

Abstract
The use of capillary electrophoresis as a tool to conduct immunoassays has been an area of increasing interest over the last decade. This approach combines the efficiency, small sample requirements, and relatively high speed of CE with the selectivity of antibodies as binding agents. This review examines the various assay formats and detection modes that have been reported for these assays, along with some representative applications. Most CE immunoassays in the past have employed homogeneous methods in which the sample and reagents are allowed to react in solution. These homogeneous methods have been conducted as both competitive binding immunoassays and as non-competitive binding immunoassays. Fluorescent labels are most commonly used for detection in these assays, but enzyme labels have also been utilized for such work. Some additional work has been performed in CE immunoassays with heterogeneous methods in which either antibodies or an analog of the analyte is immobilized to a solid support. These heterogeneous methods can be used for the selective isolation of analytes prior to their separation by CE or to remove a given species from a sample/reagent mixture prior to analysis by CE. These CE immunoassays can be used with a variety of detection modes, such as fluorescence, UV/visible absorbance, chemiluminescence, electrochemical measurements, mass spectrometry, and surface plasmon resonance.
Keywords: immunoassay, capillary electrophoresis, CE immunoassay, affinity capillary electrophoresis, review
1. INTRODUCTION
Over the last decade there has been increasing interest in using capillary electrophoresis (CE) as a tool for performing immunoassays. An immunoassay can be defined as a technique in which antibodies or antibody-related substances are used as selective binding agents for chemical detection [1-4]. The combined used of CE with these binding agents will be referred to here as a “CE immunoassay”. This group of methods can be classified as a subset of a broader range of techniques known as affinity capillary electrophoresis (ACE), in which biological ligands are used in CE for the selective binding of target analytes. Several recent reviews have discussed the theory, methodology and use of ACE [5-10]. This current review will focus on the use of CE-based immunoassays and the various approaches that have been created for conducting such assays.
The key reagent in any type of immunoassay is the antibody or antibody-related agent that is used to specifically bind the desired target [2]. Antibodies are glycoproteins produced by the body’s immune system in response to a foreign agent, or antigen. Even though antibodies interact with antigens through non-covalent interactions, the large variety of such interactions that occur for even a single antigen makes antibodies quite specific for their targets and often gives them association equilibrium constants in the range of 105 – 1012 M−1. As a result of their high specificity and strong binding, antibodies are often used as analytical reagents for the measurement and detection of analytes in complex mixtures such as blood, plasma, urine and food [1-4]. This same feature makes antibodies attractive for use in CE.
By definition, a CE immunoassay makes use of an antibody or antibody-related substance as a binding agent. However, many of the same assay formats that have been described in CE for work with antibodies can be modified to use other selective binding agents. When agents other than antibodies are employed for this purpose, the general method that is produced is referred to as a “CE binding assay”. Aptamers are one class of agents that have been utilized in place of antibodies in CE binding assays. Aptamers consist of single-stranded DNA or RNA sequences that can bind to a target molecule with high affinity. Aptamers can be developed by using a process known as the systematic evolution of ligands by exponential enrichment (SELEX) [11-13]. Advantages of using aptamers in binding assays include the ability to produce aptamers in an inexpensive and automated matter (once their sequence has been determined), their good stability, their small size (i.e., with molecular weights of a few thousand Daltons), and the ease with which they can be modified with tags such as fluorescent labels [14]. Along with aptamers, the interactions of enzymes with specific substrates has also been explored for use in CE binding assays [15].
The concept of using antibodies in capillary electrophoresis to produce a “CE immunoassay” was first introduced by Nielsen et al. in 1991, when the separation of antibody-antigen complexes by CE was demonstrated [16]. Two years later, Schultz and Kennedy reported immunoassays based on both competitive and non-competitive formats for the measurement of insulin using fluorescein isothiocyanate (FITC) as a label [17]. The detection limit in this second example was 2 nM, and the mass detection limit was 2.8 × 10−19 mol. This work indicated that several immunoassay formats could be successfully employed in CE for the study of analytes of biological interest.
Compared to conventional immunoassays, CE immunoassays offer several possible advantages. Two important advantages of CE immunoassays are their ease of automation and their relatively fast separation of antibodies, analytes and/or antibody-analyte complexes [18]. In addition, CE immunoassays tend to consume only small amounts of sample and reagents while still allowing the detection of trace amounts of analyte in a sample [19]. This last feature has made CE immunoassays appealing for work in detecting analytes from single cells and in the creation of microanalytical systems [20,21]. The detection of multiple analytes in a single run is also feasible in a CE immunoassay, due to the high resolving power of modern capillary electrophoresis systems [20,21]. Unfortunately, CE immunoassays tend to give poorer concentration-based limits of detection than solid-phase immunoassay techniques such as an enzyme-linked immunosorbent assay (ELISA) [22]. This is a result of the analytes and antibodies both being present in solution in many CE immunoassay formats, while in an ELISA a surface reaction can be used to pre-concentrate analytes for measurement and detection. In addition, CE immunoassays require more complicated instrumentation than an ELISA or most other traditional immunoassays and generally have a lower sample throughput [22].
CE immunoassays can be either homogenous or heterogeneous in nature. In a homogeneous immunoassay, both the analyte and antibody are present in a liquid phase. As complexes form between the analyte and antibody, differences in electrophoretic mobility can be used for the separation of these species by CE prior to their detection. In a heterogeneous immunoassay, an immobilized antibody or an immobilized analog of the analyte is used as part of the assay system. This review will discuss these techniques and will give examples of their applications. Practical factors to consider in the development and use of CE immunoassays will also be presented. In addition, this review will examine the various approaches that have been reported for detection in CE immunoassays. Although the emphasis in this review will be placed on immunoassays that are performed using a traditional CE system, many of the principles and formats that will be discussed can also be adapted for use with a microanalytical CE system.
2. DETECTION & LABELS IN CE IMMUNOASSAYS
Many CE immunoassays make use of chemical labels to enhance the detection of analytes. Although a variety of labels are used in traditional immunoassays [1-4], in CE immunoassays these labels are generally based on fluorescent tags or enzymes. Labels and tags can be attached to antibodies or analyte analogs through numerous methods (see reviews in Refs. [4,23-25]). Detection schemes that have been used with these labels have included laser-induced fluorescence, ultraviolet/visible absorbance detection, electrochemical detection, mass spectrometry, and chemiluminescence detection.
2.1. Fluorescence Detection and Labels
One of the most common means for detection in CE immunoassays is laser-induced fluorescence (LIF). This detection mode can provide extremely low limits of detection, with measurements in the yoctomole (10−24 mol) range being possible in some cases [26]. This mode of detection can be used with fluorescent tags that are used either to label an analyte for direct detection or as a basis for indirect detection by placing these labels on an antibody or analyte analog. Disadvantages of LIF detection can include a high background noise due to Rayleigh scattering, Raman scattering, or fluorescent impurities in solvents. Examples of CE immunoassays which have utilized LIF detection can be found in a large number of references, including most of those shown in Table 1 [14,15,18,19,21,27-75].
Table 1.
Examples of Homogeneous Competitive Binding Immunoassays using CE
| Analyte | Detection Limit | Other Assay Parameters |
Label | Reference |
|---|---|---|---|---|
| Glucagon | 4 nM (640 zmol) |
Injection volume, 0.16 nL |
Fluorescein | [18] |
| Insulin | 5.5 nM | ------- | Fluorescein | [18] |
| Insulin | 3 nM (6 fg) |
Secretion monitoring from single islets of Langerhans |
Fluorescein | [20,57,70] |
| Methadone | 51 nM (16 ng/mL) |
Multianalyte assay | Fluorescein | [21] |
| Opiatesa | 49 nM –morphine (14 ng/mL) |
Fluorescein | [21] | |
| Benzoylecgonine | 69 nM (27 ng/mL) |
Fluorescein | [21] | |
| Amphetaminesa | 200 nM – amphetamine (27 ng/mL) |
Fluorescein | [21] | |
| Theophylline | 1.4 M (0.26 mg/L) |
Range, 0 – 40 mg/L | Fluorescein | [68] |
| Amphetamine & amphetamine analogsa |
600 nM- amphetamine (80 ng/mL) |
Screening assay | Fluorescein | [76] |
| Clenbuterol | 2.5 nM (0.7 ng/mL) |
Linear range, 1 – 200 ng/mL |
Fluorescein | [30] |
| Prion proteinb | 80 ng/mL | --------- | Fluorescein | [79] |
| Vasopressin | nM range | --------- | Fluorescein | [32] |
| Hirudin | 14.2 nM | Linear range, 10 – 80 nM |
Fluorescein | [36] |
| Methamphetamine | 100 nM (20 ng/mL) |
Linear range, 50 – 1000 ng/mL |
Fluorescein | [43,45] |
| Methionine- enkeppalinb |
Not reported | Covers clinically- relevant range |
Fluorescein | [31] |
| Neuropeptide Y | 40 pM | Range, 40 pM – 1 nM | Fluorescein | [29] |
| Glycoalkaloidsb | Not reported | --------- | Fluorescein | [39,40] |
| Staphylococcal enterotoxin A |
3 aM | Range, 0.3 – 6.5 nM | Fluorescein | [56] |
| Cortisol | 30 nM (1 g/dL) |
Clinically relevant ranges for serum, 1 – 60 g/dL |
Fluorescein | [28] |
| Morphine | 140 nM (40 ng/mL) |
Linear range, 50 – 1000 ng/mL |
Fluorescein | [50] |
| Scrapie prion proteinb |
2 fmol | --------- | No label | [53] |
| Estriol | 109 pM (31.2 pg/mL) |
Linear range, 0.050 – 5 ng/mL |
Fluorescein | [55] |
| Methotrexateb | 5 pg | Range, 5 – 250 pg | Fluorescein | [58] |
| Prion proteinb | 15 amol | Range, 15 – 150 amol | Fluorescein | [59] |
| Cyclosporine A | 0.9 nM (0.9 amol) |
--------- | Fluorescein | [44] |
| 2,4-Dichloro- phenoxyacetic acid |
45 nM (1 ng) |
Linear range, 5 ppb – 1000 ppb |
Fluorescein | [62] |
| Glucagon | 20 pM | Linear range, 50 – 1300 pM |
Fluorescein | [67] |
| Human serum albumin |
300 nM (0.02 mg/mL) |
Range, 0.02 – 1.0 mg/mL |
Cy5 | [63] |
| Secretory immuno- globulin Aa |
2.6 nM – IgA dimer (1 μg/mL) |
Range, 1 – 8 μg/mL | Cy5 | [33] |
| Benzo[a]pyrene diol epoxide-DNA adductb |
0.1 μg/mL | Range, 0.1 - 10 μg/mL | Tetramethyl rhodamine |
[35] |
| Digoxigenin | 1.1 nM (0.42 mg/mL) |
Range, 0.42 – 10.42 ng/mL |
B-Phycoerythrin | [49] |
| Medroxyprogestero ne |
0.9 nM | Linear range, 2.0–50 nmol/L |
HRP | [89] |
| Thyroxine | 3.8 nM (23.2 amol) |
Linear range, 3.0 – 50 μg/L |
HRP | [94] |
| Human serum albumin |
0.1 μM | Range, 0.2 – 1.2 μM | ILITC | [98] |
| Vancomycin | 680 pM (0.98 ng/mL, 1.1 fg) |
Range, 2 – 3 orders of magnitude |
No label | [72] |
| Estradiol | 310 pM (2100 molecules) |
-------- | Fluorescein | [73] |
| IgE | 46 pM | Linear range, 5 orders of magnitude |
FITC | [14,75] |
| Thrombin | 40 nM | --------- | Fluorescein | [14,75] |
| HIV-1b | --------- | Linear range, up to 50 nM |
Fluorescein | [74] |
| Dorzolamide | 16.5 pM | ---------- | Fluorescein | [15] |
When for than one analyte was being examined by a given method, the concentration limit of detection that is listed is for a representative member of the analyte class.
Insufficient information was given in the cited reference to determine the concentration limit of detection for this analyte.
Labels that can be employed with fluorescence detection are by far the most common types of tags that are utilized in CE immunoassays. There are several characteristics that are desired for such a label to provide adequate limits of detection in a CE immunoassay, or in any other type of immunoassay [2,4]. First, this label should have a high quantum yield (ideally, a maximum of ΦF = 1.0). Second, the conjugation of this label to its target should require mild conditions and result in a stable product. Third, this conjugation process should not disturb the ability of the labeled target to bind to antibodies or related agents that will be employed in the CE immunoassay.
Several fluorescent labels meet these qualifications and have been used in CE immunoassays. Fluorescein and its derivatives are the most common fluorescent labels that have been used in CE immunoassays [14,15,17,18,20,21,27-32,34,36,39,40,43-45,50,51,53-62,67-71,73-80]. However, other fluorescent dyes have also been used in such methods. Examples of these other dyes include Cyanine 5 [33,47,63,81], tetramethylrhodamine [35,64,65], quantum dots [66], NN382 (i.e., a near infrared fluorescent dye) [38], green fluorescent protein [46], and B-phycoerythrin [49].
Fluorescein is usually used in the form of fluorescein isothiocyanate (FITC) to label chemicals for CE immunoassays. Part of the reason for the popularity for fluorescein as a label in CE is that its excitation wavelength range of 488–495 nm closely matches the emission wavelength of an argon laser (488 nm) [23]; this makes it possible to use this label with a system that can carry out laser-induced fluorescence detection. The quantum yield for fluorescein can be as high as 0.75 under ideal conditions, but its intensity fades when this compound is dissolved in a buffer, is exposed to light, or is stored for extended periods of time. Furthermore, the pH of the surrounding solution can greatly influence the fluorescence intensity of this label. At a pH below 7, the fluorescence of this agent can be quenched by 50%. When FITC is used as a labeling agent, this tag will have maximum excitation at 494 nm and maximum emission at 520 nm [23]. Since isothiocyanates are capable of forming stable products when reacted with primary amines, FITC is a valuable reagent for modifying ε-and N-terminal amines in proteins and peptides (see Figure 1).
Figure 1.

Reaction of fluorescein isothiocyanate (FITC) with a compound that contains a primary amine group. In this reaction, the nucleophilic amine attacks the central carbon on the isothiocyanate group, resulting in a thiourea linkage between FITC and chemical that originally contained the primary amine (e.g., a protein or peptide) [23].
An example of a CE immunoassay which has utilized LIF detection and a FITC label is a method that has been reported for the detection of clenbuterol [30]. In this assay a labeled analog of clenbuterol was prepared by tagging this compound with FITC. A fixed amount of this labeled analog was then mixed with clenbuterol from samples and allowed to compete for a limited number of antibodies during a 5 min incubation step. This mixture was then separated by CE and detected by LIF as part of a homogeneous competitive binding immunoassay. The CE separation step required 8 min, with a 2 min rinsing step being used between injections. A typical electropherogram that was obtained for this assay is given in Figure 2. This assay had a linear range that extended up to 200 ng/mL. The lower limit of detection was 0.7 ng/mL.
Figure 2.

Electropherograms for a competitive binding immunoassay performed by CE in which clenbuterol tagged with FITC was used as the labeled analog. In (A) only labeled clenbuterol was injected onto the CE system, as represented by peak 1. In (B) a mixture of labeled clenbuterol, anti-clenbuterol antibodies, and a sample containing clenbuterol was injected, where the immune complex between the labeled clenbuteron and the antibodies is represented by peak 2. This figure is reproduced with permission from Ref. [30].
The near infrared fluorescent dyes Cyanine 5 (Cy5) and NN382 have been employed as labels in CE immunoassays for analytes such as angiotensin II [47,82]. These dyes are desirable as labels because relatively little background absorption or emission exists in many samples over the wavelength ranges that are associated with the fluorescence of these agents. Cyanine dyes such as NN382 and Cy5 (see Figure 3) have excitation bands in the range of 600–900 nm, with longer excitation wavelengths being made possible by adding a vinyl (–CH=CH–) group to their polymethine chain. Some limitations of dyes like Cy5 are the lack of a large variety of derivatives for their use in labeling analytes and their low fluorescence quantum yields. These dyes also tend to aggregate in an aqueous solution [82], which can result in a decrease in fluorescence intensity.
Figure 3.
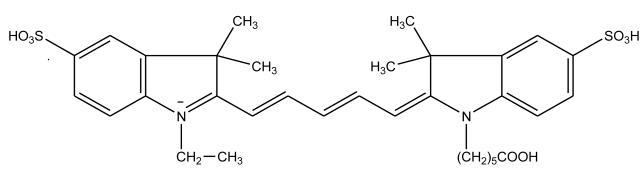
Structure of Cyanine 5 (Cy5), an example of a near infrared fluorescent dye [82].
B-Phycoerythin is a third type of fluorescent label that has been used in CE immunoassays. This tag is a fluorescent protein that has an absorbance maximum at 545 nm, a molar absorptivity of 2.4 × 106 M−1 cm−1, and a quantum yield of 0.98 [83]. The fluorescence of B-phycoerythrin has maximum emission at 575 nm [84]. This protein has been used as a label in a CE immunoassay for digoxigenin, allowing a detection limit of 2.5 ng/mL to be obtained for this analyte [49]. One useful feature that has been reported for B-phycoerythin is it has an isoelectric point of 5.5, which makes it possible to separate this label from antibodies by CE when using a borate buffer.
Quantum dots have been used as fluorescent labels in a CE immunoassay for human immunoglobulin M [66]. Quantum dots consist of nanometer-sized particles that have a semiconductor core [85]. When quantum dots are utilized in a CE immunoassay, the quantum dots act as both fluorescent tags and electrophoretic mobility modifiers, which can aid in the separation of sample and reagent components such as antibodies and antibody-analyte complexes (note: in general, any labeling agent can also act as a mobility modifier if it results in a change in the charge and/or size of the labeled species). This ability to modify electrophoretic mobility is, in part, due to the large size of the quantum dots (i.e., 10–20 nm). This property can be particularly important in work that involves large analytes because it may help in the separation process. Another advantage of quantum dots is the ability to tune their optical properties (e.g., the excitation and emission wavelengths) by changing their size [86]. This feature might be valuable in the future for the measurement of multiple analytes in a single run by using several types of antibodies that have each been tagged with a different type of quantum dot.
Tetramethylrhodamine is another dye which has been used as a fluorescent label in CE immunoassays. This dye can be excited by using two different laser sources: excitation at 488 nm using an argon ion laser or at 546 nm using a He-Ne laser [65]. However, this dye does have a small Stokes shift with an absorbance maximum at 540 nm and an emission maximum at 567 nm that must be considered [87]. This dye has been utilized as a label in CE immunoassays for detecting DNA damage [35], measuring staphylococcal enterotoxin B [64], and detecting recombinant growth hormone [65].
2.2. Enzyme Labels
Enzymes are a second group of labels that are often employed in CE immunoassays. These labels are detected by adding substrates that are converted into products that can easily be monitored. A large advantage to using enzymes as immunoassay labels is their ability to act as catalysts. This means that many product molecules can be generated by a single copy of an enzyme. This catalytic ability gives an amplified signal that is determined by the amount of available substrate, the reaction time, and the number of enzyme labels that are present. With this approach, the detection limit of a CE immunoassay can be lowered by increasing the amount of substrate or increasing the reaction time; however, altering these factors can also lead to an increased background signal or analysis time. Many techniques have been used in CE immunoassays to detect and measure enzyme labels, including electrochemical, fluorescence, absorbance, and chemiluminescence detection.
When choosing an enzyme label for possible use in a CE immunoassay, one must consider several items [1-4]. First, the desired limit of detection needs to be determined, with enzymes that have high turnover numbers giving the best results. In addition, it is necessary to have a substrate for the enzyme that is relatively stable, making it possible to maximize the sensitivity of the method and amount of product that is generated. The enzyme chosen for use as a label should also be easy to detect, stable under the assay conditions, and not present in the original sample. This enzyme label should not contain any remaining reactive groups after it has been conjugated to the analyte or antibodies, and it should be possible to store this label for long periods of time. Ideally, the enzyme label should also be available at a low cost in a pure and homogenous form.
The enzyme horseradish peroxidase (HRP) meets most of these criteria and has been the most common enzyme label employed in CE immunoassays. HRP is a holoenzyme with a molecular weight of 40 kDa. It catalyzes the oxidation of a variety of compounds by hydrogen peroxide or related agents. HRP has been conjugated to both large proteins (i.e., antibodies) and small molecules (i.e., antigens) for use in CE immunoassays [3]. It is most commonly used along with either chemiluminescent detection [88-92] or amperometric detection [93,94]. Examples of analytes that have been detected by CE immunoassays using enzyme labels are hepatitis B surface antigen [88], medroxyprogesterone [89], tumor marker CA125 [90], clenbuterol [91], bone morphogenic protein-2 [92], tumor marker CA15-3 [93], and thyroxine [94].
Another enzyme-related system that has been used in CE immunoassays is microperoxidase. Microperoxidase exists in two forms: microperoxidase-11 (MP11) and microperoxidase-8 (MP8). Both of these forms are obtained by the hydrolytic digestion of cytochrome c and exhibit peroxidase activity [95]. MP11 contains the heme prosthetic group of cytochrome c and amino acids 11 to 21, while MP8 contains residues 14 to 21 [96,97]. Both of the cytochrome c fragments can catalyze a variety of reactions involving hydrogen peroxide oxidation. Microperoxidase has been used for detection in a CE immunoassay for human serum albumin (HSA) [98]. In this assay, the microperoxidase was added to the migration buffer and isoluminol isothiocyanate (i.e., the compound which is oxidized) was the label.
2.3. Chemiluminescence
Chemiluminescence can be defined as the production of light as a result of a chemical reaction. This is yet another approach that has been used for detection in CE immunoassays [88-91,98,99]. In this method, an antibody or analog of the analyte is tagged with a label that is capable of producing light through chemiluminescence. This light-producing reaction is initiated as the labeled compound exits the CE system, giving rise to the release of light for detection of the labeled compound and, thus, the analyte.
The use of a chemiluminescent label in CE immunoassays has several advantages. First, many chemiluminescent reactions are quite rapid, which allows for fast detection. These reactions can also provide good limits of detection and have low background signals, with detection in the zeptomole (10−21 mol) range sometimes being possible [92]. The main limitation of this approach is it does require the use of a post-capillary reactor to initiate the chemiluminescence reaction. Furthermore, it is necessary in this approach to carefully optimize the concentrations of the reagents needed for the chemiluminescence reaction so that they provide the best possible detection and reproducibility for the system.
Luminol (i.e., 5-amino-2,3-dihydro-1,4-phthalazinedione) can be used as a chemiluminescent agent in reactions that are catalyzed by HRP. The reaction that is involved in this process is illustrated in Figure 4. In this process, an excited state molecule of 3-aminophthalate is formed through the reaction of luminol with hydrogen peroxide under basic conditions. This excited molecule releases visible light as it falls to ground state. An enhancer such as p-iodophenol can be used to improve the sensitivity of this chemiluminescent reaction. By adding an enhancer to the reaction mixture, greater than a 1000-fold increase in sensitivity can be achieved [100].
Figure 4.
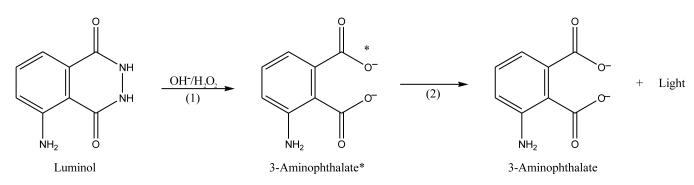
Production of light by luminol through chemiluminescence. The asterisk represents a molecule in the excited state.
An example of a CE immunoassay in which HRP was used as an enzyme label with chemiluminescent detection is a method which was reported for determining the amount of the synthetic hormone medroprogesterone acetate (MPA) in pork tissue [89]. In this assay, MPA was labeled with HRP to create a labeled analog. This labeled analog was then used in a homogeneous competitive binding immunoassay format. Separation of the immunocomplex and labeled analog in the sample/reagent mixture was performed in 8 min by CE. The linear range of this method was 2.0-50 nM and the detection limit was 0.9 nM for MPA. An assay for protein isoforms in ERK protein also utilized HRP and chemiluminescence for detection [101].
Several other chemiluminescent labels have been used for detection in CE immunoassays. For example, in an assay for HSA, a labeled analog was prepared by tagging HSA with isoluminol isothiocyanate (ILITC) [98]. This reaction again involved a process that was catalyzed by microperoxidase and that made use of luminol-based chemiluminescence. A mixture of labeled HSA, a sample containing HSA, and anti-HSA antibodies was prepared and incubated for 60 min prior to injection. The separation of these components by CE took another 24 min to conduct. Microperoxidase was included in the running buffer and a solution of 400 mM hydrogen peroxide was added in the CE detection cell to provide detection of the labeled HSA as it migrated into this cell. The detection limit of this assay was 10 nM for HSA and the linear range extended from 0.02 – 5 M.
2.4. Electrochemical Detection
Electrochemical detection has several advantages over other techniques for monitoring CE immunoassays. These advantages include the ease with which electrochemical detection can be combined with CE as well as the low cost, selectivity, and sensitivity of this detection method. Electrochemical detection involves the use of a label or compound that can undergo either oxidation or reduction. This detection is generally performed by measuring the change in current that occurs when this agent is oxidized or reduced at a constant applied potential. Selectivity is achieved by selecting the potential at which this oxidation or reduction occurs. Additional selectivity can be obtained by using a chemically modified electrode [102] or multiple electrodes for detection [103].
Electrochemical detection can be used in combination with an enzyme label that catalyzes the formation or consumption of an electrochemically-active product. For instance, electrochemical detection and enzyme labels were used in a CE immunoassay that was developed to determine the concentration of thyroxine in human serum [94]. In this assay, HRP was utilized to prepare a labeled analog of thyroxine for use in a competitive binding immunoassay format. This HRP label was used to catalyze the oxidation of 3,3,5,5-tetramethylbenzidine in a reaction capillary that was part of the CE system. The product was reduced at a carbon fiber microdisk bundle electrode (see reactions in Figure 5). Using this method, a detection limit of 23.2 attomol (or 3.8 nM) was reported for thyroxine. Other applications of electrochemical detection in CE immunoassays have been described for the tumor markers CA125 [104] and CA15-3 [93,105].
Figure 5.

Scheme for electrochemical detection based on (a) oxidation of 3,3,5,5-tetramethylbenzidine (TMB) in the presence of horseradish peroxidase (HRP), followed by (b) the reduction of this oxidized form back to the initial form of TMB at an electrode. This scheme makes it possible to detect the HRP label through its production of the oxidized form of TMB. This figure is adapted from Ref. [94].
2.5. Ultraviolet/Visible Absorbance
Absorbance detectors are common in CE and provide a non-destructive means for detecting many types of analytes. The one limitation of this approach is that it has relatively poor detection limits compared to LIF, which limits the use of absorbance detection to analytes or labeled analogs that have moderate or relatively high concentrations in an injected sample or sample/reagent mixture. This detection mode also requires that the analyte or label have a chromophore with a reasonably high molar absorptivity at the wavelength to be monitored during the assay.
Absorbance detection was employed in a CE assay that used anti-bovine serum albumin (BSA) antibodies as model analytes [106]. This assay was performed without the use of labels. Excess BSA was mixed with a sample that contained anti-BSA antibodies and this mixture was incubated for 10 min. The free BSA and BSA-antibody complexes were then separated in 35 min by using sodium dodecyl sulfate and capillary gel electrophoresis (CGE). The limit of detection reported for this method was in the low M range for the anti-BSA antibodies. Other CE immunoassays using absorbance detection have been developed to monitor human recombinant growth hormone [16] and HSA [99].
2.6. Mass Spectrometry
Mass spectrometry is another method which can be used for detection in CE immunoassays. Some care must be taken when utilizing mass spectrometry for this purpose because the buffer concentrations that are employed for the CE immunoassay need to be minimized to prevent salt build-up in the mass spectrometer (e.g., on an electrospray nozzle). In addition, the limited amount of sample which can be transferred to the mass spectrometer can lead to poor detection limits. Nevertheless, the combination of CE immunoassays with mass spectrometric detection can be a powerful technique for analyte determination.
An example of this technique is a method which was created to detect urinary codeinoids such as codeine, dihydrocodeine, and dihydrocodeine-6-glucuronide, morphine, morphine-3-glucuronide, and ethylmorphine (see Figure 6) [107]. Prior to this work, a CE immunoassay was unable to identify these compounds in urine since other compounds in the sample had similar migration times. By coupling the CE immunoassay with mass spectrometry, the peak for each analyte could be identified by using the mass-to-charge (m/z) ratios of its corresponding ions. The resulting CE immunoassay could identify the urinary codeinoids and detect them at concentrations in the range of 100 – 200 ng/mL. These concentrations were found to be adequate for use in toxicological work. Other reports that have combined CE immunoassays with mass spectrometry have been described for the analysis of methadone, amphetamines, morphine, and benzoylecgonine in urine [108] and the measurement of morphine and amphetamine [109].
Figure 6.

Electropherograms (left), mass spectra for parent ions (middle), and tandem mass spectral data for daughter ions obtained for dihydrocodeine (DHC) and codeine (COD) in a CE immunoassay. The sample in this example consisted of urine that was spiked with 10 g/mL of both COD and DHC. This figure is reproduced with permission from Ref. [107].
2.7. Surface Plasmon Resonance
Surface plasmon resonance (SPR) can also be used for detection in CE immunoassays. In SPR, large molecules that adsorb onto a surface can be detected by examining the change in the local index of refraction for the surface (which contains a layer of metal to provide a measurable response). One example of using SPR as a detection method was reported by Whelan and Zare [110]. In this work, an SPR sensor was used to monitor changes in refractive index as a function of time to create an electropherogram. Immunoglobulin G (IgG) was detected with this approach, giving a response with a dynamic range that covered 3 orders of magnitude in concentration and resulted in a detection limit of 2 fmol. SPR has also been proposed as a universal detection method for CE immunoassays by Waseda et al. [111].
3. HOMOGENEOUS IMMUNOASSAYS
There are many formats that have been considered for combining the use of CE with antibodies or related binding agents. One format that is often used for this purpose is a homogeneous immunoassay. The term “homogeneous immunoassay” refers to an immunoassay in which the analyte, antibodies, and all other reacting species are present in the solution phase [112]. Two types of homogeneous immunoassays have been used in CE. These two types are 1) a competitive binding immunoassay and 2) a non-competitive immunoassay.
3.1. Competitive Binding Immunoassays
A competitive binding immunoassay is one way in which a homogeneous assay format can be carried out by CE. This type of immunoassay is used to detect and measure an analyte by examining its competition with a fixed amount of a labeled analog for binding sites on antibodies (see Figure 7). A sample containing the analyte is first mixed with a known and fixed amount of labeled analog and combined with a limiting amount of antibodies that can bind to both of these agents. The presence of only a small amount of antibodies creates a situation in which the analyte and labeled analog must compete for binding sites. As a result, the presence of an analyte will affect the amount of the labeled analog that can bind to the antibodies. This means that the amount of bound or non-bound labeled analog that is later separated and detected by CE will provide an indirect measure of the amount of analyte that was present in the original sample.
Figure 7.

Scheme for a homogeneous competitive binding immunoassay in CE. The sample, labeled analog of the analyte and a limited amount of antibodies are first incubated and allowed to react. The components of this mixture are then separated and detected by CE. The amount of analyte in the sample is determined by measuring the relative amount of the labeled analog that is bound to the antibodies (or that remains free in solution) and by comparing this relative amount to that which is obtained when performing the same assay with standards that contain known amounts of the analyte.
Figure 8 shows an example of a calibration curve that was obtained in CE when using a homogeneous competitive binding immunoassay for the detection of neuropeptide Y and other agents in cell samples [30]. In this type of calibration curve, the concentration of analyte in the sample will be directly proportional to the measured change in the concentration of the free, non-bound form of the labeled analog but inversely proportional to the amount of labeled analog that is bound to the antibodies. Some competitive binding immunoassays performed by CE have been used to detect both the free labeled analog and the bound labeled analog, with a ratio of these terms then be used to construct a calibration curve for the analyte [18,29,50]. However, many competitive binding immunoassays in CE detect only the free labeled analog because it will generally appear as a sharp, well-defined peak.
Figure 8.

Calibration curve for a homogeneous competitive immunoassay performed by using CE for the determination of neuropeptide Y (NPY). In this assay, the concentration of neuropeptide Y in a standard or sample is plotted on the x-axis while the y-axis shows the ratio of the bound (B) versus free (F) fractions of a labeled analog of neuropeptide Y. This figure is reproduced with permission from Ref. [30].
The detection limits of competitive CE immunoassays depend upon three main factors: the concentration of antibody binding sites, the concentration of the labeled analyte analog, and the affinity of the antibody for the analyte. Information on the optimization of these parameters can be found in Ref. [73]. In this study, it was found that an antibody-to-labeled analog ratio of 1:2 was optimum for maximizing the dynamic range and minimizing the detection limit. It was also determined that work at low antibody concentrations requires the use of antibodies that have a high affinity for the analyte (e.g., antibodies with an association equilibrium constant of 1012 M−1 were found to be more effective than those with a value of 108 M−1).
The competitive binding immunoassay is currently the most popular format for a CE-based immunoassay. A recent example of such an assay was developed for detecting the antibiotic vancomycin in serum, giving a limit of detection of 0.98 ng/mL or 1.1 fg vancomycin per injection [72]. In this assay, the labeled analog was vancomycin that had been tagged with a fluorescent tracer. Another application used CE and a competitive binding immunoassay to measure medroxyprogesterone acetate in animal tissues, giving a limit of detection of 0.9 nmol/L and a linear range of 2.0–50 nM [89]. Other applications of competitive binding CE immunoassays can be found in Table 1 and in the literature [18,20,21,28-33,35,36,39,40,43-45,49,50,53,55-59,62,63,67-70,73,76,79,94,98].
In preparing the labeled analog for a competitive binding immunoassay, care must be taken to ensure that the labeled analog retains the ability to bind the antibodies that will be used to recognize and bind the desired analyte. It is also generally desired for the antibodies to have similar binding constants for the analyte and labeled analogs to provide good competition between these compounds and effective analyte quantification. In addition, this label should give a signal that is not produced by any of the original components of the sample, and the sample matrix should not have any adverse effects on the detection of this label.
Competitive binding assays in CE have several advantages and disadvantages versus non-competitive immunoassays (see Section 3.2). For instance, the separation of the bound and non-bound labeled species is often easier in a competitive binding assay, especially when working with an analyte such as a drug that is small compared with antibodies (i.e., a system in which a separation may be difficult to perform by the non-competitive immunoassay format). In addition, only an analog of the analyte needs to be labeled in a competitive binding format, which avoids the difficult task of producing a single homogeneous preparation of labeled antibodies or antibody fragments that is often encountered in the non-competitive format. However, the non-competitive format also has some important advantages versus the competitive binding immunoassay in CE. As an example, a non-competitive CE immunoassay tends to have a lower limit of detection and a larger dynamic range; this format is also usually better able to distinguish between cross-reacting species than when using a competitive binding immunoassay [65].
3.2. Non-competitive Immunoassays
Non-competitive immunoassays performed by CE are part of a broader set of techniques known as affinity probe capillary electrophoresis (APCE), in which a labeled binding agent is used for analyte measurement [5]. One characteristic of these assays is they do not involve any competition between the analyte and other agents for binding sites on antibodies. In most non-competitive immunoassays that are performed using CE, the sample is first incubated with a known excess of labeled antibodies or Fab fragments and the analyte is allowed to bind to these antibodies or antibody fragments (see Figure 9). This mixture now contains labeled immune complexes of the analyte and non-complexed labeled antibodies/antibody fragments. Next, this mixture is injected onto a CE system for separation of the free antibodies/antibody fragments from their immune complexes with the analyte. A change in the signal for either the labeled immune complex or the remaining excess of labeled antibodies/antibody fragments is then measured and used for analyte quantification. Figure 10 shows a typical calibration curve for this method [27]. A non-competitive immunoassay can also be modified and used with a labeled antigen for the detection of antibodies or antibody fragments [17].
Figure 9.

Scheme for a homogeneous non-competitive immunoassay in CE. In this method the sample is mixed and incubated with labeled antibodies prior to separation by CE. The immunocomplex (i.e., the labeled antibodies that are bound to the analyte) is then separated from the non-bound, labeled antibody on the basis of differences in electrophoretic mobility. The relative size of the immunocomplex peak can then be used to determine how much analyte was present in the original sample. The same approach can be used with a labeled analog of an analyte to determine the amount of antibodies or Fab fragments for this analyte that are present in a sample.
Figure 10.

Calibration curve for a homogeneous non-competitive immunoassay performed by CE for the determination of digoxin. This assay made use of labeled antibody fragments as the binding agents. This figure is reproduced with permission from Ref. [27].
In a non-competitive CE immunoassay, the separation of the labeled immune complexes and labeled antibodies/antibody fragments is performed based on the difference in their electrophoretic mobilities. Depending on the size and charge of the analyte in the immune complex, this change in mobility may be small and not provide for an adequate separation in some cases. In these situations an alternative format such as a competitive CE immunoassay can instead be employed (see Section 3.1). The use of Fab fragments instead of intact antibodies is useful in a non-competitive immunoassay because these fragments possess only single binding regions for analytes; as a result, Fab fragments result in a less complex mixture of immune complexes than is obtained when using intact antibodies.
One recent example of a non-competitive immunoassay that made use of CE was a method reported for measuring the tumor marker CA125 [90]. In this assay, antibodies capable of binding CA125 were labeled with the enzyme HRP and detected using chemiluminescence. The detection limit for CA125 in this assay was 1.0 × 10−12 M, with a linear range that extended from 2.5 × 10−11 to 1.0 × 10−9 M. After a 2 h incubation of the labeled antibodies with patient samples, the immune complexes which formed between CA125 and the antibodies were separated from the non-bound antibodies and detected within 4 min. Other analytes that have been measured by CE and a non-competitive immunoassay format include bovine recombinant prion protein [78], digoxin [27], recombinant human growth hormone [65], bovine serum albumin [106], tumor marker CA15-3 [93], and anti-insulin antibodies [38]. Other non-competitive assays have utilized aptamers as binding agents for the detection of IgE [14,75], thrombin [14,75], and type 1 human immunodeficiency virus (HIV-1) [74]. A non-competitive format has also been used with the enzyme carbonic anhydrase for the measurement of dorzolamide [15].
The limit of detection for a non-competitive CE immunoassay will be determined by 1) non-specific binding of the labeled antibodies or antibody fragments, 2) the detector response for the label that is attached to these reagents [113], and 3) the binding constant of the antibody/antibody fragments for the analyte (the latter often being the most important of these factors) [14]. A linear calibration curve that covers several orders of magnitude in analyte concentration can often be constructed in this type of assay by measuring the peak area or height for the labeled immune complex and plotting this result versus analyte concentration. As is true in the competitive binding format (see Section 3.1), a pre-incubation step is required in this method prior to the CE analysis. During this pre-incubation, a large excess of the labeled antibodies/antibody fragments can be used to promote binding of the analyte at low concentrations, thus helping to increase the rate of immune complex formation [114]. However, using too large of an excess of these labeled agents can also increase non-specific binding and lead to poor limits of detection.
Care must be taken when labeling antibodies or antibody fragments for a non-competitive immunoassay to avoid tagging these agents at or near their binding sites for the analyte. Tagging at such sites could result in a total or partial loss of binding activity for these agents. This effect, in turn, can lead to a decrease in the amount of analyte that can bind and be detected. These factors are of concern in many general methods for labeling antibodies, which often involve amine-based coupling methods that can involve modification sites throughout an antibody’s structure (see reviews in Refs. [4,23-25]). More selective modification can be accomplished by attaching labels to regions on an antibody such as its carbohydrate residues, which are typically located in the stem region of the antibody and away from its binding sites [4,23].
An alternative approach to minimize loss of activity is to use affinity protection chromatography to produce the labeled antibodies or antibody fragments (see Figure 11) [115]. In this method, the analyte or an appropriate analog is immobilized onto a solid support and the desired antibodies or antibody fragments for this analyte are allowed to bind to the support. These antibodies/antibody fragments are then tagged with a labeling agent while they are still bound to the immobilized analyte. This approach ensures that the labels are attached away from the binding sites of the antibodies/antibody fragments. Once the labeling process has been completed, a running buffer is used to disrupt interactions between the antibodies/antibody fragments and the support, with care being taken to avoid using conditions that will irreversibly damage the labeled binding agent. The labeled antibodies/antibody fragments are then allowed to regenerate prior to their use in a non-competitive immunoassay.
Figure 11.

General scheme for affinity protection chromatography. To ensure the label on an antibody or affinity ligand does not interfere with analyte binding, an analog of the analyte is immobilized onto a solid support. The antibody is then applied and allowed to bind the analyte. The reaction to label the antibody is then performed. Once the reaction is complete, the antibody can be eluted from the solid support. This figure is adapted from Ref. [115].
Another item to consider when implementing a non-competitive CE immunoassay is the electrophoretic mobility of the labeled antibodies (or antibody fragments) versus the resulting complex that will form between these antibodies and the analyte. It is desirable in a non-competitive immunoassay to have a labeled binding agent that has a homogeneous electrophoretic mobility. This property will allow the binding agent to migrate as a single narrow peak during the CE separation, making it easier to resolve this peak from the immune complex that is formed between the binding agent and the analyte. One problem when using native antibodies and Fab fragments for such an assay (e.g., as based on polyclonal antibodies) is that they generally have a range of electrophoretic mobilities [27]. The use of monoclonal antibodies helps, to some extent, to provide a more homogeneous set of mobilities and well-defined labeling reactions. Another route to improve the homogeneity of mobilities for these binding agents is to use a single chain variable fragment (scFv), which is a small construct made from an antibody that retains a complete binding site. An scFv can be prepared by using recombinant protein expression methods to obtain the variable-region domains of antibody heavy and light chains (i.e., VH and VL) [116]. This latter approach has been used in CE to create a non-competitive immunoassay for digoxin that made it possible to measure this drug down to a level of 400 × 10−15 M [27].
There are several factors to consider when choosing a label for a non-competitive immunoassay in CE. First, this label should give a signal that is not produced by any of original components of the sample. For instance, a fluorescent label can be used as long as its excitation and emission wavelengths are not similar to those for any of the proteins or other possible components within the sample. Second, it is important to consider the effect that the sample matrix may have on the label’s signal. This is especially true when employing fluorescent tags or chemiluminescent agents, which can be either quenched or enhanced by the sample matrix.
4. HETEROGENEOUS IMMUNOASSAYS
The second general type of CE immunoassay is one that utilizes a heterogeneous binding format. A heterogeneous immunoassay is a method in which either the analyte, an analog of the analyte, or antibody-related binding agents are immobilized onto a solid support [1-3]. Because this class of methods employs an immobilized antibody or analyte analog as a stationary phase, they represent hybrid techniques that combine immunoaffinity chromatography or affinity chromatography with the methods of CE or capillary electrochromatography [4,117-119].
4.1. Methods Based on Immobilized Antibodies
An important type of heterogeneous immunoassay in CE is an approach in which immobilized antibodies are used in a capillary to extract a class of analytes from the sample. After these analytes have been extracted, they are released from the antibodies and separated by CE (note: this release step often involves injecting a plug of buffer which has a lower pH than the buffer that was used for sample extraction) [19,120,121]. This approach can be used to examine a group of structurally-related compounds that all bind to the same immobilized antibodies or to a mixed bed of several antibodies. The high affinity of many antibodies makes this approach useful for isolating and concentrating trace substances prior to their analysis by CE [120]. However, this approach does require that appropriate conditions can be found to release the analytes once they have been captured by the immobilized antibodies, which can be a challenge when antibodies with very high affinities are used for such work.
A recent example of this approach involved the use of a microchip-based system and a mixed bed of antibodies to extract and measure various inflammatory biomarkers (see Figure 12) [19]. This method made use of twelve types of antibodies that were immobilized on a disposable glass fiber disk and used to extract the desired biomarkers prior to their separation by CE on a microchip. This approach was capable of detecting and measuring twelve different inflammatory biomarkers, providing levels of detection in the range of 0.5 to 1.1 pg/mL. Other immunoextraction assays have been used with CE to measure cytokines [121], neuropeptides [120] and atrazine [80].
Figure 12.


Analysis of several analytes using a CE immunoassay performed on a microchip. The figure in (a) shows an electropherogram that was produced by this method when using mixed bed of twelve antibodies to concentrate the analytes prior to their separation and detection. The peaks represent the following analytes: 1) transforming growth factor-beta, 2) interleukin-6, 3) interleukin-1 beta, 4) interferon gamma, 5) macrophage inflammatory protein 1 alpha, 6) macrophage chemoattractant protein 1, 7) tumor necrosis factor-alpha, 8) calcitonin gene-related peptide, 9) neuropeptide Y, 10) interleukin 8, 11) vasoactive intestinal peptide, and 12) substance P. The system in (b) shows the microchip-based system that was used to perform this CE immunoassay by using on-line immunoextraction and labeling for multi-analyte detection. These figures are reproduced with permission from Ref. [19].
The expected concentration of an analyte in a typical sample should be considered in this approach because this factor will determine whether detection is feasible with the amount of sample that is available. This factor can be examined experimentally by first injecting several standards onto the immunoaffinity support and estimating the lower limit of detection that will be obtained in the CE method at a given sample volume. The sample volume can then be increased to decrease the detection limit as required, as has been described for the use of immunoextraction in HPLC [122]. If it is not possible to obtain the desired detection range with a reasonable sample size, then an alternative analysis approach should be employed.
When using immobilized antibodies to extract an analyte, antibodies with a large association equilibrium constant for the analyte are generally used to retain this compound while letting other sample components pass through the system. If this association equilibrium constant is sufficiently high and the time spent within the immunoextraction support is relatively short, binding by the analyte can often be considered irreversible under the application conditions. It is this effect which makes it possible to use such a support to concentrate analytes from even large sample volumes [122]. Removal of the analyte from the immunoextraction support is later accomplished by using a running buffer that will disrupt non-covalent interactions between the antibody and the analyte, such as by altering the pH of the running buffer [19,120,121].
Since the capillary in a CE immunoassay is often reused for multiple samples, it is necessary to choose conditions for an immunoextraction support that will not cause irreversible damage to the antibodies that are placed on this support and used within a CE system. If these antibodies are damaged during the release of a captured analyte, the immunoextraction component of the CE method will show a loss of activity and/or have an increase in non-specific binding over time. The presence of these effects can be detected and monitored by routinely injecting control samples or calibration standards onto the CE system. In one study using immunoextraction with CE, antibodies placed within an injection port were used up to 25 times before showing any significant loss in activity [120].
If antibodies are used to extract several analytes, it is desirable for all of these analytes to be quantitatively extracted during the application step. The degree of this extraction can be adjusted by using a slower application rate for the sample or by increasing the amount of immobilized antibodies. It is especially important when analyzing multiple chemicals to have the total amount of all desired analytes in the sample to be much less than the antibody binding capacity. In one study, the minimum mole ratio of antibodies to analyte that was found to give an acceptable extraction efficiency was 15:1 in work with clinical samples [121].
4.2. Methods Based on Immobilized Analogs of the Analyte
A second type of heterogeneous immunoassay that can be carried out by CE is one that utilizes an immobilized analog of the analyte to extract antibodies and separate these from their immune complexes with the analyte. Work with these immobilized analogs has been combined with labeled antibodies for analyte detection, as is illustrated in Figure 13. This method is particularly useful when detecting small analytes for which the change in electrophoretic mobility between the antibodies and their immune complexes with the analyte may not be sufficient for the separation of these species by CE alone in a non-competitive immunoassay format.
Figure 13.

Scheme for a heterogeneous method of separating immune complexes from free labeled antibodies. In this method, an excess of labeled antibodies is mixed with the sample and incubated to allow for the formation of immune complexes. These immune complexes are later separated from the remaining non-bound antibodies by extracting these non-bound antibodies through the use of a support that contains an immobilized form of the analyte. The immune complexes pass through this support non-retained and pass on to the rest of the CE system for detection.
This approach has been used in CE along with a non-competitive immunoassay to measure the protein HSA [71]. This technique made use of immobilized HSA to bind to labeled anti-HSA antibodies after these antibodies had been mixed with a sample that contained soluble HSA. When this method was used along with FITC as a label, a limit of detection of 9.0 nM was obtained for HSA in a separation time of 5 min. After each analysis, the immobilized HSA support was washed for 5 min with a pH 2.5 buffer to remove the retained antibodies, allowing this support to be used for multiple sample injections.
A related method known as probed isoelectric focusing (PIF) has recently been reported, in which protein signaling was measured in less than 25 cells by resolving and detecting isoforms of the ERK protein [101]. In this method, proteins in a given sample were first separated by using isoelectric focusing. Once separated, the capillary containing these separated analytes was exposed to UV light to attach the proteins to photoactive moieties on the inner surface of the capillary. A wash solution was then passed through the capillary, followed by the application of solutions that contained primary antibodies (able to bind to the given proteins) and secondary antibodies (tagged with HRP and able to bind to the primary antibodies). Once the primary and secondary antibodies were selectively adsorbed within the capillary at regions containing the immobilized proteins, chemiluminescent reagents were passed through the capillary to react with the HRP. The resulting light was then detected through the capillary wall and used to signal the presence of each desired protein [101].
A number of factors should be considered when designing or optimizing an immobilized analyte analog for a CE immunoassay. To create the immobilized analog support for this method, the analyte may need to be derivatized to create a reactive group which is capable of being used for immobilization within the CE capillary. In addition, this immobilization process must not interfere with interactions between the analog and antibodies if these antibodies are to be extracted away by this analog from their immune complexes. For small immobilized analogs, a spacer arm must be used between the analog and the support to allow the analog to be easily accessible to antibodies for binding. Also, the immobilized analog will need to be present in a sufficient excess to allow all or most of the free antibodies in the sample/antibody mixture to be extracted. These requirements are similar to those reported for the use of immobilized analogs for post-column immunodetection in chromatographic-based immunoassays [4,117-119].
5. SUMMARY
This review discussed the main types of CE immunoassays and highlighted the advantages and disadvantages of each format. The most popular type of CE immunoassay is the homogeneous competitive binding assay, which is capable of determining the concentrations of both large and small analytes. Another homogeneous format that has been described for CE immunoassays is the non-competitive binding immunoassay. Heterogeneous immunoassay methods that make use of either immobilized antibodies or immobilized analogs of the analyte have also been reported in CE. Although a variety of detection methods exist for CE immunoassays, the use of fluorescent labels and laser-induced fluorescence is by far the most popular detection method for these methods. However, work is also progressing in the use of enzyme labels and detection schemes that are based on techniques such as chemiluminescence, absorbance detection, electrochemical measurements, and mass spectrometry or surface plasmon resonance.
The speed and efficiency of capillary electrophoresis, plus its ability to work with small samples, make CE immunoassays particularly valuable for work in areas such as the study of single cells or in the development of assays based on microanalytical systems (see Figure 12) [19, 20]. This latter area is appealing as a means for overcoming some of the disadvantages of CE immunoassays versus more conventional, higher-throughput methods for performing immunoassays (e.g., ELISA). Many of the same basic formats and principles as described in this review for traditional CE immunoassays can also be applied to these microanalytical systems (see Ref. [123] for a review of affinity microchip separations and Ref. [124] for a review of detection methods for microchip separations). It is expected that the field of CE immunoassays in these and other areas will continue to develop and expand into new applications. This development should be made possible by further advances in the assay formats and detection schemes for these methods.
ACKNOWLEDGEMENT
This work was supported in part by the National Institutes of Health under grant R01 GM044931.
SYMBOLS & ABBREVIATIONS
- ACE
Affinity capillary electrophoresis
- APCE
Affinity probe capillary electrophoresis
- AQC
6-Aminoquinolyl-N-hydroxysuccinimidyl carbamate BSA Bovine serum albumin
- CE
Capillary electrophoresis
- CGE
Capillary gel electrophoresis
- Cy5
Cyanine 5
- FITC
Fluorescein isothiocyanate
- HIV-1
Type 1 human immunodeficiency virus
- HRP
Horseradish peroxidase
- HSA
Human serum albumin
- ILITC
Isoluminol isothiocyanate
- LIF
Laser-induced fluorescence
- MPA
Medroprogesterone acetate
- MP8
Microperoxidase-8
- MP11
Microperoxidase-11
- SPR
Surface plasmon resonance
- scFv
Single chain variable fragment
- SELEX
Systematic evolution of ligands by exponential enrichment
REFERENCES
- [1].Hage DS. Anal. Chem. 1999;71:294R–304R. doi: 10.1021/a1999901+. [DOI] [PubMed] [Google Scholar]
- [2].Price CP, Newman DJ, editors. Principles and Practice of Immunoassay. Stockton Press; New York: 1997. p. 650. [Google Scholar]
- [3].Butler JE. Immunochemistry of Solid-Phase Immunoassay. CRC Press; Boca Raton, FL: 1991. [Google Scholar]
- [4].Moser AC, Hage DS. In: Handbook of Affinity Chromatography. 2nd Edition Hage DS, editor. CRC Press; Boca Raton, FL: 2006. pp. 789–836. [Google Scholar]
- [5].Amundsen LK, Siren H. Electrophoresis. 2007;28:99–113. doi: 10.1002/elps.200500962. [DOI] [PubMed] [Google Scholar]
- [6].Heegaard NHH, Schou C. In: Handbook of Affinity Chromatography. 2nd Edition Hage DS, editor. CRC Press; Boca Raton, FL: 2006. pp. 699–735. [Google Scholar]
- [7].Lin S, Hsu S-M. Anal. Biochem. 2005;341:1–15. doi: 10.1016/j.ab.2005.02.023. [DOI] [PubMed] [Google Scholar]
- [8].Schmalzing D, Nashabeh W. Electrophoresis. 1997;18:2184–2193. doi: 10.1002/elps.1150181209. [DOI] [PubMed] [Google Scholar]
- [9].Schmalzing D, Buonocore S, Piggee C. Electrophoresis. 2000;21:3919–3930. doi: 10.1002/1522-2683(200012)21:18<3919::AID-ELPS3919>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- [10].Yeung WSB, Luo GA, Wang QG, Ou JP. J. Chromatogr. B. 2003;797:217–228. doi: 10.1016/s1570-0232(03)00489-6. [DOI] [PubMed] [Google Scholar]
- [11].Ellington AD, Szostak JW. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- [12].Tuerk C, Gold L. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- [13].Klug SJ, Famulok M. Mol. Biol. Rep. 1994;20:97–107. doi: 10.1007/BF00996358. [DOI] [PubMed] [Google Scholar]
- [14].German I, Buchanan DD, Kennedy RT. Anal. Chem. 1998;70:4540–4545. doi: 10.1021/ac980638h. [DOI] [PubMed] [Google Scholar]
- [15].Tim RC, Kautz RA, Karger BL. Electrophoresis. 2000;21:220–226. doi: 10.1002/(SICI)1522-2683(20000101)21:1<220::AID-ELPS220>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- [16].Nielsen RG, Rickard EC, Santa PF, Sharknas DA, Sittampalam GS. J. Chromatogr. 1991;539:177–185. doi: 10.1016/s0021-9673(01)95371-3. [DOI] [PubMed] [Google Scholar]
- [17].Schultz NM, Kennedy RT. Anal. Chem. 1993;65:3161–3165. [Google Scholar]
- [18].German I, Kennedy RT. J. Chromatogr. B. 2000;742:353–362. doi: 10.1016/s0378-4347(00)00180-8. [DOI] [PubMed] [Google Scholar]
- [19].Phillips TM, Wellner EF. Electrophoresis. 2007;28:3041–3048. doi: 10.1002/elps.200700193. [DOI] [PubMed] [Google Scholar]
- [20].Tao L, Kennedy RT. Anal. Chem. 1996;68:3899–3906. doi: 10.1021/ac960560+. [DOI] [PubMed] [Google Scholar]
- [21].Caslavska J, Allemann D, Thormann W. J. Chromatogr. A. 1999;838:197–211. doi: 10.1016/s0021-9673(99)00115-6. [DOI] [PubMed] [Google Scholar]
- [22].Heegaard NHH, Kennedy RT. Electrophoresis. 1999;20:3122–3133. doi: 10.1002/(SICI)1522-2683(19991001)20:15/16<3122::AID-ELPS3122>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- [23].Hermanson GT. Bioconjugate Techniques. Academic Press; New York: 1996. [Google Scholar]
- [24].Knorre DG, Vlassov VV. Affinity Modification of Biopolymers. CRC Press; Boca Raton, FL: 1989. p. 269. [Google Scholar]
- [25].Underberg WJM, Waterval JCM. Electrophoresis. 2002;23:3922–3933. doi: 10.1002/elps.200290010. [DOI] [PubMed] [Google Scholar]
- [26].Chen DY, Dovichi NJ. J. Chromatogr. B. 1994;657:265–269. doi: 10.1016/0378-4347(94)00014-x. [DOI] [PubMed] [Google Scholar]
- [27].Hafner FT, Kautz RA, Iverson BL, Tim RC, Karger BL. Anal. Chem. 2000;72:5779–5786. doi: 10.1021/ac000853+. [DOI] [PubMed] [Google Scholar]
- [28].Schmalzing D, Nashabeh W, Yao X-W, Mhatre R, Regnier FE, Afeyan NB, Fuchs M. Anal. Chem. 1995;67:606–612. doi: 10.1021/ac00099a019. [DOI] [PubMed] [Google Scholar]
- [29].German I, Roper MG, Kalra SP, Rhinehart E, Kennedy RT. Electrophoresis. 2001;22:3659–3667. doi: 10.1002/1522-2683(200109)22:17<3659::AID-ELPS3659>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- [30].Zhou J, Xu X, Wang Y. J. Chromatogr. B. 2007;848:226–231. doi: 10.1016/j.jchromb.2006.10.027. [DOI] [PubMed] [Google Scholar]
- [31].Babu CVS, Chung BC, Lho DS, Yoo YS. J. Chromatogr. A. 2006;1111:133–138. doi: 10.1016/j.chroma.2005.06.031. [DOI] [PubMed] [Google Scholar]
- [32].Han K-Y, Ban E, Yoo YS. J. Chromatogr. A. 2003;1013:215–220. doi: 10.1016/s0021-9673(03)01432-8. [DOI] [PubMed] [Google Scholar]
- [33].Liu C-M, Tung K-H, Chang T-H, Chien C-C, Yen M-H. J. Chromatogr. B. 2003;791:315–321. doi: 10.1016/s1570-0232(03)00249-6. [DOI] [PubMed] [Google Scholar]
- [34].Ban E, Ryu J-C, Yoo YS. Microchem. J. 2001;70:211–217. [Google Scholar]
- [35].Carnelley TJ, Barker S, Wang H, Tan WG, Weinfeld M, Le XC. Chem. Res. Toxicol. 2001;14:1513–1522. doi: 10.1021/tx0100946. [DOI] [PubMed] [Google Scholar]
- [36].Ban E, Nam HS, Yoo YS. J. Chromatogr. A. 2001;924:337–344. doi: 10.1016/s0021-9673(01)00829-9. [DOI] [PubMed] [Google Scholar]
- [37].Zaugg S, Zhang X, Sweedler J, Thormann W. J. Chromatogr. B. 2001;752:17–31. doi: 10.1016/s0378-4347(00)00507-7. [DOI] [PubMed] [Google Scholar]
- [38].Sowell J, Parihar R, Patonay G. J. Chromatogr. B. 2001;752:1–8. doi: 10.1016/s0378-4347(00)00508-9. [DOI] [PubMed] [Google Scholar]
- [39].Driedger DR, LeBlanc RJ, LeBlanc EL, Sporns P. J. Agri. Food Chem. 2000;48:4079–4082. doi: 10.1021/jf000157a. [DOI] [PubMed] [Google Scholar]
- [40].Driedger DR, LeBlanc RJ, LeBlanc EL, Sporns P. J. Agri. Food Chem. 2000;48:1135–1139. doi: 10.1021/jf990680t. [DOI] [PubMed] [Google Scholar]
- [41].Jiang G, Attiya S, Ocvirk G, Lee WE, Harrison DJ. Biosens. Bioel. 2000;14:861–869. doi: 10.1016/s0956-5663(99)00056-1. [DOI] [PubMed] [Google Scholar]
- [42].Wan Q-H, Le XC. J. Chromatogr. A. 1999;853:555–562. doi: 10.1016/s0021-9673(99)00711-6. [DOI] [PubMed] [Google Scholar]
- [43].Choi J, Kim C, Choi MJ. Electrophoresis. 1998;19:2950–2955. doi: 10.1002/elps.1150191626. [DOI] [PubMed] [Google Scholar]
- [44].Ye L, Chris Le X, Xing JZ, Ma M, Yatscoff R. J. Chromatogr. B. 1998;714:59–67. doi: 10.1016/s0378-4347(98)00091-7. [DOI] [PubMed] [Google Scholar]
- [45].Choi J, Kim C, Choi MJ. J. Chromatogr. B. 1998;705:277–282. doi: 10.1016/s0378-4347(97)00527-6. [DOI] [PubMed] [Google Scholar]
- [46].Korf GM, Landers JP, O’Kane DJ. Anal. Biochem. 1997;251:210–218. doi: 10.1006/abio.1997.2242. [DOI] [PubMed] [Google Scholar]
- [47].Pritchett TJ, Evangelista RA, Chen F-TA. J. Cap. Electrophor. 1995;2:145–149. [Google Scholar]
- [48].Evangelista RA, Michael JM, Chen FTA. Am. Clin. Lab. 1995;14:27–28. [PubMed] [Google Scholar]
- [49].Chen F-TA, Pentoney SL., Jr. J. Chromatogr. A. 1994;680:425–430. doi: 10.1016/0021-9673(94)85139-5. [DOI] [PubMed] [Google Scholar]
- [50].Mi J-Q, Zhang X-X, Chang W-B. J. Immunoassay Immunochem. 2004;25:57–70. doi: 10.1081/ias-120027226. [DOI] [PubMed] [Google Scholar]
- [51].Jackman R, Schmerr MJ. Electrophoresis. 2003;24:892–896. doi: 10.1002/elps.200390112. [DOI] [PubMed] [Google Scholar]
- [52].Zaugg S, Thormann W. J. Pharm. Biomed. Anal. 2001;24:785–799. doi: 10.1016/s0731-7085(00)00546-x. [DOI] [PubMed] [Google Scholar]
- [53].Schmerr MJ, Jenny A. Electrophoresis. 1998;19:409–414. doi: 10.1002/elps.1150190308. [DOI] [PubMed] [Google Scholar]
- [54].Mi JQ, Qi XH, Zhang XX, Chang WB. Chin. Chem. Lett. 2004;15:943–946. [Google Scholar]
- [55].Su P, Zhang X-X, Wang Y-C, Chang W-B. Talanta. 2003;60:969–975. doi: 10.1016/S0039-9140(03)00179-6. [DOI] [PubMed] [Google Scholar]
- [56].Lam MT, Wan QH, Boulet CA, Le XC. J. Chromatogr. A. 1999;853:545–553. doi: 10.1016/s0021-9673(99)00677-9. [DOI] [PubMed] [Google Scholar]
- [57].Schultz NM, Huang L, Kennedy RT. Anal. Chem. 1995;67:924–929. doi: 10.1021/ac00101a020. [DOI] [PubMed] [Google Scholar]
- [58].Suzuki Y, Arakawa H, Maeda M. Anal. Sci. 2003;19:111–115. doi: 10.2116/analsci.19.111. [DOI] [PubMed] [Google Scholar]
- [59].Schmerr MJ, Jenny AL, Bulgin MS, Miller JM, Hamir AN, Cutlip RC, Goodwin KR. J. Chromatogr. A. 1999;853:207–214. doi: 10.1016/s0021-9673(99)00514-2. [DOI] [PubMed] [Google Scholar]
- [60].Schmalzing D, Nashabeh W, Fuchs M. Clin. Chem. 1995;41:1403–1406. [PubMed] [Google Scholar]
- [61].Bornemann C, Burggraef T, Heimbuechel G, Hanisch F-G, Winkels S. Anal. Bioanal. Chem. 2003;376:1074–1080. doi: 10.1007/s00216-003-2038-3. [DOI] [PubMed] [Google Scholar]
- [62].Rogers KR, Apostol AB, Brumley WC. Anal. Lett. 2000;33:443–453. [Google Scholar]
- [63].Miki S, Kaneta T, Imasaka T. J. Chromatogr. B. 2001;759:337–342. doi: 10.1016/s0378-4347(01)00228-6. [DOI] [PubMed] [Google Scholar]
- [64].Lam MT, Boulet CA, Chris Le X. Anal. Chim. Acta. 2002;457:21–28. [Google Scholar]
- [65].Shimura K, Karger BL. Anal. Chem. 1994;66:9–15. doi: 10.1021/ac00073a004. [DOI] [PubMed] [Google Scholar]
- [66].Feng H.-t., Law W-S, Yu L-J, Li SF-Y. J. Chromatogr. A. 2007;1156:75–79. doi: 10.1016/j.chroma.2006.12.077. [DOI] [PubMed] [Google Scholar]
- [67].German I, Kennedy RT. Anal. Chem. 2000;72:5365–5372. doi: 10.1021/ac000549g. [DOI] [PubMed] [Google Scholar]
- [68].Chiem NH, Harrison DJ. Clin. Chem. 1998;44:591–598. [PubMed] [Google Scholar]
- [69].Lourenco PC, Schmerr MJ, MacGregor I, Will RG, Ironside JW, Head MW. J. Gen. Virol. 2006;87:3119–3124. doi: 10.1099/vir.0.81935-0. [DOI] [PubMed] [Google Scholar]
- [70].Tao L, Aspinwall CA, Kennedy RT. Electrophoresis. 1998;19:403–408. doi: 10.1002/elps.1150190307. [DOI] [PubMed] [Google Scholar]
- [71].Giovannoli C, Anfossi L, Baggiani C, Giraudi G. J. Chromatogr. A. 2007;1155:187–192. doi: 10.1016/j.chroma.2007.02.056. [DOI] [PubMed] [Google Scholar]
- [72].Lam MT, Le XC. Analyst. 2002;127:1633–1637. doi: 10.1039/b206531b. [DOI] [PubMed] [Google Scholar]
- [73].Taylor J, Picelli G, Harrison DJ. Electrophoresis. 2001;22:3699–3708. doi: 10.1002/1522-2683(200109)22:17<3699::AID-ELPS3699>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- [74].Pavski V, Le XC. Anal. Chem. 2001;73:6070–6076. doi: 10.1021/ac0107305. [DOI] [PubMed] [Google Scholar]
- [75].Buchanan DD, Jameson EE, Perlette J, Malik A, Kennedy RT. Electrophoresis. 2003;24:1375–1382. doi: 10.1002/elps.200390176. [DOI] [PubMed] [Google Scholar]
- [76].Ramseier A, Caslavska J, Thormann W. Electrophoresis. 1998;19:2956–2966. doi: 10.1002/elps.1150191627. [DOI] [PubMed] [Google Scholar]
- [77].Zhang XX, Li J, Gao J, Sun L, Chang WB. J. Chromatogr. A. 2000;895:1–7. doi: 10.1016/s0021-9673(00)00590-2. [DOI] [PubMed] [Google Scholar]
- [78].Yang W-C, Schmerr MJ, Jackman R, Bodemer W, Yeung ES. Anal. Chem. 2005;77:4489–4494. doi: 10.1021/ac050231u. [DOI] [PubMed] [Google Scholar]
- [79].Yang W.-c., Yeung ES, Schmerr MJ. Electrophoresis. 2005;26:1751–1759. doi: 10.1002/elps.200410202. [DOI] [PubMed] [Google Scholar]
- [80].Ensing K, Paulus A. J. Pharm. Biomed. Anal. 1996;14:305–315. doi: 10.1016/0731-7085(95)01607-4. [DOI] [PubMed] [Google Scholar]
- [81].Miki S, Kaneta T, Imasaka T. J. Chromatogr. A. 2005;1066:197. doi: 10.1016/j.chroma.2005.01.037. [DOI] [PubMed] [Google Scholar]
- [82].Gomez-Hens A, Aguilar-Caballos MP. Trends Anal. Chem. 2004;23:127–136. [Google Scholar]
- [83].Glazer AN, Hixson CS. J. Biol. Chem. 1977;252:32–42. [PubMed] [Google Scholar]
- [84].Wu M, Goodwin PM, Ambrose WP, Keller RA. J. Phys. Chem. 1996;100:17406–17409. [Google Scholar]
- [85].Bruchez M, Jr., Moronne M, Gin P, Weiss S, Alivisatos AP. Science. 1998;281:2013–2016. doi: 10.1126/science.281.5385.2013. [DOI] [PubMed] [Google Scholar]
- [86].Alivisatos AP. J. Phys. Chem. 1996;100:13226–13239. [Google Scholar]
- [87].Hemmila IA. Application of Fluorescence in Immunoassays. Wiley; New York: 1991. [Google Scholar]
- [88].Zhang Y, Zhang Z, Yang F. J. Chromatogr. B. 2007;857:100–107. doi: 10.1016/j.jchromb.2007.07.006. [DOI] [PubMed] [Google Scholar]
- [89].Peng C-F, Huo T-M, Liu L-Q, Chu X-G, Xu C-L. Electrophoresis. 2007;28:970–974. doi: 10.1002/elps.200600290. [DOI] [PubMed] [Google Scholar]
- [90].Wang J, Ren J. Electrophoresis. 2005;26:2402–2408. doi: 10.1002/elps.200410246. [DOI] [PubMed] [Google Scholar]
- [91].Ji X, He Z, Ai X, Yang H, Xu C. Talanta. 2006;70:353–357. doi: 10.1016/j.talanta.2006.02.053. [DOI] [PubMed] [Google Scholar]
- [92].Wang J, Huang W, Liu Y, Cheng J, Yang J. Anal. Chem. 2004;76:5393–5398. doi: 10.1021/ac049891+. [DOI] [PubMed] [Google Scholar]
- [93].He Z, Gao N, Jin W. J. Chromatogr. B. 2003;784:343–350. doi: 10.1016/s1570-0232(02)00823-1. [DOI] [PubMed] [Google Scholar]
- [94].He Z, Jin W. Anal. Biochem. 2003;313:34–40. doi: 10.1016/s0003-2697(02)00508-0. [DOI] [PubMed] [Google Scholar]
- [95].Munro OQ, Marques HM. Inorg. Chem. 1996;35:3752–3767. doi: 10.1021/ic9502842. [DOI] [PubMed] [Google Scholar]
- [96].Mondelli R, Scaglioni L, Mazzini S, Bolis G, Ranghino G. Magn. Reson. Chem. 2000;38:229–240. [Google Scholar]
- [97].Ricoux R, Korri-Youssoufi H, Mahy J-P. J. Biol. Sci. 2005;5:44–49. [Google Scholar]
- [98].Tsukagoshi K, Jinno N, Toguchi K, Nakajima R. Bull. Chem. Soc. Japan. 2005;78:1791–1794. [Google Scholar]
- [99].Tsukagoshi K, Indou H, Sawanoi K, Oguni T, Nakajima R. Bull. Chem. Soc. Japan. 2004;77:1353–1357. [Google Scholar]
- [100].Thorpe GHG, Kricka LJ, Moseley SB, Whitehead TP. Clin. Chem. 1985;31:1335–1341. [PubMed] [Google Scholar]
- [101].O’Neill RA, Bhamidipati A, Bi X, Deb-Basu D, Cahill L, Ferrante J, Gentalen E, Glazer M, Gossett J, Hacker K, Kirby C, Knittle J, Loder R, Mastroieni C, MacLaren M, Mills T, Nguyen U, Parker N, Rice A, Roach D, Suich D, Voehringer D, Voss K, Yang J, Yang T, Vander Horn PB. Proc. Natl. Acad. Sci. U.S.A. 2006;103:16153–16158. doi: 10.1073/pnas.0607973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Vandaveer WR, Pasas SA, Martin RS, Lunte SM. Electrophoresis. 2002;23:3667–3677. doi: 10.1002/1522-2683(200211)23:21<3667::AID-ELPS3667>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- [103].Martin RS, Gawron AJ, Lunte SM, Henry CS. Anal. Chem. 2000;72:3196–3202. doi: 10.1021/ac000160t. [DOI] [PubMed] [Google Scholar]
- [104].He Z, Gao N, Jin W. Anal. Chim. Acta. 2003;497:75–81. [Google Scholar]
- [105].He ZH, Jin WR. Chin. Chem. Lett. 2002;13:1090–1092. [Google Scholar]
- [106].Ou JP, Chan STH, Yeung WSB. J. Chromatogr. B. 1999;731:389–394. doi: 10.1016/s0378-4347(99)00234-0. [DOI] [PubMed] [Google Scholar]
- [107].Wey AB, Caslavska J, Thormann W. J. Chromatogr. A. 2000;895:133–146. doi: 10.1016/s0021-9673(00)00636-1. [DOI] [PubMed] [Google Scholar]
- [108].Thormann W, Caslavska J, Ramseier A, Siethoff C. J. Microcol. Sep. 2000;12:13–24. [Google Scholar]
- [109].Tsai JL, Wu W-S, Lee H-H. Electrophoresis. 2000;21:1580–1586. doi: 10.1002/(SICI)1522-2683(20000501)21:8<1580::AID-ELPS1580>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- [110].Whelan RJ, Zare RN. Anal. Chem. 2003;75:1542–1547. doi: 10.1021/ac0263521. [DOI] [PubMed] [Google Scholar]
- [111].Waseda S, Shimosaka T, Uchiyama K, Hobo T. Chem. Lett. 1999:1195–1196. [Google Scholar]
- [112].Chaiken I, Rose S, Karlsson R. Anal. Biochem. 1992;201:197–210. doi: 10.1016/0003-2697(92)90329-6. [DOI] [PubMed] [Google Scholar]
- [113].Christopoulos TK, Diamandis EP. Immunoassay. Academic Press; San Diego, CA: 1996. [Google Scholar]
- [114].Masseyeff RF, Albert WH, Staines NA. Methods of Immunological Analysis. VCH; New York: 1992. [Google Scholar]
- [115].Attiya S, Dickinson-Laing T, Cesarz J, Giese RD, Lee WE, Mah D, Harrison DJ. Electrophoresis. 2002;23:750–758. doi: 10.1002/1522-2683(200203)23:5<750::AID-ELPS750>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- [116].Neri D, Petrul H, Roncucci G. Cell. Biophys. 1995;27:47–61. doi: 10.1007/BF02822526. [DOI] [PubMed] [Google Scholar]
- [117].Hage DS, Clarke W. Separation Techniques in Clinical Chemistry. 2003:361–387. [Google Scholar]
- [118].Hage DS, Nelson MA. Anal. Chem. 2001;73:198A–205A. [PubMed] [Google Scholar]
- [119].Hage DS, Phillips TM. In: Handbook of Affinity Chromatography. 2nd Edition Hage DS, editor. CRC Press; Boca Raton, FL: 2006. pp. 127–172. [Google Scholar]
- [120].Phillips TM, Wellner E. J. Chromatogr. A. 2006;1111:106–111. doi: 10.1016/j.chroma.2006.01.102. [DOI] [PubMed] [Google Scholar]
- [121].Phillips TM. Electrophoresis. 2004;25:1652–1659. doi: 10.1002/elps.200305873. [DOI] [PubMed] [Google Scholar]
- [122].Rollag JG, Beck-Westermeyer M, Hage DS. Anal. Chem. 1996;68:3631–3637. [Google Scholar]
- [123].Phillips TM. In: Handbook of Affinity Chromatography. 2nd Edition Hage DS, editor. CRC Press; Boca Raton, FL: 2006. pp. 763–787. [Google Scholar]
- [124].Uchiyama K, Nakajima H, Hobo T. Anal. Bioanal. Chem. 2004;379:375–382. doi: 10.1007/s00216-004-2616-z. [DOI] [PubMed] [Google Scholar]