

Abstract
The structure–activity relationship (SAR) for a novel class of 1,2,4-triazole antagonists of the human A2A adenosine receptor (hA2AAR) was explored. Thirty-three analogs of a ligand that was discovered in a structure-based virtual screen against the hA2AAR were tested in hA1, A2A, and A3 radioligand binding assays and in functional assays for the A2BAR subtype. As a series of closely related analogs of the initial lead, 1, did not display improved binding affinity or selectivity, molecular docking was used to guide the selection of more distantly related molecules. This resulted in the discovery of 32, a hA2AAR antagonist (Ki 200 nM) with high ligand efficiency. In light of the SAR for the 1,2,4-triazole scaffold, we also investigated the binding mode of these compounds based on docking to several A2AAR crystal structures.
Keywords: 1,2,4-Triazole; A2A adenosine receptor; antagonist; molecular docking; structure−activity relationship
Extracellular adenosine regulates numerous physiological processes via activation of four G protein-coupled receptors (GPCRs).1 The A1, A2A, A2B, and A3 adenosine receptor (AR) subtypes display varying affinities for adenosine and act via different signaling pathways. The A2A and A2BAR subtypes are primarily coupled to Gs and thereby increase intracellular cAMP levels, whereas the A1 and A3ARs inhibit cAMP production via activation of Gi. The human (h) A2AAR is expressed in both the periphery and the central nervous system (CNS). The extracellular adenosine concentration increases in response to cell stress or damage, and activation of the hA2AAR protects tissues by reducing inflammation.2 In the CNS, a postsynaptic striatal hA2AAR regulates the effects of other neurotransmitters via interactions with D2 dopamine receptors and metabotropic glutamate receptor-5.3 There is a growing interest in the hA2AAR as a drug target. Agonists are explored as anti-inflammatory drugs, and antagonists are developed for the treatment of neurodegenerative disorders such as Parkinson’s disease.4,5
Until recently, drug discovery efforts targeting the hA2AAR have been limited to ligand-based medicinal chemistry approaches.6,7 Many compound series that display high affinity for the A2A subtype have been developed based on adenosine or naturally occurring antagonists, e.g. caffeine.8 In late 2008, the determination of the first atomic-resolution structures of the hA2AAR9 led to an increasing interest in the use of structure-based approaches in ligand discovery. One of these, the molecular docking method, can be used to computationally screen large chemical libraries against the binding site of a protein.10 Two independent docking screens that were carried out against the first crystal structure of the hA2AAR were remarkably successful, with hit-rates of 35 and 41%, respectively.11,12
The starting point of this study, compound 1, was discovered based on a docking screen of 1.4 million compounds against the first high-resolution crystal structure of the hA2AAR (Table 1, Figure 1A). The molecule was ranked as number 88 based on its score for complementarity to the orthosteric site and was selected for experimental evaluation together with 19 other compounds from the in silico screen. Seven of these molecules were shown to bind to the A2AAR with inhibition constant (Ki) values lower than 10 μM. Among these, compound 1 was one of the most potent and represented a novel class of 1,2,4-triazole antagonists.11 Herein we explore the structure–activity relationship (SAR) for 1,2,4-triazole antagonists by testing a series of 33 analogs of 1 in radioligand binding assays against the A1, A2A, and A3AR subtypes and functional assays for the A2B receptor. Molecular docking to hA2AAR receptor crystal structures (PDB accession codes 3eml(9) and 3pwh(13)) was used to guide the selection of compounds for experimental testing and to investigate the binding modes of the ligands.
Table 1. Binding Affinities of a Series of 1,2,4-Triazole Derivatives at the Human A1, A2A, and A3ARs Measured in Radioligand Binding Assays and Percent Inhibition of cAMP Accumulation in the Presence of 300 nM NECA in a Functional Assay for the A2BAR.
|
Ki (μM) or % inhibition at 10 μMa |
|||||||
|---|---|---|---|---|---|---|---|
| compd | R | X | Y | A1 | A2A | A3 | A2B |
| 1 | CH3 | 3-CH3 | H | 19% | 1.2 ± 0.1 | 3.0 ± 0.2 | 18% |
| 2 | CH3 | 3-Cl | H | 1.1 ± 0.1 | 1.0 ± 0.2 | 2.6 ± 0.5 | 28% |
| 3 | CH3 | H | H | 5.2 ± 0.8 | 1.0 ± 0.1 | 3.3 ± 0.6 | 23% |
| 4 | CH3 | 4-CH3 | H | 38% | 6.4 ± 0.9 | 1.4 ± 0.2 | –6% |
| 5 | CH3 | 4-Cl | H | 3.9 ± 0.4 | 4.0 ± 0.9 | 2.5 ± 0.1 | 51% |
| 6 | CH3 | 2-F | H | 3.7 ± 0.4 | 1.0 ± 0.2 | 2.6 ± 0.7 | 31% |
| 7 | (CH3)2 | 4-Cl | H | 41% | 8.8 ± 0.6 | 0.5 ± 0.1 | 6% |
| 8 | CH2CH3 | H | H | 1.5 ± 0.5 | 1.4 ± 0.2 | 1.9 ± 0.3 | –9% |
| 9 | H | 3-CH3 | H | 2.3 ± 0.7 | 2.0 ± 0.4 | 1.5 ± 0.3 | 8% |
| 10 | H | H | H | 20% | 1.8 ± 0.5 | 2.3 ± 0.1 | 16% |
| 11 | H | 2-CH3 | H | 16% | 1.7 ± 0.3 | 1.0 ± 0.1 | 13% |
| 12 | H | 2,6-CH3 | H | 49% | 1.7 ± 0.5 | 5.5 ± 0.3 | 43% |
| 13 | H | 3-(CH2)3-4 | H | 46% | 3.9 ± 0.2 | 2.2 ± 0.8 | 9% |
| 14 | H | 3,5-CH3 | H | 1.6 ± 0.7 | 3.2 ± 0.8 | 2.7 ± 0.9 | –5% |
| 15 | 3.3 ± 0.9 | 2.2 ± 1.0 | 1.9 ± 0.2 | 21% | |||
| 16 | 40% | 3.4 ± 0.7 | 8.3 ± 0.6 | 40% | |||
| 17 | 8.1 ± 0.7 | 5.0 ± 0.1 | 4.6 ± 0.9 | 7% | |||
| 18 | H | H | 4-F | 2% | 3.5 ± 0.6 | 1.5 ± 0.3 | –10% |
| 19 | H | 2-F | 4-CH3 | 2% | 2% | 50% | 11% |
| 20 | H | 2-OCH3 | 4-CH3 | 8% | 49% | 1.0 ± 0.1 | –10% |
| 21 | H | H | 4-CH3 | 6.5 ± 1.5 | 3.6 ± 0.4 | 3.2 ± 0.4 | 23% |
| 22 | CH3 | H | 2-CH3 | 6% | 26% | 48% | –3% |
| 23a | CH3 | H | 3-CH3 | 8% | 2.0 ± 0.1 | 5.9 ± 1.2 | 5% |
| 23b | CH3 (R) | H | 3-CH3 | 12% | 1.8 ± 0.3 | 6.3 ± 1.4 | |
| 23c | CH3 (S) | H | 3-CH3 | 13% | 23% | 32% | |
| 24 | 37% | 38% | 5.3 ± 1.5 | –15% | |||
Measured in three independent experiments.
Figure 1.

(A–D) Predicted binding modes for the A2AAR crystal structure with PDB accession code 3eml: (A) 1, (B) 27, (C) 32, (D) 33. (E) Alignment of two A2AAR crystal structures, PDB accession codes 3eml and 3pwh. Key residues are shown in sticks (orange, 3eml; white, 3pwh). (F–H) Predicted binding modes for the A2AAR crystal structure with PDB accession code 3pwh: (F) 1, (G) 27, (H) 32. The binding site is shown in white ribbons with selected side chains shown in sticks. Ligands are depicted with orange carbon atoms. Black dotted lines indicate hydrogen bonds.
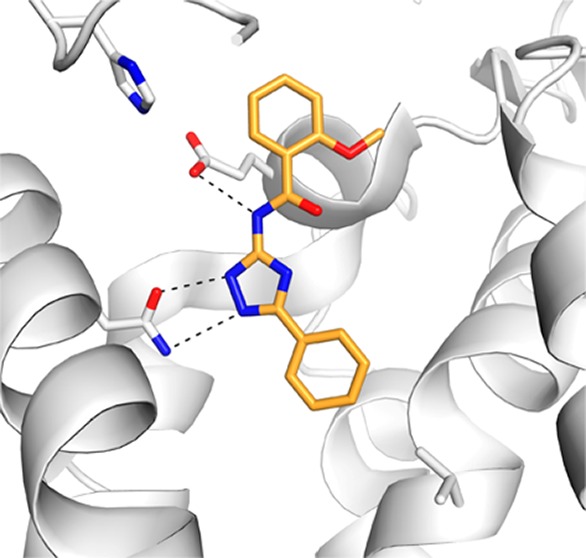
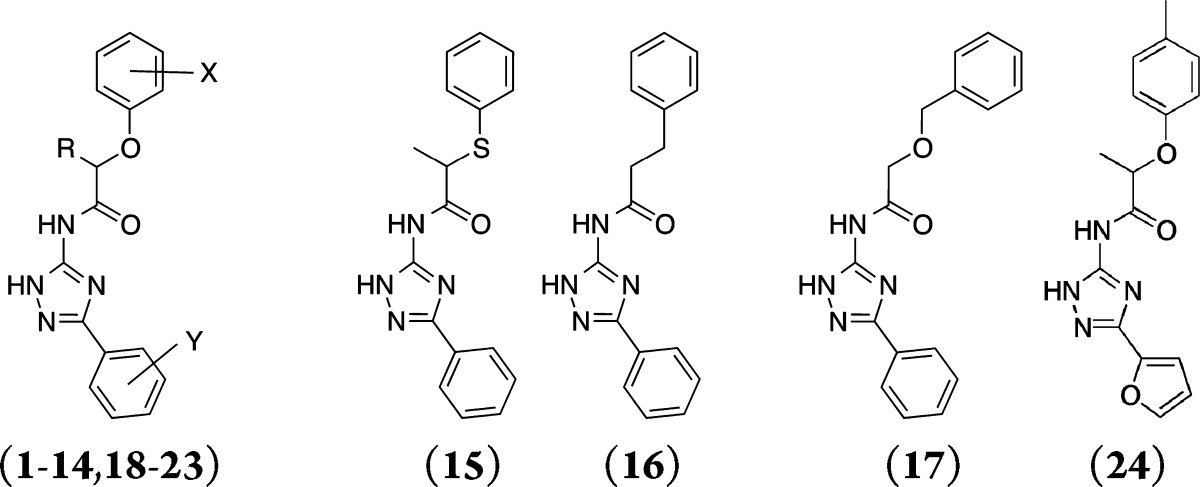
A series of closely related derivatives of compound 1 was first tested to obtain an SAR that could serve as a starting point for further structural optimization (Table 1, compounds 1–24). Compound 1 displayed a Ki of 1.2 μM in binding to the hA2AAR. This differed from that reported previously by a factor of 6,11 which was likely due to differences in the experimental conditions. Replacing the 3-methyl group on the phenoxy ring of compound 1 with either a chloride or hydrogen (2 and 3, respectively) did not change hA2AAR affinity, while para-substituted compounds displayed 3–5-fold reductions of the binding affinity (4–5). Modifications at the chiral center did not improve affinity in the series (7–8). The 1,2,4-triazole scaffold was further explored starting from compound 9, which lacked the chiral center of compound 1 and displayed a Ki of 2 μM at the hA2AAR. Mono- and dimethyl substitutions on the phenoxy ring (10–14) did not significantly improve the affinity. In agreement with observations for compounds 2–5, ortho and meta substitutions on the phenoxy ring did not affect binding, while para-substitutions led to a 2-fold loss of affinity. Replacing the phenoxy group with a phenylsulfanyl, benzyl, or benzyloxy group (15–17) led to 2–4-fold reductions of affinity compared to compound 1. The predicted binding mode of compound 1 (Figure 1A) suggested that the phenoxy group interacted with residues in the extracellular loops (ELs). Although several residues in EL2 have been shown to be critical for ligand binding to the hA2AAR,14,15 the inherent flexibility of the GPCR loop regions makes it difficult to relate the SAR to the predicted binding modes for these analogs, a point to be discussed below. In the next step, substitutions on the 3-phenyl of the triazole ring were tested. Methyl substitutions in the para (19–21) and ortho (22) positions reduced hA2AAR affinity, while the meta-methylated analog (23a) displayed a Ki of 2 μM. The reduction of affinity observed for all compounds with substitutions on the 3-phenyl ring suggested that this part of the ligand was buried in a sterically limited pocket, consistent with the predicted binding mode of compound 1. Docking of compounds 1 and 19 suggested that a para substituent clashed with Val84 at the bottom of the orthosteric site (Figure 1A). This would push compound 19 upward toward the extracellular side and reduce favorable hydrogen bond interactions with Asn253. To test the possible benefit of a smaller substituent, the 3-phenyl was replaced with a furyl ring (24), but this led to reduced binding affinity. A possible reason for the loss of activity for compound 24 is that the smaller size of the furyl ring leads to a loss of van der Waals interactions in the orthosteric site. As the initial hit and several analogs were chiral (1–6, 8, 15, 22, 23a–c, 24), the pure enantiomers (23b and 23c) were synthesized for compound 23a. This revealed that the active form of these ligands is the R-form, which is consistent with the docking predictions for both compound 23 and the initial hit in ref (11) (1, Figure 1A).
As the 23 close analogs of compound 1 did not display improved affinity, focus was placed on exploring more distantly related molecules. In this step, molecular docking calculations with DOCK3.616−18 were used to guide selection of molecules from commercially available libraries. Docking of compounds 1–24 to the hA2AAR crystal structure revealed that the phenoxy group was solvent exposed in several cases, and this conclusion was also supported by the affinity of several analogs being unaffected by substitutions of the phenyl ring. Based on these observations, it appeared likely that the phenoxy group did not contribute significantly to binding. To test this hypothesis, we explored compounds where the phenoxy group was replaced by a series of small aliphatic substituents (25–28). The ethyl- and propyl-substituted compounds displayed the same levels of affinity as the close analogs of compound 1, confirming our prediction that the phenoxy group was not essential for binding (Table 2). The propyl substituted compound (27) displayed a Ki of 1.3 μM at the A2AAR, which was equipotent to compound 1 (Figure 1B). However, it should be noted that 27 was significantly smaller than compound 1, making it a more promising lead structure. The ligand efficiency (LE)19 of compound 27, calculated as its free energy of binding divided by the number of heavy atoms of the molecule, was 0.48, which put this fragment-sized compound in a promising range for further optimization.20 In comparison, compound 1 displayed essentially the same affinity, but had a ligand efficiency of only 0.34 per atom.
Table 2. Binding Affinities of a Series of 1,2,4-Triazole Derivatives at the Human A1, A2A, and A3ARs Measured in Radioligand Binding Assays and Percent Inhibition of cAMP Accumulation in the Presence of 300 nM NECA in a Functional Assay for the A2BAR.

|
Ki (μM) or %
inhibition at 10 μMa |
|||||
|---|---|---|---|---|---|
| compd | R | A1 | A2A | A3 | A2B |
| 25 | CH3 | 18% | 43% | 0.3 ± 0.01 | 10% |
| 26 | CH2CH3 | 39% | 2.5 ± 0.6 | 0.3 ± 0.2 | 8% |
| 27 | (CH2)2CH3 | 0.8 ± 0.2 | 1.1 ± 0.2 | 0.2 ± 0.04 | 5% |
| 28 | 3.2 ± 0.7 | 5.9 ± 0.5 | 3.1 ± 0.7 | –7% | |
| X | |||||
| 29 | H | 1.9 ± 0.3 | 0.4 ± 0.02 | 0.3 ± 0.2 | 72% |
| 30 | 4-CH3 | 1.3 ± 0.4 | 0.3 ± 0.1 | 0.1 ± 0.04 | 29% |
| 31 | 4-Cl | 31% | 2.8 ± 0.8 | 0.2 ± 0.1 | –2% |
| 32 | 2-OCH3 | 0.5 ± 0.1 | 0.2 ± 0.06 | 0.6 ± 0.2 | 16% |
| 33 | 22% | 0.2 ± 0.04 | 0.3 ± 0.2 | 10% | |
| 34 | 29% | 1.0 ± 0.2 | 1.0 ± 0.3 | –6% | |
Measured in three independent experiments.
To explore the possibility to further improve affinity while retaining relatively high ligand efficiency, another series of commercially available analogues was docked to the hA2AAR orthosteric binding site. A set of compounds with substituted phenyl rings that docked in the same overall binding mode as the other ligands was identified (Table 2, 29–34). Compared to compound 1, the molecules had a more compact structure and did not extend as far toward the ELs. Compound 29 displayed a Ki of 0.4 μM, a 3-fold improvement compared to 27. This compound also retained a good LE of 0.44. A series of substitutions at the benzamide ring was explored, and the 2-methoxy substituted analog (32) led to another 2-fold improvement of affinity to a Ki of 200 nM (Figure 1C). Interestingly, replacing the amide of 32 with a urea group resulted in compound 33, which also displayed a Ki of 200 nM at the A2AAR (Figure 1D).
In parallel to our efforts to identify a potent 1,2,4-triazole antagonist of the hA2AAR, the 34 compounds were also screened at the hA1, A3, and A2BAR subtypes. Compound 1 was quite selective for the hA2A and A3AR with only 19% inhibition of A1AR radioligand binding at 10 μM. The functional assays carried out for the A2BAR subtype showed no significant activity for compound 1 or any of the close analogs (Table 1). An unusual property of compounds 1–24 was that most of them bound with affinities in the 1–5 μM range at the A2A and A3AR subtypes, but 14 of them had ≤50% inhibition of the A1AR at 10 μM. Based on sequence identity in the binding pocket, one would expect similar affinities for the A1 and A2A subtypes; the binding sites of the A1 and A2A subtypes differ by only a few residues in the orthosteric site, while 10 out of 20 binding cavity residues are unique to the A3 receptor.21 Based on a comparison of the A2AAR crystal structure to models of the A1 and A3 subtypes, we identified that Leu167 in EL2 (Glu and Gln in A1 and A3, respectively) and Met270 in helix 7 (Thr and Val in A1 and A3, respectively) were likely responsible for the observed A2A and A1 selectivity. The 1,2,4-triazole series was predicted to mainly have nonpolar interactions with Leu167 and Met270 in the A2AAR, and in both cases, the most polar residue in this position is found in the A1 subtype, which may reduce binding to this receptor (Figure S1, Supporting Information). As the size of the ligands was reduced, affinities typically increased at all subtypes for several of the most potent compounds (e.g., compounds 27, 30, and 32). To further improve A2AAR selectivity, it is likely necessary to increase the compound size and extend substituents further toward nonconserved residues in the outer regions of the orthosteric site.
Subsequent to the testing of the 34 compounds described here, several new high-resolution structures of the A2AAR have been reported.13 Interestingly, two of these are cocrystallized with the same antagonist, but the orientations of the ligand and residues in the ELs differ. Since the docking screen was carried out against a rigid receptor structure, the structural reorganization at the opening of the orthosteric site could significantly affect the predicted binding modes of the ligands. For this reason, representative ligands in the series were docked to an alternative crystal structure of the A2AAR (PDB accession code 3pwh(13)) to further investigate the binding mode of the 1,2,4-triazole antagonists. Docking to the first hA2AAR crystal structure (PDB accession code 3eml(14)) favored a conformation where the 1,2,4-triazole and amide nitrogens hydrogen bonded to Asn253 and Glu169, respectively (Figure 1A). In the alternative antagonist-bound crystal structure (PDB accession code 3pwh(13)), a hydrogen bond between the side chains of Glu169 and His264 was broken, which opened a hydrophobic pocket (Figure 1E). The predicted binding modes for three representative compounds to the alternative crystal structure are shown in Figure 1F–H. Compounds 1 and 32 were predicted to bind in the same overall binding mode in both structures, but compound 27 docked in an alternative conformation. In the case of compound 27, the amide nitrogen interacts directly with Asn253 instead of Glu169, and an internal hydrogen bond was formed between the triazole ring and the amide carbonyl (Figure 1G). This binding mode was not accessible in the crystal structure used in the docking screen, because the ligands would clash with Glu169 in EL2. To test if this second binding mode was also energetically favored for 1 and 32, docking calculations with restricted conformational sampling parameters were carried out for these two compounds. Both compounds 1 and 32 did fit in the alternative conformation (Figure 2). However, the docking energy of the alternative conformation was less favorable by more than 7 kcal/mol, in support of the first binding mode. The docking energies also tend to favor the first crystal structure (PDB accession code 3eml(9)) because of the strong electrostatic interaction energy between the ligands and Glu169. However, because the internal energy contribution of a receptor is notoriously difficult to estimate and is not taken into account in the docking energy calculations, the relative free energy of binding for the two conformations could not be calculated accurately. Thus, it was not possible to conclude if only one, or both, receptor conformations were accessible for the 1,2,4-triazole series. In the case of the most potent 1,2,4-triazole ligand, compound 32, both predicted binding modes appeared reasonable. In the first structure, the methoxy-substituent potentially could form hydrogen bonds with two backbone nitrogens in EL2 (Figure 1C), and in the alternative structure (Figure 1H), the same group was buried in a hydrophobic pocket created by the conformational reorganization. For this reason, it is likely advantageous to use an ensemble of crystal structures in lead optimization to identify the receptor conformation(s) that are most relevant for a given ligand of interest.
Figure 2.
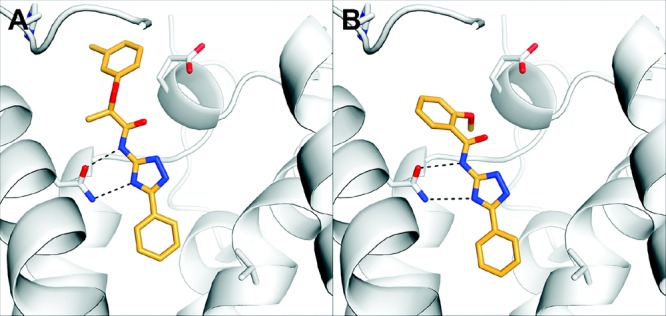
Alternative binding modes of compounds (A) 1 and (B) 32 to the A2AAR crystal structure with PDB accession code 3pwh. The A2AAR binding site is shown in white ribbons with selected side chains shown in sticks. Ligands are depicted with orange carbon atoms. Black dotted lines indicate hydrogen bonds.
The present study explores the SAR for a novel class of 1,2,4-triazole antagonists. None of the close analogs of the initial hit, 1, displayed improved potency at the hA2AAR, but molecular docking calculations were used here to interpret the SAR and guide the selection of more distantly related compounds for experimental testing. This led to the discovery of compound 32, with a Ki of 200 nM and a more favorable ligand efficiency of 0.42. The molecular docking calculations highlighted the need to consider several receptor conformations in lead optimization, which will help to guide further development of the 1,2,4-triazole series.
Glossary
Abbreviations
- AR
adenosine receptor
- CNS
Central Nervous System
- EL
extracellular loop
- GPCR
G protein-coupled Receptor
- LE
ligand efficiency
- SAR
Structure–Activity Relationship
Supporting Information Available
Descriptions of the molecular modeling and experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
J.C. performed the molecular modeling studies. D.K.T., K.P., and Z.-G.G. carried out the experiments. The manuscript was written by J.C. and K.A.J.
Supported by the NIDDK Intramural Res. Program (to K.A.J.) and the Knut and Alice Wallenberg Foundation (to J.C.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fredholm B. B.; IJzerman A. P.; Jacobson K. A.; Linden J.; Müller C. E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors-An Update. Pharmacol. Rev. 2011, 63, 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn M. R.; Vance C. O.; Morschl E.; Wilson C. N. Adenosine receptors and inflammation. Handb. Exp. Pharmacol. 2009, 215–69. [DOI] [PubMed] [Google Scholar]
- Sebastiao A. M.; Ribeiro J. A. Adenosine receptors and the central nervous system. Handb. Exp. Pharmacol. 2009, 471–534. [DOI] [PubMed] [Google Scholar]
- Jacobson K. A.; Gao Z. G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discovery 2006, 5, 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller C. E.; Jacobson K. A. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 1290–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi S.; Tikhonova I. G.; Harden T. K.; Jacobson K. A. Ligand and structure-based methodologies for the prediction of the activity of G protein-coupled receptor ligands. J. Comput.-Aided Mol. Des. 2009, 23, 747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst E.; van der Pijl R.; Mulder-Krieger T.; Bender A.; IJzerman A. P. Substructure-Based Virtual Screening for Adenosine A2A Receptor Ligands. ChemMedChem 2011, 6, 2302–2311. [DOI] [PubMed] [Google Scholar]
- Cristalli G.; Muller C. E.; Volpini R. Recent developments in adenosine A2A receptor ligands. Handb. Exp. Pharmacol. 2009, 59–98. [DOI] [PubMed] [Google Scholar]
- Jaakola V. P.; Griffith M. T.; Hanson M. A.; Cherezov V.; Chien E. Y. T.; Lane J. R.; IJzerman A. P.; Stevens R. C. The 2.6 Angstrom Crystal Structure of a Human A2A Adenosine Receptor Bound to an Antagonist. Science 2008, 322, 1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen D. B.; Decornez H.; Furr J. R.; Bajorath J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discovery 2004, 3, 935–949. [DOI] [PubMed] [Google Scholar]
- Carlsson J.; Yoo L.; Gao Z. G.; Irwin J. J.; Shoichet B. K.; Jacobson K. A. Structure-Based Discovery of A2A Adenosine Receptor Ligands. J. Med. Chem. 2010, 53, 3748–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V.; Jaakola V. P.; Lane J. R.; Lin J.; IJzerman A. P.; Yeager M.; Kufareva I.; Stevens R. C.; Abagyan R. Structure-Based Discovery of Novel Chemotypes for Adenosine A2A Receptor Antagonists. J. Med. Chem. 2010, 53, 1799–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doré A. S.; Robertson N.; Errey J. C.; Ng I.; Hollenstein K.; Tehan B.; Hurrell E.; Bennett K.; Congreve M.; Magnani F.; Tate C. G.; Weir M.; Marshall F. H. Structure of the Adenosine A2A Receptor in Complex with ZM241385 and the Xanthines XAC and Caffeine. Structure 2011, 19, 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakola V. P.; Lane J. R.; Lin J. Y.; Katritch V.; IJzerman A. P.; Stevens R. C. Ligand Binding and Subtype Selectivity of the Human A2A Adenosine Receptor, Identification and Characterization of Essential Amino Acid Residues. J. Biol. Chem. 2010, 285, 13032–13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. H.; Jiang Q. L.; Glashofer M.; Yehle S.; Wess J.; Jacobson K. A. Glutamate residues in the second extracellular loop of the human A2A adenosine receptor are required for ligand recognition. Mol. Pharmacol. 1996, 49, 683–691. [PMC free article] [PubMed] [Google Scholar]
- Lorber D. M.; Shoichet B. K. Flexible ligand docking using conformational ensembles. Protein Sci. 1998, 7, 938–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorber D. M.; Shoichet B. K. Hierarchical docking of databases of multiple ligand conformations. Curr. Top. Med. Chem. 2005, 5, 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K.; Mysinger M. M.; Huang N.; Colizzi F.; Wassam P.; Cao Y. Q. Automated Docking Screens: A Feasibility Study. J. Med. Chem. 2009, 52, 5712–5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz I. D.; Chen K.; Sharp K. A.; Kollman P. A. The maximal affinity of ligands. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 9997–10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Katritch V.; Kufareva I.; Abagyan R. Structure based prediction of subtype-selectivity for adenosine receptor antagonists. Neuropharmacology 2011, 60, 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.