radical species produced by skeletal muscle and their generation is increased during contractile activity (32). This has been reported to induce oxidative injury to muscle (43) although more recent data indicate that reactive oxygen and nitrogen species (RONS) play multiple regulatory roles in the physiology of skeletal muscles by modifying cell signaling pathways (19, 26, 54), control of gene expression by activating redox-sensitive transcription factors (24), and modulation of skeletal muscle force production (56). NO in muscle is produced by the nitric oxide synthases (NOS) (63), but there is little consensus on the prime intracellular sources for superoxide production. Early studies suggested that 2%–5% of the total oxygen (O2) consumed by mitochondria may undergo one-electron reduction with the generation of superoxide (8, 40, 48) and, based on this observation, a large number of publications relate the increase in reactive oxygen species (ROS) to the elevated O2 consumption that occurs with increased mitochondrial activity, implying potentially a 50–100-fold increase in superoxide generation in skeletal muscles subjected to aerobic contractions (17, 34, 65). Recent data from our group (51, 60) and others (4, 28, 44, 62) argue against such a substantial formation of superoxide within mitochondria and recent assessments of the rate of superoxide production by mitochondria indicate that ∼0.15% of the total O2 consumed is reduced to superoxide (62), a value that is an order of magnitude lower than the original estimates.
Innovation.
This is the first study to examine the subcellular sites (mitochondrial or extramitochondrial) that modulate cytosolic superoxide in skeletal muscle at rest and during contractions. Our data indicate that mitochondrial sources do not contribute to cytosolic superoxide, but nicotinamide adenine dinucleotide phosphate oxidase is a major source at rest and during contractile activity in skeletal muscle. These results are biologically and potentially clinically relevant and have widespread implications for the understanding of diverse scientific areas, including the responses of muscle to exercise training, age-related loss of muscle mass and function, as well as inflammatory or degenerative muscle diseases, such as the muscular dystrophies that are associated with increased levels of oxidative damage and muscle weakness.
Monitoring of dihydroethidium (DHE) oxidation may provide a reliable technique to monitor superoxide within single skeletal muscle fibers and recent data showed that contractile activity induced a significant increase in DHE oxidation due to increased superoxide within the fiber cytoplasm (60). Other studies that have examined superoxide in the mitochondrial matrix (4) or mitochondrial redox potential (44) indicate that mitochondria may not be the major source for the increased fiber ROS generation observed during contractile activity.
Skeletal muscle mitochondria produce superoxide from both complexes I and III of the electron transport chain (47). Superoxide generated at complex I appears to be released into the mitochondrial matrix, but complex III releases superoxide to both sides of the inner mitochondrial membrane (IMM)—the matrix and the mitochondrial intermembrane space (MIS) (47). Studies that have investigated mitochondrial superoxide formation or changes in mitochondrial redox potential have used MitoSOX Red or mito-roGFP constructs, but these probes accumulate in the mitochondrial matrix and do not respond to changes in superoxide or the redox potential in the MIS. Although superoxide is a relatively membrane-impermeable anion (54), studies of isolated mitochondria indicate that channels in the outer mitochondrial membrane (OMM) can facilitate diffusion of superoxide from the MIS to the cytosol (11, 27). Thus, the increase in cytoplasmic superoxide observed during contractile activity might be related to superoxide released across the OMM, although this possibility does not appear to have been examined in skeletal muscle.
Additional extramitochondrial sites for superoxide production within skeletal muscle have been proposed, including reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzymes (30, 45), enzymes of the phospholipase A2 family (26, 49), and xanthine oxidase (25), but their contribution during contractile activity has not been fully evaluated.
The aims of the current study were therefore to: (i) identify the primary source(s) that contribute(s) to cytosolic superoxide in mature skeletal muscle fibers at rest and following a period of contractile activity, (ii) determine the effect of increasing frequency of contractions on fiber NO and superoxide, and (iii) define whether mitochondria within single muscle fibers release superoxide from the MIS or mitochondrial matrix to the cytoplasm of skeletal muscles at rest and following contractions.
Results
Single skeletal muscle fibers loaded with 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate and dihydroethidium
Figure 1A(i), B(i), and C(i) shows bright-field images of single muscle fibers displaying good morphology and well-defined striations along the sarcolemma. 4-Amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) fluorescence from a fiber at rest appears homogeneous throughout the fiber, indicating no subcellular localization of this probe (Fig. 1A[ii], [iii]). The unreacted form of dihydroethidium (DHE) appeared to accumulate in the cytosol (Fig. 1B[ii], [iii]) but, upon reaction with superoxide, the oxidized form of DHE indicated by ethidium (E+) fluorescence was predominantly localized to nuclei, shown by the colocalization of 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) with E+ fluorescence (Fig. 1C[iv]).
FIG. 1.

DAF-FM DA and DHE to assess changes in NO and superoxide at rest and following contractile activity. (A) Confocal images of a single isolated fiber from the FDB muscle after 16 h in culture under bright field (i); fluorescent image following loading with DAF-FM DA (ii); merged image of (i) and (ii)–(iii). (B) Confocal images of a single isolated fiber under bright field (i); fluorescent image showing blue fluorescence from nonoxidized DHE (ii); merged image of (i) and (ii)–(iii). (C) Confocal images of a single isolated fiber under bright field (i); fluorescent image showing DAPI staining (ii); fluorescent image showing ethidium, the oxidized form of DHE (iii), and a merged image of (ii) and (iii)–(iv). Original magnification: 60×(scale bar=30 μm). (D) Schematic illustration of the protocols for electrical stimulations at different intensities. The stimuli train was constant in all contraction protocols. The time at rest between repetitions varied according to the intensity of each protocol. DAF-FM DA, 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate; DAPI, 4′,6-diamidino-2-phenylindole dihydrochloride; DHE, dihydroethidium; FDB, flexor digitorum brevis.
To determine the effect of increasing intensity of contractions on superoxide and NO activities, single muscle fibers were subjected to three different contractile activity protocols that are shown schematically in Figure 1D.
Intracellular NO and superoxide in single skeletal muscle fibers following contractile activity protocols of different intensities
Resting noncontracted flexor digitorum brevis (FDB) fibers loaded with 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM DA) showed no significant changes in the rate of change in fluorescence over the time course of the study (Fig. 2A). Fibers subjected to the Moderate and Low contraction protocols showed a significant increase in the rate of change in DAF-FM fluorescence by a mean of ∼75% following contractions indicating an increase in NO production (Fig. 2A). DHE oxidation was observed to increase in an intensity-dependent manner in DHE-loaded fibers following muscle contractions (Fig. 2B) with the group of fibers subjected to the High, Moderate, and Low contraction protocols showing a 5.8-, 4.1-, and 2.8-fold increase relative to resting fibers (Fig. 2C).
FIG. 2.

Changes in DAF-FM fluorescence and DHE oxidation following contractile activity. (A) Rate of change in DAF-FM fluorescence from resting FDB fibers and fibers subjected to contractile activity of different intensities over the 10–20 min period. *p<0.05 compared with values from the same group prior to contractions 7–10 fibers in each group). (B) Relative change in DHE oxidation from resting FDB fibers and fibers subjected to contractile activity of different intensities over the 10–20-min period. *p<0.05 compared with fibers from the contracted groups at the corresponding time point. #p<0.05 compared with fibers subjected to the Low contraction protocol (n=7–8 fibers in each group). (C) Rate of change in DHE oxidation from resting FDB fibers and fibers subjected to a 10-min period of contractile activity with different intensities. *p<0.05 compared with fibers from the resting group. #p<0.05 compared with fibers subjected to the Low contractile activity protocol (n=7–8 fibers in each group).
Contribution of mitochondria to cytosolic superoxide in single skeletal muscle fibers at rest and during contractile activity
Changes in superoxide in the mitochondrial matrix were monitored using MitoSOX Red. This probe selectively accumulated in the mitochondria of single muscle fibers as shown by the colocalization of MitoTracker Green FM with MitoSOX Red fluorescence. The colocalization coefficients were as follows: Pearson's correlation (Rr)=0.51, Manders's overlap (R)=0.78, Manders's colocalization coefficient for channel 1 (Mred)=0.91, and Manders's colocalization coefficient for channel 2 (Mgreen)=0.99 (Fig. 3A[v]). MitoSOX Red fluorescence was not detectable in the nuclei of muscle cells (Fig. 3A[v]).
FIG. 3.

Release of superoxide from intact mitochondria following treatment with Ant A does not occur through the mPTP or iMAC. (A) Confocal images of a single isolated fiber under bright field (i); fluorescent image following loading with DAPI (ii); fluorescence from MitoTracker Green FM (15 nM) (iii); fluorescent image from MitoSOX Red (iv); and a merged image of (ii)–(iv)–(v). Original magnification: 60×(scale bar=30 μm). (B) Relative change in DHE oxidation from isolated FDB fibers either untreated or treated with Ant A (5 or 10 μM) at 30 min. *p<0.05 compared with values from fibers treated with 5 μM Ant A or control vehicle-treated (V.control) fibers at the same time point (n=7–8 fibers in each group). (C) Relative change in MitoSOX Red fluorescence from isolated FDB fibers either untreated or treated with Ant A (5 or 10 μM) at 30 min. *p<0.05 compared with values from fibers treated with 5 μM Ant A and control vehicle-treated (V.control) fibers at the same time point (n=6–8 fibers in each group). (D) Relative change in DHE oxidation from control fibers and fibers loaded with Ant A (5 μM) at 30 min. Fibers were either treated with vehicle only (V.control) or treated with the mitochondrial-targeted SS-31 peptide at 10 or 100 μM (n=6 fibers in each group). (E) Relative change in DHE oxidation from control fibers and fibers loaded with Ant A (5 μM) at 30 min. Fibers were either treated with vehicle only (V.control) or treated with the mPTP (CsA) or iMAC (4Cl-DZP) inhibitors. *p<0.05 compared with values from fibers in all other Ant A–treated groups and control vehicle-treated (V.control) fibers at the corresponding time points (n=6–8 fibers in each group). (F) Relative change in MitoSOX Red fluorescence from control fibers and fibers treated with Rot at 30 min. A group of fibers was also treated with SS-31 peptide (10 μM). *p<0.05 compared with values from control vehicle-treated (V.control) fibers at the same time point. #p<0.05 compared with values from Rot-treated fibers incubated in the presence of SS-31 peptide at the corresponding time point (n=6–9 fibers in each group). (G) Relative change in DHE oxidation from control fibers and fibers treated with Rot at 30 min. 4Cl-DZP, 4′-chlorodiazepam; Ant A, antimycin A; CsA, cyclosporin A; iMAC, innermembrane anion channel; mPTP, mitochondrial permeability transition pore; Rot, rotenone.
Skeletal muscle mitochondria release superoxide to the cytosol of fibers following treatment with antimycin A
To examine mitochondria as a potential source of superoxide detected in the cytosol, fibers were treated with the electron transport chain inhibitors—Antimycin A (Ant A) and rotenone (Rot). Treatment of fibers with either 5 or 10 μM Ant A at 30 min induced a dose-dependent increase in DHE oxidation compared with control untreated fibers, indicating release of superoxide from mitochondria to the cytosol (Fig. 3B). MitoSOX Red–loaded fibers also showed a dose-dependent increase in fluorescence following addition of Ant A, indicating an increase in superoxide in the mitochondrial matrix (Fig. 3C).
Release of superoxide from mitochondria following treatment with Ant A does not occur through the mitochondrial permeability transition pore or the innermembrane anion channel
Previous reports have shown that superoxide can be released from the mitochondrial matrix through the mitochondrial permeability transition pore (mPTP) (1) and the innermembrane anion channel (iMAC) (41). To identify whether either of these two channels contributed to the extramitochondrial superoxide release seen following treatment with Ant A, fibers were pretreated with the SS-31 peptide, a mitochondrial-targeted antioxidant peptide that accumulates on the IMM and can scavenge superoxide to form tyrosine hydroperoxide (15, 41). Treatment with SS-31 at either 10 or 100 μM had no effect on the Ant A–stimulated increase in DHE oxidation (Fig. 3D). The potential role of the mPTP and iMAC in release of superoxide was further examined using inhibitors of the two potential pores/channels (cyclosporin A [CsA] and 4′-chlorodiazepam [4Cl-DZP]) but neither was found to reduce DHE oxidation in Ant A–treated fibers (Fig. 3E). Together these data suggest that superoxide released following treatment with Ant A did not derive from the mitochondrial matrix. The role of matrix superoxide was further examined by treatment of fibers with the complex I inhibitor Rot. This treatment increased MitoSOX Red oxidation, indicating increased superoxide generation in the matrix (Fig. 3F), but had no effect on DHE oxidation (Fig. 3G).
Channels of the OMM mediate the diffusion of superoxide from the MIS to the cytosol of skeletal muscle fibers
To identify channels that might play a role in the diffusion of superoxide from the MIS to the cytosol of single muscle fibers, mitochondrial and cytosolic fractions from gastrocnemius (GTN) muscles were prepared. Figure 4A illustrates the relative purity of the mitochondrial and cytosolic fractions obtained and Figure 4B illustrates the protein contents of the three voltage-dependent anion channel (VDAC) isoforms. In the presence of dextran sulfate (DS), a VDAC inhibitor, the increase in DHE oxidation observed following treatment with Ant A at 30 min was partially inhibited over the next 20 min, but reached a similar level by the end of the experiment compared with values from fibers loaded with Ant A only (Fig. 4C). No effect on baseline DHE oxidation was observed following treatment of fibers with DS alone at 30 min (Fig. 4D). Potential effects of inhibition of VDAC by 4,4′-diisothiocyano-2,2′-disulfonic acid stilbene could not be assessed since this agent was found to affect fiber viability at IC50 values of 0.2 mM (data not shown in detail). The protein expression of the Bax channel in fibers is shown in Figure 4E. Following treatment with the Bax channel blocker (Bax CB), fibers showed a slightly lower rate of increase in DHE oxidation (by ∼8%) during the 40–50 min period following addition of Ant A (Fig. 4F). Fibers pretreated with the Bax CB and VDAC inhibitor (DS) together showed a further reduction in fluorescence, with the relative increase in DHE oxidation being statistically lower (∼13%) during the 20–50 min period following Ant A treatment compared with fibers treated with Ant A only (Fig. 4F). The increase in DHE oxidation in the Bax CB and DS group was lower over the 30–90 min time course compared with the Ant A–treated fibers (Fig. 4F) but this did not occur in fibers treated with DS only (Fig. 4C), suggesting that both channels might play a synergistic role in mediating the diffusion of superoxide from the MIS across the OMM. Inhibition or blocking of these two OMM channels did not completely prevent extramitochondrial superoxide release following addition of Ant A (Fig. 4F). To ensure that data were not influenced by any confounding direct interactions between Ant A and DHE, unrelated to effects of Ant A on the electron transport chain, control experiments were conducted in a cell-free medium but these showed no effect of Ant A on DHE or MitoSOX Red oxidation (data not shown in detail).
FIG. 4.

Channels of the OMM mediate the diffusion of superoxide from the MIS to the cytosol of fibers following treatment with Ant A. (A) Example of Western blots for GAPDH and cytochrome oxidase IV (COXIV) to illustrate the purity of the extracted Mito F and Cyto F fractions obtained from the GTN muscle. (B) Representative Western blots of VDAC1, VDAC2, and VDAC3 proteins in Cyto F and Mito F fractions from GTN muscle, in lysate from single isolated fibers from the FDB muscle (fibers), and in whole GTN muscle. (C) Relative change in DHE oxidation from control fibers and fibers loaded with Ant A (5 μM) at 30 min. Fibers were either treated with vehicle alone or with DS. *p<0.05 compared with fibers treated with DS at the corresponding time point (n=7 fibers in each group). (D) Relative change in DHE oxidation from isolated FDB fibers either untreated or treated with DS at 30 min (n=7–8 fibers in each group). (E) Representative Western blot of Bax protein in cytosolic (Cyto F) and mitochondrial (Mito F) fractions from GTN muscle, in lysate from single isolated FDB muscle fibers, and in whole GTN muscle. (F) Relative change in DHE oxidation from control fibers and fibers loaded with Ant A (5 μM) at 30 min. Fibers were either untreated or treated with Bax CB or with Bax CB and DS. *p<0.05 compared with values from Ant A–treated fibers preincubated with Bax CB and DS at the same time point. #p<0.05 compared with values from fibers treated with Ant A in the presence of Bax CB at the same time point (n=6–7 fibers in each group). (G) Rate of change in DHE oxidation from resting fibers and fibers subjected to the Moderate contraction protocol for a period of 10 min. Contracted fibers were either untreated, treated with DS, or treated with DS and Bax CB. *p<0.05 compared with values from fibers at rest (n=9–11 fibers in each group). Bax CB, Bax channel blocker; Cyto F, cytosolic fractions; DS, dextran sulfate; GTN, gastrocnemius; Mito F, mitochondrial fractions; VDACs, voltage-dependent anion channels.
The effect of VDAC (DS) and Bax (Bax CB) inhibitors was assessed in fibers following a 10-min contraction period but no differences in DHE oxidation were observed between the contracted groups (Fig. 4G).
Identifying the cytosolic sources of superoxide production in single skeletal muscle fibers at rest and during contractile activity
Effect of inhibition of calcium-independent phospholipase A2 enzyme and NADPH oxidase(s) on cytosolic superoxide
The presence of calcium-independent phospholipase A2 enzyme (iPLA2) in the cytosolic compartment of skeletal muscles was confirmed by Western blotting (Fig. 5A) but no effect of the selective iPLA2 inhibitor, bromoenol lactone (BEL), on DHE oxidation was observed either in quiescent (Fig. 5B) or contracted fibers (Fig. 5C).
FIG. 5.
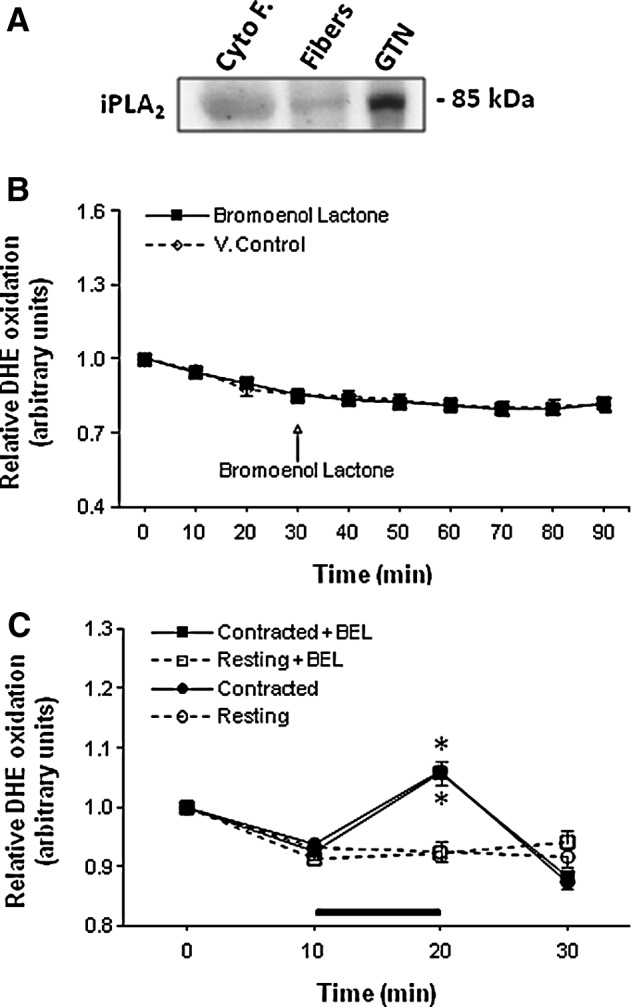
Effect of IPLA2 inhibition on cytosolic superoxide following contractile activity. (A) Representative Western blot of iPLA2 protein in Cyto F from GTN muscle, in lysate from single isolated FDB muscle fibers, and in whole GTN muscle. (B) Relative change in DHE oxidation from isolated FDB fibers either untreated or treated with BEL (0.6 μM) at 30 min (n=7–10 fibers in each group). (C) Relative change in DHE oxidation from resting FDB fibers and fibers subjected to the Moderate contraction protocol over the 10–20-min period. Fibers were either untreated or treated with BEL (0.6 μM). *p<0.05 compared with values from fibers of the same group prior to contractions (n=7–8 fibers in each group). BEL, bromoenol lactone; iPLA2, calcium-independent phospholipase A2.
The mRNA and protein expression of NADPH oxidase subunits in single skeletal muscle fibers of the FDB muscle is shown in Figure 6A and B. Further experiments were also undertaken to investigate whether the NOX4 or NOX2 subunits were present in mitochondrial fractions from GTN muscles and demonstrated the presence of NOX4 in this fraction (Fig. 6C, D). The role of NADPH oxidase complexes in producing superoxide under resting conditions was initially assessed using the nonspecific flavoprotein inhibitor diphenyleneiodonium chloride (DPI) (Fig. 6E). Resting muscle fibers treated with DPI at 30 min of the experimental period showed a rapid increase in fluorescence compared with control nontreated fibers (Fig. 6E). In addition, DPI was also observed to prevent the fibers from contracting following field stimulation. An alternative NADPH oxidase inhibitor, 4-(2-aminoethyl)-benzenesulfonyl fluoride inhibitor, was also found to inhibit muscle contractions (data not shown in detail). Fibers were therefore incubated in the presence of apocynin (APO), a nonspecific NADPH oxidase inhibitor. Quiescent muscle fibers showed a reduction in fluorescence by ∼10% over the 50–90 min time course compared with vehicle-treated fibers (Fig. 6F). APO was also found to decrease DHE oxidation following a period of contractile activity by ∼70% compared with untreated fibers (Fig. 6G, H). Recent data have suggested that APO can also act as a scavenger for reaction products of hydrogen peroxide (H2O2), in vascular cells that lack myeloperoxidase (MPO) or produce low amounts of ROS (29), and hence the potential inhibitory effects of gp91ds-tat, a specific peptide inhibitor of NADPH oxidases, were also examined (Fig. 6I, J). Resting fibers showed a decline in fluorescence by a mean of 18% compared with vehicle-treated fibers and fibers treated with the control peptide scrambled-tat (Fig. 6I). Treatment of fibers with gp91ds-tat also prevented the increase in DHE oxidation in response to a 10-min period of contractions (Fig. 6J), but the scrambled-tat-treated fibers also showed a lower rate of increase in DHE oxidation than the vehicle-treated control group (Fig. 6J).
FIG. 6.


NADPH oxidase is expressed in skeletal muscle fibers and regulates changes in superoxide following contractile activity. (A) RT-PCR amplification of NOX2, NOX4, Rac1, p67phox, p47phox, p22phox, p40phox, and GAPDH transcripts in single isolated fibers from the FDB muscle. The PCR products correspond to the amplicon sizes shown in Table 1. (B) Representative Western blots of NOX2, NOX4, Rac1, p67phox, p47phox, p22phox, and p40phox proteins in lysate from single isolated FDB muscle fibers and whole GTN muscle. Appropriate positive controls (PC) are shown: lysate from mouse heart for NOX2, NOX4, and p40phox; lysate from mouse liver for p67phox and p22phox; human platelet extract for Rac1 and Raw macrophage 264.7 whole cell lysate for p47phox. *p40phox was immunoprecipitated; see the section “Materials and Methods” for details. (C, D) Representative Western blots to show detection of NOX4, but not NOX2 proteins in Mito F from GTN muscle compared with lysate from whole GTN muscle. (E) Relative change in DHE oxidation from isolated FDB fibers either untreated or treated with DPI at 30 min. *p<0.05 compared with control-untreated fibers at the same time points (n=6–8 fibers in each group). (F) Relative change in DHE oxidation from isolated FDB fibers either untreated or treated with APO at 30 min. *p<0.05 compared with control vehicle-treated (V.control) fibers at the same time points (n=9 fibers in each group). (G) Relative change in DHE oxidation from resting FDB fibers and fibers subjected to the Moderate contraction protocol over the 10–20-min period. Fibers were either untreated or treated with APO. *p<0.05 compared with values from fibers of the same group prior to contractions. #p<0.05 compared with contracted fibers treated with APO at the corresponding time point (n=14–17 fibers in each group). (H) Rate of change in DHE oxidation from resting fibers and fibers subjected to a 10-min period of contractile activity. Fibers were either untreated or treated with APO. *p<0.05 compared with fibers from both resting groups. #p<0.05 compared with contracted fibers treated with APO (n=14–17 fibers in each group). (I) Relative change in DHE oxidation from isolated FDB fibers either untreated or treated with gp91ds-tat or scrmb-tat at 30 min. *p<0.05 compared with vehicle-treated (V.control) fibers and fibers treated with scrmb-tat at the corresponding time points (n=6–7 fibers in each group). (J) Rate of change in DHE oxidation from resting FDB fibers and fibers subjected to the Moderate contraction protocol for a period of 10 min. Contracted fibers were either untreated or treated with gp91ds-tat or scrmb-tat. *p<0.05 compared with values from fibers of the resting group. #p<0.05 compared with contracted fibers treated with gp91ds-tat. †p<0.05 compared with values from contracted fibers treated with scrmb-tat (n=7 fibers in each group). APO, apocynin; DPI, diphenyleneiodonium chloride; RT-PCR, real-time–polymerase chain reaction.
NADPH oxidase subunits are located on the plasma membrane and transverse tubules of single skeletal muscle fibers
To determine the cellular location of NADPH oxidase subunits, immunocytochemistry of single isolated fibers from the FDB muscle was undertaken (Fig. 7A–D). The regulatory subunits (p40phox, p47phox, p67phox, and Rac1) were found to be localized on, or in close proximity to the, sarcolemma, but immunofluorescence from p40phox and p67phox was also observed in the cytosolic compartment of the muscle fibers (Fig. 7A). The catalytic subunits (NOX2 and NOX4) and the small membrane-bound integral p22phox subunit were localized to the plasma membrane of the muscle fibers, as indicated by colocalization (yellow staining) with caveolin-3, the muscle-specific caveolin isoform, present in sarcolemmal caveolae (Fig. 7B). A striated pattern of staining for all three subunits was also observed in close proximity to the sarcolemma, potentially due to expression in the transverse tubules (T-tubules) (Fig. 7B). To examine this possibility, the membrane-bound subunits of the NADPH oxidase complex from single muscle fibers were co-immunostained with the α1s subunit of the dihydropyridine receptor (Fig. 7C). Confocal images showed a high degree of colocalization (yellow staining), strongly suggesting that the NADPH oxidase components NOX2 (Rr=0.60, R=0.67, Mred=0.85, Mgreen=0.96), NOX4 (Rr=0.98, R=0.98, Mred=0.98, Mgreen=1), and p22phox (Rr=0.92, R=0.97, Mred=1, Mgreen=0.92) were also expressed on the T-tubule membrane in skeletal muscle fibers (Fig. 7C).
FIG. 7.



NOX-catalytic subunits are located on the plasma membrane and T-tubules of skeletal muscle fibers. (A) Immunocytochemistry of single isolated fibers from the FDB muscle showing the expression of p40phox, p47phox, p67phox and Rac1 subunits of the NADPH oxidase complex. (B) Immunocytochemistry for the NOX2, NOX4 and p22phox NADPH oxidase components. Fibers were co-immunostained with an antibody to Caveolin-3 (red staining) to demonstrate sarcolemmal colocalization (yellow staining). (C) Fibers were immunostained using antibodies against NOX2, NOX4 and p22phox and co-immunostained with an antibody to α1s DHPR (red staining) to demonstrate T-tubular colocalization (yellow staining). (D) Immunocytochemistry of single isolated fibers showing the subcellular location of the cytosolic NADPH oxidase subunits p40phox and p67phox at rest and following a 10-min period of moderate contractions. Nuclei (blue staining) were stained with DAPI. (E) Profile of the distribution of fluorescence from immunostaining for p40phox across the single resting and contracted fibers shown in (D). α1s, subunit of dihydropyridine receptor; T-tubules, transverse tubules.
The effect of contractions on p40phox and p67phox translocation in single skeletal muscle fibers
The effect of contractions on p40phox and p67phox protein translocation is shown in Figure 7D. Fluorescent distribution analysis showed no evidence for contraction-induced translocation of the p67phox protein (not shown in detail), but fluorescent distribution analysis for p40phox (Fig. 7E) showed an increased proportion of the fiber fluorescence at the sarcolemma and a relative reduction in the cytosolic compartment of contracted fibers compared with quiescent fibers. These data suggest that NADPH oxidase activation in skeletal muscle fibers during contractions may involve translocation of p40phox to the plasma membrane.
Discussion
Methodological considerations
We have previously utilized electron-spin resonance spectroscopy (53), fluorescence-based microscopic assays (51, 55, 60), microdialysis (12), and high-performance liquid chromatography techniques (53, 60) to assess intracellular and extracellular changes in RONS produced by skeletal muscle. In the current study, we utilized fluorescence imaging microscopy to allow monitoring of real-time changes in radical species in single isolated muscle fibers and to examine potential subcellular pathways that are involved in the regulation of cytosolic superoxide content at rest and during contractile activity. Within subcellular compartments of skeletal muscle, superoxide will be rapidly dismutated to H2O2 or undergo chemical reactions to generate products, such as peroxynitrite [see ref. (60) for a discussion], but superoxide per se can be detected in subcellular regions, such as the mitochondria or cytoplasm, due to its poor membrane permeability. Superoxide has also previously been claimed to directly induce changes in muscle function and degeneration during aging (60).
Assessment of E+ fluorescence following MitoSOX Red and DHE loading as a measure of superoxide anion radical in cellular compartments has been criticized and recent studies have identified 2-hydroxyethidium (2-OH-E+) as a specific product of the reaction of DHE and MitoSOX Red with superoxide (71). We have previously evaluated the use of DHE oxidation and showed that E+ and 2-OH-E+ followed the same pattern of change in resting and contracted fibers from wild-type mice and mice lacking SOD1 (60). Previous work also showed that the anticipated increase in DHE oxidation following contractions was completely abolished following loading of fibers with the superoxide scavengers Tiron or Tempol (60). Thus, we argue that the technique used in this study, based on monitoring E+ fluorescence from single muscle cells, is capable of detecting changes in superoxide production at rest but also in response to a physiological stimulus such as contractile activity.
NO and superoxide production following contractile activity protocols of different intensities
The increase in RONS activity during exercise appears to be in major part due to generation by contracting skeletal muscle (32, 54) and one objective of the present study was to determine the effect of increasing frequency of contractions on muscle fiber NO and superoxide activity. We previously showed that contractile activity induces an increase in intracellular DAF-FM fluorescence in mouse skeletal muscle fibers due to increased NO generation (55). In the present study an increase in DAF-FM fluorescence was seen from fibers subjected to all contraction protocols, but the increase reached statistical significance only for the Moderate and Low contraction protocols.
For DHE-loaded fibers, those subjected to the High contraction protocol exhibited the largest increase in DHE oxidation and those subjected to the Low protocol had the least. This finding is in agreement with a previous study that demonstrated that intracellular ROS generation in primary myotubes was higher following an intense electrical stimulation protocol than that with a moderate stimulation protocol (61). The increase in DHE oxidation immediately postcontractions was followed by a reduction in fluorescence at 10 min following the contractile activity period. Early studies investigating the electrochemical reduction of E+ reported DHE as the final product (31) and the reduction of E+ to DHE has been reported in cell free (6) and cellular systems (23). Work from our group (unpublished data) has also shown that the reduction of E+ at 10 min postcontractions is related to an increase in DHE as assessed by changes in blue fluorescence.
Contribution of mitochondria to cytosolic superoxide in single skeletal muscle fibers
We reasoned that the isolated single mature skeletal muscle fiber approach would provide a valid model to study the role of mitochondria in regulating changes in cytosolic superoxide. These fibers contain an intact mature mitochondrial matrix in situ and retain excitability to electrical stimuli, hence facilitating the study of ROS generation following contractions in real time. The current approach indicated that the rapid increase in superoxide observed following contractions likely originated from nonmitochondrial sources since the change in DHE oxidation reflects reaction with superoxide within the cytosolic compartment of the muscle cells (33). Superoxide is a membrane-impermeable anion, but recent data have indicated that superoxide can diffuse out of isolated mitochondria through the mPTP (1), the iMAC (15), and channels located on the OMM (11, 27).
Treatment of fibers with the electron transport chain inhibitors Ant A and Rot increased the oxidation of matrix-localized MitoSOX Red, but only Ant A caused an increase in cytosolic DHE oxidation, supporting the possibility that only superoxide within the MIS can contribute to cytosolic changes. The lack of any role for matrix superoxide in oxidation of DHE in the cytoplasm was further supported by the lack of any effect of SS-31 peptide that scavenged superoxide within the mitochondrial matrix as shown by the reduction of MitoSOX Red fluorescence following addition of Rot. These data provide evidence that superoxide generated by complex III and released into the matrix does not escape from intact mitochondria in muscle fibers. These findings are consistent with previous reports that failed to show any extramitochondrial superoxide release from mitochondria in response to the addition of Rot (47). Inhibitors of iMAC or mPTP were also ineffective in reducing cytosolic DHE oxidation in Ant A–treated fibers, further indicating a lack of a role for these channels in release of superoxide from the mitochondrial matrix to the cytosol of muscle fibers.
A role for OMM channels in release of superoxide from the MIS to the cytoplasm was supported by the inhibitory effects of VDAC and Bax inhibitors on DHE oxidation following Ant A treatment, and inhibition of VDACs resulted in a greater reduction in DHE oxidation than that of Bax inhibition, potentially because VDACs are the major channels of the OMM responsible for the passage of proteins and solutes between the OMM and cytoplasm (42). Neither VDAC nor Bax inhibition fully prevented the extramitochondrial superoxide release induced by Ant A, suggesting that either of the inhibitors failed to completely close the respective channels or other channels, such that peptide-sensitive channel or the translocase of outer membrane (11) might also mediate the release of superoxide across the OMM.
Despite the effects of VDAC and Bax channel inhibitors seen on DHE oxidation in Ant A–treated fibers, no similar effects were seen following contractions, indicating that the increase in cytosolic superoxide during contractile activity does not derive from mitochondria. DHE oxidation following contractions increased by ∼20%, whereas the change in fluorescence in response to Ant A increased by ∼90%. Thus, the observed increase in superoxide following addition of Ant A may be considered “nonphysiological” compared with that seen following a period of contractile activity. Hence, we propose that intact mitochondria do not release superoxide to the cytoplasm of skeletal muscle fibers at rest or in response to contractile activity, but can release superoxide under conditions where mitochondrial superoxide production is grossly excessive.
Contribution of iPLA2 enzymes to cytosolic superoxide in skeletal muscle fibers
Recent work (26) demonstrated that iPLA2 enzyme can modulate cytosolic oxidant activity in skeletal muscle cells indicated by a reduction in 2′,7′-dichlorodihydrofluorescin oxidation (a general indicator of ROS activities) following inhibition of iPLA2 (26). In the present study no effects of iPLA2 inhibitors on DHE oxidation were seen. The previous study (26) used BEL at 10 μM whereas in the present study 0.6 μM was used since preliminary studies indicated that higher concentrations affected the viability of muscle fibers (data not shown in detail). BEL at 0.6 μM has previously been shown to inhibit iPLA2 activity by 100% (2). Thus, current data do not support the possibility that cytosolic superoxide levels at rest or during contractions are modulated by the activity of the iPLA2 enzymes although the higher concentrations of inhibitor used by other researchers could not be examined in the model. Zuo et al. demonstrated that PLA2 enzymes in skeletal muscle do not directly produce superoxide but can regulate the activity of the lipoxygenases by modulating the release of arachidonic acid (72).
Expression of NADPH oxidase and its contribution to cytosolic superoxide in skeletal muscle fibers
Skeletal muscles have previously been shown to express NADPH oxidase(s) (22, 30, 45, 67), but little information is available regarding the role and regulation of this complex in generation of superoxide in muscles. Our initial results show mRNA and protein expression of NADPH oxidase subunits in single isolated fibers from the FDB muscle. Others have identified various NADPH oxidase subunits in mouse and rabbit skeletal muscles (30, 67), but this appears to be the first report to demonstrate the expression of NADPH oxidase components in a pure muscle fiber preparation devoid of all nonmyogenic cells.
Inhibitor studies support a role for NADPH oxidase in contributing to cytosolic superoxide production. Resting fibers treated with APO and gp91ds-tat inhibitors showed a decrease in DHE oxidation compared with control fibers, suggesting that the complex is active and modulates superoxide generation under resting conditions. Similarly, fibers subjected to contractile activity showed a significantly lower increase in DHE oxidation following treatment with APO whereas gp91ds-tat treatment abolished the increase in response to contractions. Scrambled-tat-pretreated fibers also showed a decrease in fluorescence following contractions, but some previous studies have also observed this (57). Gp91ds-tat appeared to be more effective than APO, which can be attributed to the different inhibitory properties of the compounds.
Recent evidence suggests that APO might require activation by peroxidases (69) and can also act as an antioxidant for peroxide-dependent ROS in cellular systems that lack MPO, or produce low amounts of ROS (29). The proposed inhibitory mechanism of action of APO on NADPH oxidases is to prevent the translocation of p47phox and p67phox to the membrane-located catalytic subunits (46) whereas gp91ds-tat peptide, the most potent inhibitor (IC50: 3 μM), has been reported to selectively inhibit NOX2 oxidase (14), although early studies suggested that the peptide additionally inhibited NOX1 and NOX4 homologues (9, 57) by preventing their interaction with p47phox (57). The inhibitory effects of both APO and gp91ds-tat imply that p47phox is a critical component of the NADPH oxidase complex and can regulate the activity of the enzyme.
Treatment of resting fibers with the nonspecific inhibitor DPI showed a paradoxical increase in DHE oxidation. The mechanism by which DPI induced increased DHE oxidation in the current study is unclear, but DPI has been reported to inhibit flavoenzymes in addition to NADPH oxidases, such as NOS, xanthine oxidase, and NADPH-cytochrome P450 reductase (5, 39, 52), and hence it can affect cellular processes unrelated to NADPH oxidase. Data on the regulation of intracellular ROS by DPI are controversial, with both inhibitory and stimulatory actions of DPI being reported. Some studies indicate that DPI can stimulate the production of ROS (37, 58) and reactive nitrogen species (5), affecting cellular redox status indicated by increased levels of lipid peroxidation (58), DNA damage (39, 52), increased glutathione disulfide (58), apoptosis (5, 39, 52), and increased nitration of tyrosine residues in cellular proteins (5). Some previous studies have also reported an increased oxidation of ROS-sensitive probes, including dihydrorhodamine 123 (5) and 2′,7′-dichlorodihydrofluorescein diacetate (5, 52), following addition of DPI. Potential mechanisms through which DPI might stimulate RONS production have been proposed, including increased mitochondrial superoxide production via inhibition of complex I (37, 38). Formation of phenyl radicals during NADPH oxidase inhibition by DPI has also been reported (39, 50).
The data presented imply that NOX2 is likely to be the predominant oxidase system contributing to cytosolic superoxide generation in muscle fibers, since most recent data indicate that the gp91ds-tat peptide selectively inhibits the assembly of NOX2 oxidase (14). Immunocytochemistry of single isolated fibers from the FDB muscle revealed that NOX isoforms and subunits were localized on, or in close proximity to, the plasma membrane of muscle fibers although p40phox and p67phox were also present in the cytosolic compartment of quiescent muscle fibers. Further experiments supported previous findings (22, 30) of apparent localization of these subunits to the T-tubules. In addition, NOX4 was found to be present in skeletal muscle mitochondria. Recent reports have shown that NOX4 localized to cardiac (3, 15, 36) and liver (7) mitochondria. NOX4 has been predicted to localize on the IMM (7) and produce H2O2 (66) but recent reports have shown that it can also regulate changes in superoxide within the mitochondrial matrix (3, 7, 15, 36). These findings, however, have not been examined in skeletal muscle; thus, future studies are warranted to examine the role of NOX4 in skeletal muscle mitochondria.
In phagocytic cells, NADPH oxidase activation requires the translocation of the cytosolic regulatory subunits to cytochrome b558, the catalytic core of the enzyme (20, 35). Fluorescent distribution analysis showed no evidence for translocation of the p67phox protein following contractions but p40phox-immunostained fibers showed an increased proportion of fluorescence at the membrane and a reduction in the cytoplasmic compartment following contractions, which suggests that NADPH oxidase activation in single skeletal muscle fibers from young mice in response to contractions may involve translocation of p40phox to the plasma membrane. Although the role of p40phox has been controversial, studies in phagocytes indicate that on stimulation p40phox translocates to cytochrome b558 (20) and functions as a positive regulator of the superoxide-producing phagocyte oxidase by enhancing recruitment of p67phox and p47phox to the membrane (35). In addition, lack of p40phox in homozygotic knockout mice reduced superoxide production in both in vivo and in vitro models (21) and decreased the expression of p67phox (21, 64).
Physiological implications
Previous studies indicated that myotube depolarization induced superoxide generation through activation of NADPH oxidase (22) and data presented here support this general proposal. The presence of NADPH oxidase that is localized to the sarcolemma and T-tubules together with the effects of increasing frequency of contractions on superoxide suggest that repeated membrane depolarization may regulate the activity of skeletal muscle NADPH oxidase. A potential physiological role for this process was proposed by Hidalgo et al. who suggested that the superoxide generated by NADPH oxidase can stimulate calcium release from the sarcoplasmic reticulum through oxidative modification of the ryanodine receptor (30).
There is also emerging evidence of interplay between ROS generated by NADPH oxidases and that by mitochondria; for instance, NADPH oxidase generated superoxide triggering mitochondrial ROS formation. Studies in vascular tissue have shown that NOX-derived cytosolic ROS can trigger mitochondrial ROS formation (10) by opening the mitochondrial ATP-sensitive potassium channels, leading to changes in the mitochondrial membrane potential (18). Conversely, increased mitochondrial H2O2 release has been reported to activate NADPH oxidase via a protein kinase C–dependent pathway, resulting in increased cytosolic superoxide production (16, 18). The concept that NADPH oxidase activation can trigger mitochondrial ROS formation and vice verse is also supported by in vivo pharmacological (70) and transgenic approaches (68) that have shown that mitochondrial ROS can regulate the expression of the NADPH oxidase components. The location of NOX4 in skeletal muscle mitochondria that is reported in the current work also supports the concept of interplay between the NADPH oxidases and mitochondrial ROS sources, although it is unknown whether this cross-talk plays any role in skeletal muscle at rest or during contractile activity.
In conclusion, these data are the first to report the relative contribution of potential mitochondrial and cytosolic sources to superoxide production within the cytosolic compartment of single muscle fibers both at rest and during contractile activity. Data indicate that contractile activity increases superoxide content in an intensity-dependent manner. Experiments with isolated mature skeletal muscle fibers support the conclusion that mitochondrial sources do not contribute to cytosolic superoxide either at rest or following contractile activity. In contrast, under conditions where mitochondrial superoxide production is grossly excessive, channels of the OMM, such as VDAC and Bax, appear to be able to release superoxide from the MIS to the cytosol of skeletal muscle fibers. Inhibition of NADPH oxidase activity induced a significant decrease in DHE oxidation at rest and following contractions. Our data are therefore consistent with the proposal that NADPH oxidase is the major contributor to superoxide production both at rest and during contractile activity in skeletal muscle.
Materials and Methods
Mice
Four- to eight-month-old, female C57Bl/6 mice were used in this study. All experiments were performed in accordance with United Kingdom Home Office guidelines under the United Kingdom Animals (Scientific Procedures) Act 1986. Mice were sacrificed by cervical dislocation and the FDB muscles were rapidly removed for isolation of single muscle fibers (see paragraph “Isolation of single mature skeletal muscle fibers”). Other muscles and tissues were harvested and stored at −70°C until analysis.
Chemicals and reagents
Unless stated otherwise, all chemicals used in this study were obtained from Sigma Chemical Company (Dorset, United Kingdom).
Isolation of single mature skeletal muscle fibers
Single muscle fibers were isolated from the FDB muscles of mice as previously described (51, 55). Experiments were only performed on fibers that displayed excellent morphology and exhibited clear striations along the sarcolemma (e.g., see Fig. 1).
Use of DAF-FM DA to monitor intracellular NO in single isolated fibers
To monitor changes in NO (Fig. 1A), fibers were loaded with DAF-FM DA as previously described (55).
Use of DHE to monitor cytosolic superoxide changes in isolated fibers
Fibers were loaded with 5 μM DHE (Invitrogen, Paisley, United Kingdom), in a similar manner to DAF-FM DA. DHE is a noncharged fluorescent probe specifically sensitive to superoxide. Unreacted DHE displays “blue” fluorescence (71) in the cytoplasm of single muscle fibers (Fig. 1B), but upon oxidation, the oxidized form of DHE, namely, the E+ cations, accumulate in the nucleus and intercalate with the negatively charged DNA phosphate backbone, producing E+ fluorescence (Fig. 1C). Application of the technique to the study of single isolated mature skeletal muscle fibers has been recently reported by Sakellariou et al. (60).
Use of MitoSOX Red to monitor mitochondrial superoxide changes in isolated fibers
Fibers were loaded with 250 nM MitoSOX Red (Invitrogen), using the same protocol as for DAF-FM DA and DHE. MitoSOX Red is a derivative of DHE designed for highly selective detection of superoxide in mitochondria and exhibits fluorescence (MitoSOX Red fluorescence) on oxidation and subsequent binding to mitochondrial DNA (Fig. 3A) (59).
Microscopy and fluorescent imaging
The imaging system consisted of a Zeiss Axiovert 200M epifluorescence microscope equipped with 500/20 excitation and 535/30 emission filter set for the detection of DAF-FM fluorescence and a 510/60 nm excitation and 590 nm emission filter set for E+ and MitoSOX Red fluorescence (Carl Zeiss GmbH, Welwyn Garden City, United Kingdom). Using a×20 objective, fluorescence images were captured with a computer-controlled Zeiss MRm charged-coupled device camera (Carl Zeiss GmbH) and analyzed with the Axiovision 4.0 image capture and analysis software (Carl Zeiss GmbH, Welwyn Garden City, United Kingdom). All experiments were carried out at 25°C.
Contractile activity protocols
Contractions in single isolated muscle fibers were induced by electrical field stimulation using established techniques (51, 55, 60). Following loading, fibers remained at rest for 10 min and were then exposed to trains of bipolar square wave pulses of 2 ms in duration for 0.5 s at 50 Hz and 30 V/well. The duty cycle (i.e., proportion of time that the fiber was stimulated) was varied to modify the intensity of the contraction protocol. The High contraction protocol consisted of a 2.5-s stimulation cycle (20% duty cycle), the Moderate protocol consisted of a 5-s stimulation cycle (10% duty cycle), and the Low protocol consisted of a 10-s stimulation cycle (5% duty cycle) (Fig. 1D). The Moderate intensity protocol has been used in previous studies and shown to increase RONS activities in single isolated muscle fibers (51, 55, 60). A video of contracting FDB fibers was presented in previous work by Palomero et al. (51).
Use of pharmacological agents to identify sources of superoxide
Mitochondrial superoxide production was induced by treatment of fibers with Rot (250 nM) or Ant A (5 or 10 μM). The mitochondria-targeted SS-31 peptide (10 or 100 μM) was obtained from W.M. Keck Fdn. Biotechnology Resource Laboratory at Yale (New Haven, CT). CsA (0.5 μM), 4Cl-DZP (12 μM), DS Mr 6500–10,000 (DS; 0.1 mM), and the Bax CB [(±)-1-(3,6-dibromocarbazol-9-yl)-3-piper azin-1-yl-propan-2-ol; 5 μM; Merck Chemicals Ltd, Nottingham, United Kingdom] were used to assess the role of specific pathways in superoxide release from mitochondria. The contribution of NADPH oxidase enzymes was assessed using the NADPH oxidase inhibitors: APO (0.5 mM), DPI (10 μM; Enzo Life Sciences, Exeter, United Kingdom), gp91ds-tat, and the control peptide scrambled-tat (5 μM; Anaspec, Fremont). The iPLA2 was blocked by the selective inhibitor BEL (0.6 μM; Enzo Life Sciences). Incubation of fibers with inhibitors was commenced at 30 min prior to the start of fluorescence measurements, with the exception of the SS-31 peptide, which was added 4 h prior to measurements. Cell viability following treatments was determined by assessment of propidium iodide exclusion and control experiments were undertaken to examine any potential effects of the drugs on muscle contractions (data not shown in detail). Contractile activity was monitored and fibers that did not visibly contract during the entire contractile activity period were excluded.
RNA isolation and real-time–polymerase chain reaction analysis
Single skeletal muscle fibers from the FDB muscle were isolated, washed with cold phosphate-buffered saline (PBS), and frozen in liquid nitrogen. RNA from single muscle cells was extracted using Tri Reagent (Qiagen, Sussex, United Kingdom). All RNA samples were DNase-treated and purified using the RNeasy MinElute cleanup-kit (Qiagen). Purified RNA was utilized to generate first-strand cDNA using the iScript cDNA synthesis kit (Bio-Rad, Hertfordshire, United Kingdom). Primers for real-time polymerase chain reaction (PCR) analyses were designed (Table 1) and the optimal annealing temperature for each primer set was determined by using an annealing temperature gradient between 55°C and 62°C. Real-time PCR reactions were performed on an iCycler Detection System (Bio-Rad) using iQ SYBR Green Supermix (Bio-Rad). Specificity of the PCR products was determined by melt curve analysis and agarose gel electrophoresis.
Table 1.
Sequences of the Specific Primers Used for RT-PCR Amplification of NADPH Oxidase Subunits in Isolated Fibers from the Flexor Digitorum Brevis Muscle
| Primer name (ID) | Forward primer sequence | Reverse primer sequence | Amplicon size (bp) |
|---|---|---|---|
| NOX2 | cctgaatttcaactgtatgctga | cctgaatttcaactgtatgctga | 151 |
| NOX4 | ggatttgctactgcctccat | agtgactccaatgcctccag | 163 |
| Rac1 | gccaatgttatggtagatggaaa | tttcaaatgatgcaggactca | 151 |
| p67phox | gaccttaaagaggccttgacg | atgccaactgctcttctgct | 160 |
| p47phox | gtccctgcatcctatctgga | atgacctcaatggcttcacc | 155 |
| p22phox | gccattgccagtgtgatcta | tggtaggtggttgcttgatg | 118 |
| p40phox | tgacttcactgggaacagca | tagccagttggtggtgtcct | 184 |
| GAPDH | ccgtagacaaaatggtgaagg | tcgttgatggcaacaatctc | 109 |
NADPH, nicotinamide adenine dinucleotide phosphate; RT-PCR, real-time–polymerase chain reaction.
Western blotting of muscle proteins
Protein extracts (50 μg/sample) were separated using a standard protocol for Western blots (60). Peroxidase activity was detected using an ECL kit (Amersham International, Cardiff, United Kingdom), and band intensities were analyzed using Quantity One Software (Bio-Rad). Mitochondrial and cytosolic subcellular fractions were obtained from the same GTN muscle as previously described (13).
Immunoprecipitation of p40phox
Single isolated muscle fibers from the FDB muscle, powdered frozen muscle tissue from GTN, and heart tissue were homogenized and lysed in CelLytic-M Mammalian Cell Lysis/Extraction Reagent. Protein extracts (∼1 mg protein/sample) were incubated with 2 μg of anti-p40phox antibody (Santa Cruz Biotechnology, Middlesex, United Kingdom) overnight at 4°C. The mixture was added to 30 μl of protein G-Sepharose beads and was further incubated for 2 h at 4°C. Beads were centrifuged and washed five times with immunoprecipitation buffer and bound proteins were eluted by boiling in loading buffer (National Diagnostics, Hessle, United Kingdom) before being resolved by electrophoresis and identified by Western blotting.
Immunocytochemistry of NADPH oxidase subunits in single isolated muscle fibers
Single skeletal muscle fibers from the FDB muscle were isolated and plated on culture dishes. Cells were rinsed with warm PBS and immediately fixed in 4% paraformaldehyde for 20 min at room temperature. After three washes with PBS, fibers were permeabilized and blocked in PBS containing 0.1% Triton X-100 and 1% bovine serum albumin (BSA). After 10 h of incubation, fibers were washed with PBS and incubated overnight at 4°C with primary antibodies diluted in PBS and 1% BSA. Fibers were washed with PBS plus 1% BSA three times for 5 min, followed by incubation with appropriate secondary antibodies (Alexa Fluor 488 and 532; Invitrogen; diluted 1:800) for 1.5 h at room temperature. Fluorescence images were obtained using a C1 confocal laser scanning microscope (Nikon Instruments Europe BV, Surrey, United Kingdom) equipped with a 405 nm excitation diode laser, a 488 nm excitation argon laser, and a 543 nm excitation helium-neon laser. Emission fluorescence was detected through a set of 450/35, 515/30, and 605/15 emission filters. Using a×60 objective, fluorescence images were captured and analyzed with the EZC1 V.3.9 (12bit) acquisition software. To quantify subcellular (cytosolic and membrane) fluorescent distribution and the degree of colocalization of proteins and fluorescent probes in muscle fibers, NIH Image J software was used. The colocalization coefficients were as follows: Pearson's correlation (Rr) coefficient, Manders's overlap (R) coefficient, and Manders's colocalization coefficient for each image; channel 1 (Mred) and channel 2 (Mgreen) were calculated over the entire confocal image.
Statistical analyses
Data are presented as mean±standard error of the mean for each experiment. For multiple comparisons at any time point, analysis was done by one-way analysis of variance followed by the post hoc LSD test. Single comparisons between two experimental conditions at a single time point were undertaken using the unpaired Student's t test. Comparisons between data from individual fibers at different time points were undertaken using Student's paired t test. Data were analyzed using SPSS 18 and p-values of<0.05 were considered statistically significant.
Abbreviations Used
- α1s DHPR
α1s subunit of dihydropyridine receptor
- 2-OH-E+
2-hydroxyethidium
- 4Cl-DZP
4′-chlorodiazepam
- Ant A
antimycin A
- APO
apocynin
- Bax CB
Bax channel blocker
- BEL
bromoenol lactone
- BSA
bovine serum albumin
- CsA
cyclosporin A
- Cyto F
cytosolic fractions
- DAF-FM DA
4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate
- DAPI
4′,6-diamidino-2-phenylindole dihydrochloride
- DHE
dihydroethidium
- DPI
diphenyleneiodonium chloride
- DS
dextran sulfate
- E+
ethidium
- FDB
flexor digitorum brevis
- GTN
gastrocnemius
- H2O2
hydrogen peroxide
- iMAC
innermembrane anion channel
- IMM
inner mitochondrial membrane
- iPLA2
calcium-independent phospholipase A2
- Mgreen
Manders's colocalization coefficient for channel 2
- MIS
mitochondrial intermembrane space
- Mito F
mitochondrial fractions
- MPO
myeloperoxidase
- mPTP
mitochondrial permeability transition pore
- Mred
Manders's colocalization coefficient for channel 1
- NADPH
nicotinamide adenine dinucleotide phosphate
- NO
nitric oxide
- NOS
nitric oxide synthases
- OMM
outer mitochondrial membrane
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- R
Manders's overlap
- RONS
reactive oxygen and nitrogen species
- ROS
reactive oxygen species
- Rot
rotenone
- Rr
Pearson's correlation
- T-tubules
transverse tubules
- VDACs
voltage-dependent anion channels
Acknowledgments
The authors thank Dr. M.B. Reid (University of Kentucky Center for Muscle Biology) for the kind gift of the SS-31 peptide. Generous financial support from the Wellcome Trust (Grant No. 073263/Z/03), U.S. National Institute on Aging (Grant No. AG-20591), and the Alexander S. Onassis Public Benefit foundation (Greece) is gratefully acknowledged.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Abou-Sleiman PM. Muqit MM. Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosci. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 2.Ackermann EJ. Conde-Frieboes K. Dennis EA. Inhibition of macrophage Ca(2+)-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J Biol Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- 3.Ago T. Kuroda J. Pain J. Fu C. Li H. Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aydin J. Andersson DC. Hanninen SL. Wredenberg A. Tavi P. Park CB. Larsson NG. Bruton JD. Westerblad H. Increased mitochondrial Ca2+ and decreased sarcoplasmic reticulum Ca2+ in mitochondrial myopathy. Hum Mol Genet. 2009;18:278–288. doi: 10.1093/hmg/ddn355. [DOI] [PubMed] [Google Scholar]
- 5.Balcerczyk A. Soszynski M. Rybaczek D. Przygodzki T. Karowicz-Bilinska A. Maszewski J. Bartosz G. Induction of apoptosis and modulation of production of reactive oxygen species in human endothelial cells by diphenyleneiodonium. Biochem Pharmacol. 2005;69:1263–1273. doi: 10.1016/j.bcp.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 6.Benov L. Sztejnberg L. Fridovich I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med. 1998;25:826–831. doi: 10.1016/s0891-5849(98)00163-4. [DOI] [PubMed] [Google Scholar]
- 7.Block K. Gorin Y. Abboud HE. Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A. 2009;106:14385–14390. doi: 10.1073/pnas.0906805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boveris A. Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandes RP. A radical adventure: the quest for specific functions and inhibitors of vascular NAPDH oxidases. Circ Res. 2003;92:583–585. doi: 10.1161/01.RES.0000066880.62205.B0. [DOI] [PubMed] [Google Scholar]
- 10.Brandes RP. Triggering mitochondrial radical release: a new function for NADPH oxidases. Hypertension. 2005;45:847–848. doi: 10.1161/01.HYP.0000165019.32059.b2. [DOI] [PubMed] [Google Scholar]
- 11.Budzinska M. Galganska H. Karachitos A. Wojtkowska M. Kmita H. The TOM complex is involved in the release of superoxide anion from mitochondria. J Bioenerg Biomembr. 2009;41:361–367. doi: 10.1007/s10863-009-9231-9. [DOI] [PubMed] [Google Scholar]
- 12.Close GL. Kayani AC. Ashton T. McArdle A. Jackson MJ. Release of superoxide from skeletal muscle of adult and old mice: an experimental test of the reductive hotspot hypothesis. Aging Cell. 2007;6:189–195. doi: 10.1111/j.1474-9726.2007.00277.x. [DOI] [PubMed] [Google Scholar]
- 13.Cox B. Emili A. Tissue subcellular fractionation and protein extraction for use in mass-spectrometry-based proteomics. Nat Protoc. 2006;1:1872–1878. doi: 10.1038/nprot.2006.273. [DOI] [PubMed] [Google Scholar]
- 14.Csanyi G. Cifuentes-Pagano E. Al Ghouleh I. Ranayhossaini DJ. Egana L. Lopes LR. Jackson HM. Kelley EE. Pagano PJ. Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Radic Biol Med. 2011;51:1116–1125. doi: 10.1016/j.freeradbiomed.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai DF. Chen T. Szeto H. Nieves-Cintron M. Kutyavin V. Santana LF. Rabinovitch PS. Mitochondrial targeted antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011;58:73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta. 2010;1797:897–906. doi: 10.1016/j.bbabio.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 17.Davies KJ. Quintanilha AT. Brooks GA. Packer L. Free radicals and tissue damage produced by exercise. Biochem Biophys Res Commun. 1982;107:1198–1205. doi: 10.1016/s0006-291x(82)80124-1. [DOI] [PubMed] [Google Scholar]
- 18.Doughan AK. Harrison DG. Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 19.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 20.Dusi S. Donini M. Rossi F. Mechanisms of NADPH oxidase activation: translocation of p40phox, Rac1 and Rac2 from the cytosol to the membranes in human neutrophils lacking p47phox or p67phox. Biochem J. 1996;314(Pt 2):409–412. doi: 10.1042/bj3140409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ellson CD. Davidson K. Ferguson GJ. O'Connor R. Stephens LR. Hawkins PT. Neutrophils from p40phox−/− mice exhibit severe defects in NADPH oxidase regulation and oxidant-dependent bacterial killing. J Exp Med. 2006;203:1927–1937. doi: 10.1084/jem.20052069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Espinosa A. Leiva A. Pena M. Muller M. Debandi A. Hidalgo C. Carrasco MA. Jaimovich E. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J Cell Physiol. 2006;209:379–388. doi: 10.1002/jcp.20745. [DOI] [PubMed] [Google Scholar]
- 23.Fink B. Laude K. McCann L. Doughan A. Harrison DG. Dikalov S. Detection of intracellular superoxide formation in endothelial cells and intact tissues using dihydroethidium and an HPLC-based assay. Am J Physiol Cell Physiol. 2004;287:C895–C902. doi: 10.1152/ajpcell.00028.2004. [DOI] [PubMed] [Google Scholar]
- 24.Go YM. Jones DP. Redox control systems in the nucleus: mechanisms and functions. Antioxid Redox Signal. 2010;13:489–509. doi: 10.1089/ars.2009.3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gomez-Cabrera MC. Close GL. Kayani A. McArdle A. Vina J. Jackson MJ. Effect of xanthine oxidase-generated extracellular superoxide on skeletal muscle force generation. Am J Physiol Regul Integr Comp Physiol. 2010;298:R2–R8. doi: 10.1152/ajpregu.00142.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gong MC. Arbogast S. Guo Z. Mathenia J. Su W. Reid MB. Calcium-independent phospholipase A2 modulates cytosolic oxidant activity and contractile function in murine skeletal muscle cells. J Appl Physiol. 2006;100:399–405. doi: 10.1152/japplphysiol.00873.2005. [DOI] [PubMed] [Google Scholar]
- 27.Han D. Antunes F. Canali R. Rettori D. Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem. 2003;278:5557–5563. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- 28.Herrero A. Barja G. ADP-regulation of mitochondrial free radical production is different with complex I- or complex II-linked substrates: implications for the exercise paradox and brain hypermetabolism. J Bioenerg Biomembr. 1997;29:241–249. doi: 10.1023/a:1022458010266. [DOI] [PubMed] [Google Scholar]
- 29.Heumuller S. Wind S. Barbosa-Sicard E. Schmidt HH. Busse R. Schroder K. Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 30.Hidalgo C. Sanchez G. Barrientos G. Aracena-Parks P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S -glutathionylation. J Biol Chem. 2006;281:26473–26482. doi: 10.1074/jbc.M600451200. [DOI] [PubMed] [Google Scholar]
- 31.Hu X. Wang Q. He P. Fang Y. Spectroelectrochemistry study on the electrochemical reduction of ethidium bromide. Anal Sci. 2002;18:645–650. doi: 10.2116/analsci.18.645. [DOI] [PubMed] [Google Scholar]
- 32.Jackson MJ. Control of reactive oxygen species production in contracting skeletal muscle. Antioxid Redox Signal. 2011;15:2477–2486. doi: 10.1089/ars.2011.3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson-Cadwell LI. Jekabsons MB. Wang A. Polster BM. Nicholls DG. ‘Mild Uncoupling’ does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J Neurochem. 2007;101:1619–1631. doi: 10.1111/j.1471-4159.2007.04516.x. [DOI] [PubMed] [Google Scholar]
- 34.Kanter MM. Free radicals, exercise, and antioxidant supplementation. Int J Sport Nutr. 1994;4:205–220. doi: 10.1123/ijsn.4.3.205. [DOI] [PubMed] [Google Scholar]
- 35.Kuribayashi F. Nunoi H. Wakamatsu K. Tsunawaki S. Sato K. Ito T. Sumimoto H. The adaptor protein p40(phox) as a positive regulator of the superoxide-producing phagocyte oxidase. EMBO J. 2002;21:6312–6320. doi: 10.1093/emboj/cdf642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuroda J. Ago T. Matsushima S. Zhai P. Schneider MD. Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li N. Ragheb K. Lawler G. Sturgis J. Rajwa B. Melendez JA. Robinson JP. DPI induces mitochondrial superoxide-mediated apoptosis. Free Radic Biol Med. 2003;34:465–477. doi: 10.1016/s0891-5849(02)01325-4. [DOI] [PubMed] [Google Scholar]
- 38.Li Y. Trush MA. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun. 1998;253:295–299. doi: 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- 39.Longpre JM. Loo G. Paradoxical effect of diphenyleneiodonium in inducing DNA damage and apoptosis. Free Radic Res. 2008;42:533–543. doi: 10.1080/10715760802126692. [DOI] [PubMed] [Google Scholar]
- 40.Loschen G. Azzi A. Richter C. Flohe L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974;42:68–72. doi: 10.1016/0014-5793(74)80281-4. [DOI] [PubMed] [Google Scholar]
- 41.Maack C. Bohm M. Targeting mitochondrial oxidative stress in heart failure. J Am Coll Cardiol. 2011;58:83–86. doi: 10.1016/j.jacc.2011.01.032. [DOI] [PubMed] [Google Scholar]
- 42.Mannella CA. Conformational changes in the mitochondrial channel protein, VDAC, and their functional implications. J Struct Biol. 1998;121:207–218. doi: 10.1006/jsbi.1997.3954. [DOI] [PubMed] [Google Scholar]
- 43.McArdle A. van der Meulen JH. Catapano M. Symons MC. Faulkner JA. Jackson MJ. Free radical activity following contraction-induced injury to the extensor digitorum longus muscles of rats. Free Radic Biol Med. 1999;26:1085–1091. doi: 10.1016/s0891-5849(98)00317-7. [DOI] [PubMed] [Google Scholar]
- 44.Michaelson LP. Shi G. Ward CW. Rodney GG. Mitochondrial redox potential during contraction in single intact muscle fibers. Muscle Nerve. 2010;42:522–529. doi: 10.1002/mus.21724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mofarrahi M. Brandes RP. Gorlach A. Hanze J. Terada LS. Quinn MT. Mayaki D. Petrof B. Hussain SN. Regulation of proliferation of skeletal muscle precursor cells by NADPH oxidase. Antioxid Redox Signal. 2008;10:559–574. doi: 10.1089/ars.2007.1792. [DOI] [PubMed] [Google Scholar]
- 46.Muijsers RB. van Den Worm E. Folkerts G. Beukelman CJ. Koster AS. Postma DS. Nijkamp FP. Apocynin inhibits peroxynitrite formation by murine macrophages. Br J Pharmacol. 2000;130:932–936. doi: 10.1038/sj.bjp.0703401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muller FL. Liu Y. Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 48.Naqui A. Chance B. Cadenas E. Reactive oxygen intermediates in biochemistry. Annu Rev Biochem. 1986;55:137–166. doi: 10.1146/annurev.bi.55.070186.001033. [DOI] [PubMed] [Google Scholar]
- 49.Nethery D. Stofan D. Callahan L. DiMarco A. Supinski G. Formation of reactive oxygen species by the contracting diaphragm is PLA(2) dependent. J Appl Physiol. 1999;87:792–800. doi: 10.1152/jappl.1999.87.2.792. [DOI] [PubMed] [Google Scholar]
- 50.O'Donnell BV. Tew DG. Jones OT. England PJ. Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. Biochem J. 1993;290(Pt 1):41–49. doi: 10.1042/bj2900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palomero J. Pye D. Kabayo T. Spiller DG. Jackson MJ. In situ detection and measurement of intracellular reactive oxygen species in single isolated mature skeletal muscle fibers by real time fluorescence microscopy. Antioxid Redox Signal. 2008;10:1463–1474. doi: 10.1089/ars.2007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park SE. Song JD. Kim KM. Park YM. Kim ND. Yoo YH. Park YC. Diphenyleneiodonium induces ROS-independent p53 expression and apoptosis in human RPE cells. FEBS Lett. 2007;581:180–186. doi: 10.1016/j.febslet.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 53.Pattwell D. Ashton T. McArdle A. Griffiths RD. Jackson MJ. Ischemia and reperfusion of skeletal muscle lead to the appearance of a stable lipid free radical in the circulation. Am J Physiol Heart Circ Physiol. 2003;284:H2400–H2404. doi: 10.1152/ajpheart.00931.2002. [DOI] [PubMed] [Google Scholar]
- 54.Powers SK. Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev. 2008;88:1243–1276. doi: 10.1152/physrev.00031.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pye D. Palomero J. Kabayo T. Jackson MJ. Real-time measurement of nitric oxide in single mature mouse skeletal muscle fibres during contractions. J Physiol. 2007;581:309–318. doi: 10.1113/jphysiol.2006.125930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reid MB. Invited review: redox modulation of skeletal muscle contraction: what we know and what we don't. J Appl Physiol. 2001;90:724–731. doi: 10.1152/jappl.2001.90.2.724. [DOI] [PubMed] [Google Scholar]
- 57.Rey FE. Cifuentes ME. Kiarash A. Quinn MT. Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 58.Riganti C. Gazzano E. Polimeni M. Costamagna C. Bosia A. Ghigo D. Diphenyleneiodonium inhibits the cell redox metabolism and induces oxidative stress. J Biol Chem. 2004;279:47726–47731. doi: 10.1074/jbc.M406314200. [DOI] [PubMed] [Google Scholar]
- 59.Robinson KM. Janes MS. Pehar M. Monette JS. Ross MF. Hagen TM. Murphy MP. Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sakellariou GK. Pye D. Vasilaki A. Zibrik L. Palomero J. Kabayo T. McArdle F. Van Remmen H. Richardson A. Tidball JG. McArdle A. Jackson MJ. Role of superoxide-nitric oxide interactions in the accelerated age-related loss of muscle mass in mice lacking Cu,Zn superoxide dismutase. Aging Cell. 2011;10:749–760. doi: 10.1111/j.1474-9726.2011.00709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Silveira LR. Pereira-Da-Silva L. Juel C. Hellsten Y. Formation of hydrogen peroxide and nitric oxide in rat skeletal muscle cells during contractions. Free Radic Biol Med. 2003;35:455–464. doi: 10.1016/s0891-5849(03)00271-5. [DOI] [PubMed] [Google Scholar]
- 62.St-Pierre J. Buckingham JA. Roebuck SJ. Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 63.Stamler JS. Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81:209–237. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- 64.Tian W. Li XJ. Stull ND. Ming W. Suh CI. Bissonnette SA. Yaffe MB. Grinstein S. Atkinson SJ. Dinauer MC. Fc gamma R-stimulated activation of the NADPH oxidase: phosphoinositide-binding protein p40phox regulates NADPH oxidase activity after enzyme assembly on the phagosome. Blood. 2008;112:3867–3877. doi: 10.1182/blood-2007-11-126029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Urso ML. Clarkson PM. Oxidative stress, exercise, and antioxidant supplementation. Toxicology. 2003;189:41–54. doi: 10.1016/s0300-483x(03)00151-3. [DOI] [PubMed] [Google Scholar]
- 66.von Lohneysen K. Noack D. Jesaitis AJ. Dinauer MC. Knaus UG. Mutational analysis reveals distinct features of the Nox4-p22 phox complex. J Biol Chem. 2008;283:35273–35282. doi: 10.1074/jbc.M804200200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Whitehead NP. Yeung EW. Froehner SC. Allen DG. Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse. PLoS One. 2010;5:e15354. doi: 10.1371/journal.pone.0015354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Widder JD. Fraccarollo D. Galuppo P. Hansen JM. Jones DP. Ertl G. Bauersachs J. Attenuation of angiotensin II-induced vascular dysfunction and hypertension by overexpression of thioredoxin 2. Hypertension. 2009;54:338–344. doi: 10.1161/HYPERTENSIONAHA.108.127928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wind S. Beuerlein K. Eucker T. Muller H. Scheurer P. Armitage ME. Ho H. Schmidt HH. Wingler K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol. 2010;161:885–898. doi: 10.1111/j.1476-5381.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wosniak J., Jr. Santos CX. Kowaltowski AJ. Laurindo FR. Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid Redox Signal. 2009;11:1265–1278. doi: 10.1089/ars.2009.2392. [DOI] [PubMed] [Google Scholar]
- 71.Zielonka J. Kalyanaraman B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth. Free Radic Biol Med. 2010;48:983–1001. doi: 10.1016/j.freeradbiomed.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zuo L. Christofi FL. Wright VP. Bao S. Clanton TL. Lipoxygenase-dependent superoxide release in skeletal muscle. J Appl Physiol. 2004;97:661–668. doi: 10.1152/japplphysiol.00096.2004. [DOI] [PubMed] [Google Scholar]