

Abstract
The phenomenological diagnosis of depression is successful in increasing diagnostic reliability, but it is a classification scheme without biological bases. One subtype of depression for which evidence suggests a unique biological basis is late-life depression (LLD), with first onset of symptoms after the age of 65. LLD is common and poses a significant burden on affected individuals, caretakers, and society. The pathophysiology of LLD includes disruptions of the neural network underlying mood, which can be conceptualized as the result of dysfunction in multiple underlying biological processes. Here, we briefly review current LLD hypotheses and then describe the characteristics of molecular brain aging and their overlap with disease processes. Further, we propose a new hypothesis for LLD, the Age-by-Disease Interaction hypothesis, which posits that the clinical presentation of LLD is the integrated output of specific biological processes that are pushed in LLD-promoting directions by changes in gene expression naturally occurring during brain aging, hence leading the brain to a physiological state that is more susceptible to LLD, since additional pushes by genetic, environmental and biochemical factors may now be sufficient to generate dysfunctional states that produce depressive symptoms. We put our propositions together into a decanalization model to aid in illustrating how age-related biological changes of the brain can shift the repertoire of available functional states in a pro-depression direction, and how additional factors can readily lead the system into distinct and stable maladaptive phenotypes, including LLD. This model brings together basic research on neuropsychiatric and neurodegenerative diseases more closely with the investigation of normal aging. Specifically, identifying biological processes affected during normal aging may inform the development of new interventions for the prevention and treatment of LLD.
Keywords: late-life depression, aging, depression, geriatric, canalization, decanalization
Depression, a complex biological disease
A common and debilitating disease
According to the diagnostic and statistical manual of mental disorders, fourth edition, text revision (DSM-IV-TR), a diagnosis of Major Depressive Disorder (MDD) requires an individual to experience low mood or anhedonia plus five symptoms from a list that includes change in sleep, feelings of guilt or worthlessness, anergia, poor concentration, change in appetite, psychomotor retardation, and thoughts of death or suicide.1 MDD is a common mental illness that occurs across the lifespan, it is estimated to affect 10% to 15% of the general population in their lifetime2 and is responsible for significant disability worldwide.3 “Depression” is a more general term used to refer to depressive symptoms that cause distress and functional impairment, irrespective of whether the symptoms meet the DSM-IV-TR criteria for MDD, this is how we will use the term in this article. Given that depression is a common cause of disability, the etiology of depression is a major focus of research.
A neural network model of depression
Convergent results from disparate areas of inquiry point to a model in which the signs and symptoms of depression result from disruption of the neural network underlying mood. Multiple network models of depression have been proposed based on structural and functional data from individuals with depression.4-6 Though the models differ with respect to specifics, they all generally implicate a similar subset of brain areas in the cortical and limbic systems. The prefrontal cortex, cingulate cortex, amygdala, hippocampus, and hypothalamus are some of the areas thought to be of critical importance. Evidence suggests that each of these brain areas can serve as functional nodes,7 are necessary to the processing of emotions, and can be impaired in depression; hence are referred to here as a “neural network underlying mood” or “mood network”. Specifically, the prefrontal and cingulate cortices are implicated in the control and regulation of subcortical limbic brain regions, including the amygdala, hippocampus and hypothalamus8, and together are associated with emotional expression and experience.9 In a simplified model, it is thought that, in individuals with depression, the normal top-down inhibitory control and regulation that is exerted by these cortical areas upon the limbic areas may break down as the result of cortical hypofunction, limbic hyperactivity, or a combination of both.
Multiple biological processes contribute to depression
Neural network disruptions reflect the dysfunction of neurons and glial cells within respective areas that compose the mood network, which in turn are supported by various molecular dysfunctions. At the molecular level, there is compelling evidence for the involvement of various biological processes including, but not limited to: altered monoaminergic neurotransmission, altered stress hormone homeostasis, reduced neurotrophic support, metabolic dysregulation, inflammation, oxidative stress responses, mitochondrial function, as well as other aspects of brain plasticity, synaptic function, and calcium regulation.10 These biological processes interact at the molecular level, are modulated by genetic variants, and impacted by environmental conditions, together leading to neuronal and glial dysfunction, mood network disruption, and depressive symptoms. Hence, the biological underpinnings of depression may be varied and heterogeneous. Later in this article, we will propose that depressive symptoms may often arise for the first time in late life because of how these multiple biological processes are selectively affected, or “pushed” in a disease direction, by the complex phenomenon of aging.
Late-life depression: definition, extent, burden and hope
Among individuals 65 years of age or older, approximately 1% meet criteria of MDD, a prevalence much lower than found in younger individuals.11 However, approximately 15-25% of individuals over the age of 65 experience depressive symptoms that, while not meeting criteria for MDD, do cause significant distress and interfere with daily functioning.12 This discrepancy between formal diagnosis of MDD and clinically significant depressive symptoms likely reflects the tendency of older individuals to under-report psychiatric symptoms like those required for the DSM-IV-TR diagnosis of MDD, such as the predominance of vegetative and somatic symptoms as part of the clinical presentation of depression in older individuals, the inability of some older individuals to express depressive symptoms secondary to cognitive impairment,13 and the possibility that depression in older individuals represents its own disease entity with unique clinical presentation and pathophysiology. For the remainder of this article, the term late-life depression (LLD) will be used to refer to individuals over the age of 65 who for the first time in their lives meet criteria for MDD or experience clinically significant depressive symptoms.
The burden of LLD on the individual with the disease is significant. Individuals with LLD experience greater functional disability14 and cognitive decline15 than those without. Further, they are at increased risk of morbidity and mortality from medical illness,16 a phenomenon most likely attributable to a combination of both maladaptive health risk behaviors as well as physiologic effects of LLD. Interestingly, emerging evidence suggests that the association between medical illness and depression is bidirectional, that is, not only does depression magnify the negative consequences of medical illness, but medical illness negatively affects the course of depression;17-18 accordingly depression has been proposed as a disease of accelerated aging.19 In addition to amplifying the rates of morbidity and mortality from medical illness, LLD appears to lead to increased rates of suicide among older individuals.20 Among individuals 75 years old or older, 60% to 75% of individuals committing suicide had diagnosable depression.12
LLD can be effectively treated using pharmacotherapy, psychotherapy or both,21 and response and remission rates are comparable to those in individuals with mid-life depression.22 Successful diagnosis and treatment of LLD improves depressive symptoms and decreases suicide rates.23 Despite the demonstrated effectiveness of treatment, many obstacles remain. For example, only 40-50% of older adults respond to the first prescribed antidepressant medication.24-25 In those that do respond, response is often slow, sometimes taking up to 4 months26 and once response is achieved, relapse and recurrence are common. Approximately 60% of community-dwelling older adults with MDD who initially responded to antidepressant treatment became depressed again within 2 years unless they were maintained on antidepressant pharmacotherapy. Multiple factors including coexisting anxiety, low self-esteem, poor sleep, and coexisting medical burden have been identified that predict more difficult-to-treat depression.27
With the hope that a better understanding of the biology underlying LLD would facilitate the development of improved treatments and outcomes for those with LLD, the biological substrates of LLD are beginning to be characterized. Hence, several hypotheses for the etiology and pathophysiology of LLD have now been proposed.
Current hypotheses for biological mechanisms promoting LLD
Three influential hypotheses for mechanisms recruited in LLD are the vascular hypothesis,28inflammation hypothesis,29 and dementia prodrome hypothesis.30 These hypotheses are not mutually exclusive and data exists to support aspects of all three. Moreover, all three hypotheses are consistent with a neural network model of depression described above, as well as the idea of multi-process biological vulnerability, which we will elaborate on later in this article.
The vascular depression hypothesis is based on the observation of an unexpectedly high degree of co-morbidity between vascular disease, vascular disease risk factors, and ischemic brain lesions with depressive symptoms.28 Despite this observation being over a century old,31 the nature of the relationship remains unclear. That is, does vascular disease cause depression, depression cause vascular disease, or do both vascular disease and depression share a common etiology? Though there is some evidence for each of these scenarios, the vascular depression hypothesis assumes the former and posits that vascular disease may predispose, precipitate, or perpetuate LLD.28 One way vascular disease might lead an individual to develop LLD is by causing brain lesions that directly disrupt the neural network underlying mood and thus the capacity to regulate mood, and subsequently depressive symptoms. Though data supporting a causal link between vascular disease and LLD is difficult to obtain and thus lacking, correlative data supporting the vascular depression hypothesis include MRI studies of the brain of individuals with LLD showing increased rates of white-matter hyperintensities compared to non-depressed age-matched controls32-33 and the finding that these white-matter hyperintensities correlate with functional changes in the mood neural network.34
The inflammatory hypothesis proposes that aging- and disease-related processes result in a pro-inflammatory state that contributes to the etiology of LLD in a subset of individuals. It proposes that the pro-inflammatory state leads to changes in brain areas included in the neural network underlying mood, thus predisposing aging individuals to developing LLD.29 The inflammatory hypothesis is supported by a number of experimental observations. For example, aging leads to an exaggerated and prolonged inflammatory response in the brain, and such dysregulation of inflammatory responses leads to emotional and cognitive changes reminiscent of LLD.35-37 Further, antidepressants have been shown to reduce inflammatory markers38-40 and some anti-inflammatory agents appear to have antidepressant properties.41-43
The dementia prodrome hypothesis30 is based on the observation that older adults with depressive symptoms have a much greater risk of developing dementia upon follow-up compared to those without depressive symptoms.44-45 Multiple, non-mutually exclusive etiological causes have been proposed to explain this association between depression and dementia46 and, generally, they all propose that the processes that play an etiological role in disrupting the neural circuitry underlying cognitive function and thus lead to dementia, also disrupt the neural network underlying mood. One brain area that overlaps between the mood and cognitive neural networks that may have special importance for this hypothesis is the hippocampus. There is evidence that hippocampal atrophy, in addition to being a classic feature of many dementias and aging itself, confers vulnerability to depression.47-48 Some of the processes with presumed etiological roles could be subsumed under the vascular or inflammatory hypotheses, but others such as amyloid plaque formation49-50 are unique to this hypothesis. Interestingly, hippocampal regions have been found to be especially vulnerable to ischemia51 and to activation of the hypothalamic-pituitary-adrenal (HPA) axis, resulting from stress and chronic medical illness.52 This latter observation may be of particular relevance to the etiology of LLD relative to early-onset depression, given the well-known role of stress and HPA axis dysfunction (i.e., glucocorticoid hypersecretion) on neurotoxicity of the hippocampus and the increased length and probability of exposure to stress with advancing age.53-54 So extended disease- and/or age-related exposure to elevated glucocorticoid levels could contribute to a series of physiological changes akin to an accelerated aging process.55 Interestingly, the hippocampus is the not the only node in the mood network that undergoes structural remodeling as a result of stress-related HPA axis dysfunction. The prefrontal cortex atrophies and the amygdala increases in size during acute stress and atrophies during extended periods of stress, thus suggesting multiple routes by which stress can disrupt the mood network.56 One interpretation of the dementia prodrome hypothesis is that the neural networks underlying mood partially overlaps with areas subserving cognition, which are susceptible to subtle and/or cumulative disruption.46
The neural network model of depression and LLD
Central to all three of the above hypotheses and consistent with the putative mechanisms of each, is the concept that LLD, as discussed above with regard to depression generally, is also a disorder of mood network disruption. The clinical presentation of depression can differ dramatically among individuals, and early-onset depression and LLD consistently present differently. Using the DSM-IV-TR criteria alone, there are 227 possible symptom constellations that would warrant a diagnosis of MDD1, but only a small percentage of LLD meets this diagnostic criteria. Such heterogeneity in the presentation of depression may be explained by the nature and location of the disruption in the neural network underlying mood, as these factors would determine how the mood network may engage adaptive mechanisms and process stimuli. And, though all disruptions would conceivably disrupt the neural networks underlying mood, the unique effects of each disruption could lead to a distinct clinical picture thus providing a biological explanation for the clinical heterogeneity of depression, including variable symptomatic presentations and treatment response. Given what we know about the differences between early-onset depression and LLD (e.g., family history, neurological comorbidity), somewhat different processes would be expected to converge and give rise to mood network dysfunction and thus clinical depression. That is, the observation that LLD frequently presents clinically in a very different way from early-onset depression may be related to the fact that processes that disrupt the circuitry in older adults have a particular mechanism or anatomical propensity that is different from those that disrupt the circuitry in early-life depression, for example, the subcortical predominance of vascular lesions in older individuals or the particular vulnerability of the hippocampus in older individuals to ischemic damage and structural remodeling associated with stress.
Molecular brain aging: an etiological role in LLD?
At the gene level, a subset of genes is observed to have age-related and lifelong progressive changes in expression (at least from a cross-sectional point of view), which is selective to specific cellular functions. This observation is consistent with the idea that aging per se, like depression, reflects the integrated output of multiple and specific biological processes (described below). So, the presentation of depression on the backdrop of biological aging raises the question “How does biological aging contribute to LLD and other age-gated diseases?” Our group has begun to address this question by systematically identifying and quantifying molecular changes in the brain during aging, and by investigating the extent of overlap between age- and disease-related processes. As described below, results from these studies demonstrate that age-related gene changes are selective and greatly overlap with biological processes investigated in the context of multiple neurodegenerative and neuropsychiatric disorders. Moreover age-related changes are overwhelmingly in pro-disease directions (Table 1). These observations have led us to propose that biological aging of the brain may in fact promote age-gated diseases, including LLD, by contributing to selective disruptions of specific neuronal and glial processes, each with putative etiological roles in disease-related biological processes, including mood network disruption.57-58
Table 1.
A number of genes display similar changes in expression levels between aging and multiple neurodegenerative and neuropsychiatric disorders, including depression.
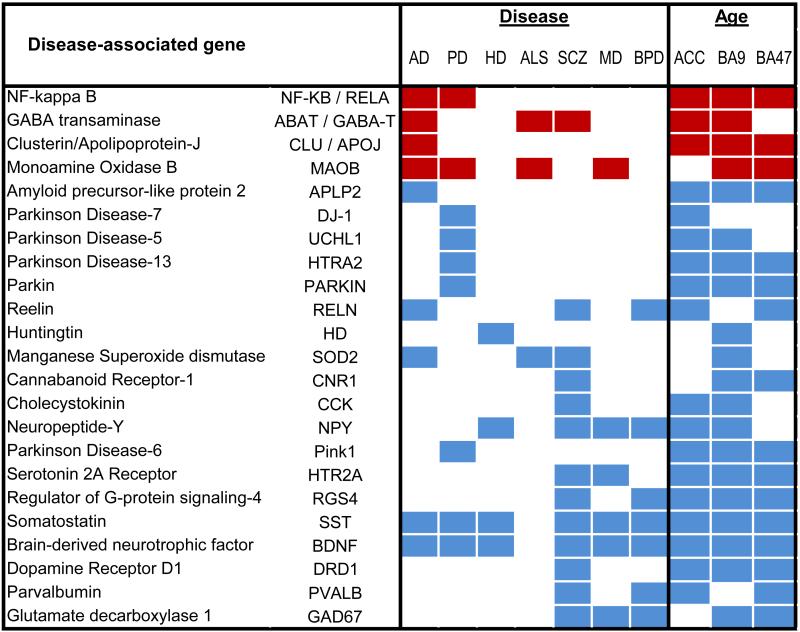
|
Red, increased expression; Blue, decreased expression; AD, Alzheimer's disease; PD, Parkinson's disease; HD, Huntington's disease, ALS, amyotrophic lateral sclerosis; SCZ, schizophrenia; MD, major depression; BPD, bipolar depression. Adapted from Glorioso et al.57
“Molecular aging” of the human brain
It has been known for some time that robust changes in gene expression occur with aging.59 The fact that age-related changes in gene expression extend to the brain may not be surprising given the body of knowledge about changes in structure and function of the brain with age. In light of the many changes seen with aging, one might hypothesize that age-related changes in gene expression reflect a general deterioration of the brain and thus a preponderance of genes would be affected. However, what is emerging from recent genome-wide studies is that the changes in gene expression with aging affect a relatively smaller number of genes than one might have expected. Studies in rodent, monkey and human brains estimate the number of genes exhibiting age-related changes to represent less than 10%, and commonly less than 5%, of the entire genome.60-66 In a study from our group, the age-related changes of a large number of genes using microarray technology were profiled in prefrontal cortex samples from human subjects aged 13-79 years.66 The data from this study identified life-long progressive changes in expression with age of up to 7.5% of the genes tested. This restricted scope of transcript changes suggests that specific cellular populations and biological processes are selectively vulnerable during aging. Interestingly, the set of age-dependent genes in our group's study was very similar to those observed in other studies,57,60-62,64,66-68 and in fact displayed high degree of conservation across cohorts and cortical brain regions.58 Expression of genes playing a role in glial-mediated inflammation, oxidative stress responses, mitochondrial function, synaptic function and plasticity, and calcium regulation have now consistently been shown across multiple studies to be affected by aging, despite differences in sample size, selection of tissue type or brain region, expression analysis platforms, and analytical methods.57,60-62,64,66-68 Of note, not only is the identity of the genes and gene classes that are affected with aging consistent among studies but so are the directions of change. Overall, age-upregulated genes are mostly of glial origin and related to inflammation and cellular defenses, while downregulated genes display mostly neuron-enriched transcripts relating to cellular communication and signaling.66 This consistency and specificity of age-related changes in fact fulfills criteria for biomarkers. Indeed, we have shown that the predicted age for a particular individual, based on regression analysis of expected age-related trajectories for those age-biomarkers (denoted as “molecular age”), is highly correlated with the chronological age of that individual.57,66
Molecular aging of the brain interacts with neuropsychiatric and neurodegenerative disease pathways
Studies of the molecular basis of neurodegenerative and neuropsychiatric disorders often describe age-related changes in genes of interest. For instance, we have reported that the variance in depression-related decrease in the neuropeptide somatostatin (SST) can be mostly explained by superimposed age-related changes58,69 (Figure 1A). Indeed, consistent with prior findings,66 normal subjects lose ~40-50% of SST expression between the age of 20 and 70 years, while subjects affected with depression appear to be on an early trajectory of age-related decline 69 (Figure 1A, top panel), and similar findings have been reported in schizophrenia70 (Figure 1A, bottom panel). Central to our proposed model for age-by-disease interaction, it is not known whether these anticipated changes reflect a disease effect or an early age-decline, which would have placed the system at increased vulnerability for depression.
Figure 1.

Many genes, such as somatostatin, show similar age- and disease-related changes in gene expression, leading to our model of how age-related changes in expression of disease-related genes could influence age of onset of disease. (A) Somatostatin expression decreases with age, and individuals with major depression (top panel) and schizophrenia (bottom panel) display lower levels of expression than control subjects. We hypothesize that decreased expression of somatostatin in psychiatric disorders may represent an early brain- and age-related molecular phenotype in these individuals, which render these subjects vulnerable to developing psychiatric diseases. (B) In a model derived from the proposed Age-by-Disease Interaction hypothesis, change in expression of disease-related genes (a decrease is shown here) across a threshold (horizontal red line) marks the onset of disease symptoms. Changes in the trajectory of age-related changes in expression of disease-related genes (Y-axis) determine the age (X-axis), or even if, an individual develops disease symptoms (vertical red arrows). Per this model, modulators (blue arrows), genetic or environmental, place subjects on an “at risk” or protected trajectory for developing mood symptoms and LLD. Adapted from Morris et al,70 Tripp et al69 and Glorioso et al.57-58
Expanding these observation to genome-wide investigations, we have reported that up to a third of age-regulated genes (>800-1000 genes) in the human brain have been otherwise associated with neurodegenerative (Alzheimer's, Parkinson's and Huntington's diseases, amyotrophic lateral sclerosis) and neuropsychiatric disorders (bipolar depression, major depression and schizophrenia).58 Not only do the genes relevant to these brain diseases show age-related changes, but the direction of the changes that occur with age is almost always in the direction thought to cause or promote diseases58 (Table 1), as exemplified by SST age- and disease-related changes shown here (Figure 1A). It is important to note that such normal age effects can go unnoticed in age-matched paired sample design, which may incorrectly suggest that changes in gene expression in depression, Alzheimer's disease, or other age-gated diseases are separate from what would be seen in normal aging. Conversely, very few (<5%) of the larger pool of genes that do not display age-dependent changes, are otherwise associated with neurodegenerative and neuropsychiatric diseases.58 Together, these data suggest that, at the single gene level, aging may promote selective changes in gene expression in ways that promote diseases. Collectively, the large number of disease-related genes affected during aging in pro-disease directions also suggest that system-level adaptations occur with age, such an age-by-disease interaction leads towards physiological states that are in fact closer to disease states than at younger ages (See Douillard-Guilloux et al71 in this issue).
Genetic determinants of the age-by-disease interaction may modulate the vulnerability to develop age-related diseases: a correlative proof of concept
The nature of external and internal events driving age-related transcript changes are mostly unknown, and the underlying “molecular clock” is somewhat of a holy grail in aging research. Interestingly, genes forming the “molecular aging” profile include numerous transcriptional regulators,57 and an individual's molecular age can deviate from its predicted trajectory, together suggesting that modulating factors may contribute, not only to age-related changes, but to their intrinsic variability. Hence, we have proposed that those individuals with older predicted molecular ages compared to their chronological age may not only display greater biological brain aging, but may also be at greater risk of age-gated brain diseases, because gene expression of disease-related genes would have proceeded further in disease-promoting directions. Conversely, subjects with younger gene trajectories or predicted molecular ages would be at lower risk and may in fact display resiliency against LLD and other late-life disorders (Figure 1B)
Environmental and genetic factors represent obvious candidate modulators of the trajectory of biological aging. In recent years, the identification of single gene mutations affecting aging and longevity in nematodes, insects and rodents has clearly demonstrated the presence of a genetic program underlying aging.72 In the mammalian brain, the course of aging parallels that of peripheral tissues, and additional mechanisms may reflect the specificities of post-mitotic differentiated neurons,73 but one might expect a certain level of conservation in basic molecular mechanisms relating to aging, including their regulation. In fact, using the above-described “molecular age” assay and focusing on a family of genes with phylogenetically-conserved age-modulatory roles (i.e. sirtuins,74), we have reported that subjects carrying a low-expressing polymorphism of the Sirtuin 5 gene had molecular ages that were older than actual chronological age, as measured in the cingulate cortex of human postmortem samples.57 We further showed that this effect was accompanied by expression changes for a set of genes whose products are localized to the mitochondria, including PINK-1 and DJ-1, two Parkinson's disease-associated genes, in ways that would promote mitochondrial dysfunction-related diseases, including the age-gated Parkinson's disease. Confirmation of this putative mechanism will require large-scale assessment of live subjects with Parkinson's disease and normal controls to assess whether either disease onset and severity, or age-related functional declines in the domains of mood, cognition, or motor function are differentially associated with this or other sirtuin 5 gene polymorphisms. Together, this (correlative) proof-of-principle study suggests that factors that affect biological aging of the brain (genetic or environmental) can potentially place an individual at higher risk for disease, through a mechanism by which it accelerates brain molecular aging, and thus promotes changes in expression of disease-relevant genes in disease-causing directions. With respect to Figure 1B, the low-expressing polymorphism of the Sirtuin 5 gene can be thought of a modulator that puts one on the “at risk” trajectory. The converse of this model is that, based on population frequencies, the “risk” allele may in fact be the major allele and thus considered normal, with the other allele conferring protection or resiliency. Hence, the assessment of risk and resiliency may in fact be built in the same model.58 Here the prediction would be that factors delaying age trajectories of gene changes may lead to younger brain molecular aging and potential resiliency towards developing functional declines and age-related disorders, including LLD (Figure 1B).
Altered biological landscape and physiological homeostasis in depression and aging: an Age-by-Disease Interaction hypothesis for LLD
The above observations of (i) parallel age and disease trajectories in gene changes, (ii) anticipated and greater extent of changes in neurodegenerative and neuropsychiatric diseases, (iii) relative paucity of disease association for age-independent genes, and (iv) putative genetic modulation of age-mediated risk, together provide a compelling rationale for investigating aging and brain disorders simultaneously. Indeed, these observations suggest that age-related changes in gene function may in fact promote vulnerability to develop a set of diseases that are otherwise described as age-gated or age-dependent, through progressive changes in gene expression for disease-related genes in directions predicted to promote those diseases. This model departs from the current framework for neurodegenerative and neuropsychiatric disorders in that it places the aging process and age-related changes in gene expression as a putative driving force in the etiology and onset of multiple brain disorders, rather than a mere clinical confounding parameter. How could such a model work?
A gene–cell–neural network feedback loops maintaining physiological homeostasis is affected in depression and aging
The physiological and functional output of any biological system or subsystem represents the vertical integration of events occurring at the levels of molecule, cell, cellular connectivity and communication, and endocrine control, among others. Constant feedback among biological scales contribute to maintaining the physiological output of the system in homeostasis, as illustrated in a simplified interactive feedback loop that incorporate genes, cells and cell assemblies (Figure 2). Modulators of this feedback loop include hormone-like factors that follow constitutive, activity-dependent, or cyclic pattern and most frequently regulate patterns of gene expression in target cells through the activation of signal transduction pathways or by direct activation of nuclear receptors. These long-acting modulators are responsible for inducing and maintaining long-term adaptive shifts in molecular balances and associated changes in biological landscapes.
Figure 2.

Integration of multiple biological scales and maintenance of neural network homeostasis: a simplified interactive gene → cell → neural network → gene feedback loop. The molecular composition (1) of particular neurons, in terms of receptors, signal transduction pathways, channel composition, etc, determine the firing properties of those cells (2). The firing patterns and local connectivity of those individual neurons determine the characteristic activity of a particular brain region (3), which, in concert with related brain areas contributes to neural network-based functional output. The critical component of the feedback loop is the constant internal molecular and cellular adaptation that occurs in response to cellular and neural network activities (4). Notably, molecular adaptations are mediated and modulated by long-acting factors such as metabolic-, stress-, and sex-related hormones and neurotrophic factors (4). The corresponding changes in gene expression and associated protein-mediated functions in turn allow for the long-term cellular adaptations necessary for fine-tuning short-term neuronal firing properties and for adapting neural activity in response to an ever-changing molecular and cellular environment, thus closing the gene → cell → neural network → gene integrative feedback loop. Over time, stable states of dynamic equilibrium are reached, which can however be set at various levels. In turn, these different stable or homeostatic biological states be conceptualized as “regional attractor states” within the broader biological landscape (See Fig.3).
An example of modulator of neural network activity that contributes to such feedback loops is brain-derived neurotrophic factor (BDNF). BDNF is essential for maintaining multiple aspects of neuron structure and function, a role that is mediated by membrane receptors and signal transduction pathways that affect the expression of specific sets of genes. Expression of the BDNF gene and release of BDNF protein are triggered by neuronal activity, but are also under constant constitutive release that follows a circadian pattern.75 It is thought that the variable kinetics of BDNF release are critical to the continuous neuronal adaptations that are required to face an energetic and functional environment that is under constant flux.76 For instance, using human postmortem samples and genetic studies in mice, we recently showed that BDNF levels are reduced in the amygdala of women affected with major depression, and that this reduction correlated with (and likely induced, based on mouse genetic studies) molecular adaptations in specific subsets of GABA-containing inhibitory neurons that target the dendrites of pyramidal neurons, a cellular compartment critical for integrating incoming information-rich signals.77 We have speculated that a state of BDNF-dependent reduced dendritic inhibition may represent a novel stable state over time, although it is maladaptive in that it may mediate increased amygdala reactivity, a neural network endophenotype frequently observed in MDD and thought to underlie the rumination or bias for negative emotions in depressed subjects.78 Supporting our age-by-disease interaction model, BDNF is also robustly downregulated with increasing age, suggesting that similar downstream changes may occur in older subjects (See Douillard-Guilloux et al71 in this issue).
Other relevant hormone-type factors that follow similar complex patterns of programmed and activity-dependent release and that have often been associated with depression-related mechanisms, include metabolic (insulin, thyroid, etc) and, sex and stress steroid hormones. These factors may originate in peripheral organs and/or brain, but notably exert their effects on neurons and other brain cells by controlling and coordinating transcriptional programs through binding and activating nuclear receptors, which directly act as transcription factors for various gene sets. Recently, we have investigated the coordination of gene transcript levels across brain areas (denoted “gene synchrony”), and reported significant changes in gene synchrony between the amygdala and the anterior cingulate cortex in subjects with major depression, compared to psychiatric control subjects.79 The interpretation of these findings was that the transcriptional regulation of those genes had changed between these two areas in the context of depression. Notably, investigating potential upstream mediators, we showed that this state of altered corticolimbic gene synchrony in depression could be explained by the combined dysregulation of several hormone-like factors previously implicated in depression, such as insulin, interleukin-1, thyroid hormone, estradiol and glucocorticoids.79 Based on these results, we have speculated on the presence of a distinct and integrated hormone-like-factor-mediated corticolimbic homeostatic, although maladaptive and pathological, state in major depression.
A decanalization perspective on changing biological landscapes to illustrate the Age-by-Disease Interaction hypothesis of LLD
The “gene → cell → neural network → gene” loop is essential for biological adaptation, so that each system or subsystem reaches an equilibrium state from which it is biologically allowed to wobble within a determined range in order to maintain physiological homeostasis in the face of a fluctuating environment. A useful concept to visualize such homeostasis and associated disturbances is provided by the “canalization” framework (Figure 3). Waddington et al 80-81 first used this conceptualization to model how genetic, environmental or other influences can interact to affect the course of development of normative phenotypes and how “decanalization” can lead to distinct and stable alternative phenotypes. In this model, the normal function of a biological system or subsystem over time is represented by a ball in the canal of the model surface, with depth and slope of the canal banks representing the magnitude of the constraining forces on normal variability (Figure 3, top left panel). “Canalizing” influences maintain progression towards the normative canal. Disruptive or “decanalizing” influences can be of two forms: temporary or structural. Temporary decanalizing effects lead the system away from the normative canal for a short duration, as the system will naturally come back to a stable state within the normative canal. Structural decanalizing effects on the other hand affect the shape of the model surface such that new canals representing alternative stable phenotypes may be formed and/or the slope of the banks of the canals representing barriers of entry into or exit from the canals may change. Indeed, a central feature of this concept is that the canalization landscape is not rigid. Development, disease processes, and aging (as we propose here) affect the topography of the landscape. Here, we define decanalizing influences, as events that induce structural changes away from a normative, optimal, and adaptive landscape profile. For instance, we can conceptualize the function of the corticolimbic neural network underlying mood as the biological system in question, where the “gene → cell → neural network → gene” loop described above allows the system to move in the canalization model within a delineated range of variable states (Figure 3, top left panel), corresponding to the natural propensity to adapt and experience states of normal, high, or low mood. In other words, the canalization landscape provides a defined repertoire of physiological/emotional states, and the depth of canals and slope of their banks represent the propensity of the individual to reside or experience the respective states.
Figure 3.

A canalization-decanalization representation of the Age-by-Disease Interaction hypothesis of LLD, and its putative effect on homeostatic states of a neural network underlying mood. This model allows for the visualization of how canalizing influences can interact to maintain normative biology, e.g. mood network function, and how decanalizing influences can lead to stable alternative, but sometimes maladaptive functioning, as for mood network dysregulation in clinical depression. Here, the normal variability over time in the functional state of the neural network underlying mood is represented by a ball, which can circulate along the canals of a model surface (or biological landscape), representing the repertoire of allowed physiological states (top left panel). Decanalizing influences lead the system away from the normative canal or affect the shape of the model surface such that canals representing alternative stable phenotypes are either made easier to enter into or formed de novo. In depression (bottom panel), decanalizing influences change the topography of the model surface such that it is easier for the “ball” to enter into existing or new canals representing alternative stable, but maladaptive, functioning of the neural network underlying mood and remain in those canals. Just as development and diseases can modify the topography or landscape of the model surface, so too does aging (top right panel). Because the biological processes disrupted in aging overlap to a significant degree with those recruited in a number of brain diseases including LLD, the areas of the model surface that change with aging are more likely to be those corresponding to disease states (red section of lansdscape). The result is that aging changes the topography of the brain biological landscape, leading to a shift in the repertoire of available physiological states in a pro-disease or LLD direction.
Effect of depression on the canalization landscape
As described earlier, a multi-system deregulation is likely to occur in depression, and variable sets of stable biological disturbances may characterize individual subjects, despite similarities in clinical presentation. Intrinsic to the canalization model is the notion that the underlying architecture of the landscape is determined and maintained by the molecular and cellular composition of the system, and that minute homeostatic changes in “gene → cell → neural network → gene” feedback loops cooperate to affect and mold the shape of the landscape, hence changing the biological landscape and associated repertoire of allowed functional states. In other words, biological changes occurring in the context of diseases (e.g., low BDNF and altered GABA inhibition, altered corticolimbic gene synchrony, etc) have long-term effects on the structure of the biological landscape, leading to altered constraints on adaptive movements within that landscape. In disease, strong attractor states are thus created by increasing the slope of the banks of existing or new canals, resulting in higher propensity to enter into and remain in those states (Figure 3, bottom panel). For instance, the core symptomatology of depression points to a critical deficit in mood regulation, specifically a high propensity for entering low mood states and a reduced ability to experience positive emotions, supporting the presence of a novel attractor state in an area of the canalization landscape corresponding to a low mood state (i.e., deep landscape groove in the bottom panel of Figure 3). As depression is often characterized by life-long chronicity and increased severity, this modified landscape is thus not expected to easily revert back to a control topography, but rather to lead to increased delineation and reduced barrier to entry into grooves corresponding to low mood states.
Effect of aging on the canalization landscape: implications for LLD
Similarly as development and diseases can modify the canalization landscape, the numerous biological changes occurring in the human brain during aging are adaptive in the way that they maintain brain functional homeostasis. This would translate in morphological changes in the canalization landscape, i.e. changes in the slopes of the banks of existing canals, or emergence of new ones (Figure 3, right panel). As previously described, age-dependent changes in the function of multiple genes affect various, yet specific, cellular and molecular pathways in directions predicting increased vulnerability to disease states; so the modifications to the landscape are most likely to affect areas corresponding to disease states (red section of lansdscape in Figure 3), with increased probability of residing in those states, through steepening of the banks of the canals or lowering barriers of entry into the canals. The result is a shift in the repertoire of available states in a pro-disease direction. Like in the original conceptualization, genetic and environmental influences, via their effects on multiple biological processes, could further push the trajectory of an individual toward a canal representing normative mood network function, or toward other canals corresponding to mood network disruption, resulting for instance in more stable states of low mood associated with clinical depression (Figure 3, right panel). So, as aging proceeds, the dynamics of changes between states are progressively affected, so that combinations of small environmental perturbations and milder genetic vulnerability could now become sufficient to lead to states of network dysfunction and depression, whereas a “younger” landscape may be “structurally” resilient to such influences. Together we propose that the characteristics of biological aging of the brain, and specifically their overlap with neurological and neuropsychiatric disease processes, correspond to changes in the canalization and biological landscape that places the system at elevated vulnerability for increased incidence and stability of disease states, including corticolimbic disruption and low mood associated with depression.
Summary, conclusions and future directions
Here we hypothesize and provide data supporting a novel putative mechanism for the development of LLD. According to this proposed Age-by-Disease Interaction hypothesis, LLD and associated symptoms may partly arise from normal changes in the expression of depression- and other disease-related genes, which occur in disease-causing directions with increasing age. This model does not preclude previously hypothesized mechanisms (e.g., vascular, inflammatory, and dementia prodrome hypotheses), but rather positions age-related changes in gene expression as the mechanism driving dysfunctions in biological processes that in turn promote LLD, including vascular, inflammatory, neurotrophic and dementia-related processes. Perhaps the most exciting implication of this model relates to how it might inform in several ways research and development of new interventions for the prevention and treatment of LLD. First, identifying the biological changes that occur during normal aging may provide valuable information about the cellular and molecular processes that may contribute to age-related brain diseases like LLD, hence providing mechanistic entry points and potential targets for early intervention. Per this model, one intervention would be to slow down the trajectory of molecular aging for critical genes, via targeting biological modulators and transcriptional regulators for instance. Candidate interventions may include known interventions such as antidepressant medications, psychotherapy, exercise, etc., since investigating how these therapeutic approaches affect molecular aging trajectories may help in optimizing their implementation with respect to timing and duration of intervention for age-dependent diseases. Understanding the “molecular clock” behind age-dependent changes in humans and model organisms is somewhat of a “holy grail” in aging research. Mechanisms related to oxidative stress, cellular metabolism and genome integrity, either through repair or maintenance of proper telomere length for instance, are current topics of research.64,82-85 The brain and neuronal systems specificities of these biological processes represent unique areas of investigation for psychiatric and or neurological disorders. Another important topic of further investigation relates to the individual variability in age-related vulnerability to develop functional declines and associated disease symptoms, including LLD. Identifying genetic and environmental factors that place individuals on accelerated or slowed-down molecular trajectories for critical genes may lead to individualized strategies aimed at promoting resilience and successful aging. Finally, for the broader fields of aging and gerontology, the implications of this hypothesis is that it brings together research on normal aging more closely with the investigation of neuropsychiatric and neurodegenerative diseases. Indeed, our data firmly support the assertion that they may in fact be related facets of similar biological processes, while also providing a putative mechanism of age-by-disease interaction.
Acknowledgement
This work was supported by National Institute of Mental Health (NIMH) MH084060 and MH093723 grants (ES). The funding agency had no role in the study design, data collection and analysis, decision to publish and preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIMH or the National Institutes of Health.
Footnotes
Conflict of interest. The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.American Psychiatric Association . Diagnostic criteria from DSM-IV-TR. American Psychiatric Association; Washington, D.C.: 2000. [Google Scholar]
- 2.Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 3.WHO . World Health Organization - The Global Burden of Disease - 2004 update. WHO Library; 2008. [Google Scholar]
- 4.Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct. 2008;213:93–118. doi: 10.1007/s00429-008-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mayberg HS. Targeted electrode-based modulation of neural circuits for depression. J Clin Invest. 2009;119:717–725. doi: 10.1172/JCI38454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L, Potter GG, Krishnan RK, et al. Neural Correlates Associated With Cognitive Decline in Late-Life Depression. Am J Geriatr Psychiatry. 2011 doi: 10.1097/JGP.0b013e31823e2cc7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gunning FM, Smith GS. Functional neuroimaging in geriatric depression. Psychiatr Clin North Am. 2011;34:403–422. viii. doi: 10.1016/j.psc.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett MR. The prefrontal-limbic network in depression: Modulation by hypothalamus, basal ganglia and midbrain. Prog Neurobiol. 2011;93:468–487. doi: 10.1016/j.pneurobio.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 9.Taylor SF, Liberzon I. Neural correlates of emotion regulation in psychopathology. Trends Cogn Sci. 2007;11:413–418. doi: 10.1016/j.tics.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 10.Belmaker RH, Agam G. Major depressive disorder. N Engl J Med. 2008;358:55–68. doi: 10.1056/NEJMra073096. [DOI] [PubMed] [Google Scholar]
- 11.Weissman MM, Leaf PJ, Tischler GL, et al. Affective disorders in five United States communities. Psychol Med. 1988;18:141–153. doi: 10.1017/s0033291700001975. [DOI] [PubMed] [Google Scholar]
- 12.Koenig HG, Blazer DG. Epidemiology of geriatric affective disorders. Clin Geriatr Med. 1992;8:235–251. [PubMed] [Google Scholar]
- 13.Wiener P, Alexopoulos GS, Kakuma T, et al. The limits of history-taking in geriatric depression. Am J Geriatr Psychiatry. 1997;5:116–125. [PubMed] [Google Scholar]
- 14.Dombrovski AY, Mulsant BH, Houck PR, et al. Residual symptoms and recurrence during maintenance treatment of late-life depression. J Affect Disord. 2007;103:77–82. doi: 10.1016/j.jad.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lenze EJ, Schulz R, Martire LM, et al. The course of functional decline in older people with persistently elevated depressive symptoms: longitudinal findings from the Cardiovascular Health Study. J Am Geriatr Soc. 2005;53:569–575. doi: 10.1111/j.1532-5415.2005.53202.x. [DOI] [PubMed] [Google Scholar]
- 16.Ganguli M, Dodge HH, Mulsant BH. Rates and predictors of mortality in an aging, rural, community-based cohort: the role of depression. Arch Gen Psychiatry. 2002;59:1046–1052. doi: 10.1001/archpsyc.59.11.1046. [DOI] [PubMed] [Google Scholar]
- 17.Katon W, Russo J, Lin EH, et al. Depression and diabetes: factors associated with major depression at five-year follow-up. Psychosomatics. 2009;50:570–579. doi: 10.1176/appi.psy.50.6.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kennedy GJ, Kelman HR, Thomas C. Persistence and remission of depressive symptoms in late life. Am J Psychiatry. 1991;148:174–178. doi: 10.1176/ajp.148.2.174. [DOI] [PubMed] [Google Scholar]
- 19.Wolkowitz OM, Reus VI, Mellon SH. Of sound mind and body: depression, disease, and accelerated aging. Dialogues Clin Neurosci. 2011;13:25–39. doi: 10.31887/DCNS.2011.13.1/owolkowitz. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Orden K, Conwell Y. Suicides in late life. Curr Psychiatry Rep. 2011;13:234–241. doi: 10.1007/s11920-011-0193-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexopoulos GS, Meyers BS, Young RC, et al. Recovery in geriatric depression. Arch Gen Psychiatry. 1996;53:305–312. doi: 10.1001/archpsyc.1996.01830040039008. [DOI] [PubMed] [Google Scholar]
- 22.Mitchell AJ, Subramaniam H. Prognosis of depression in old age compared to middle age: a systematic review of comparative studies. Am J Psychiatry. 2005;162:1588–1601. doi: 10.1176/appi.ajp.162.9.1588. [DOI] [PubMed] [Google Scholar]
- 23.Andreescu C, Reynolds CF., 3rd Late-life depression: evidence-based treatment and promising new directions for research and clinical practice. Psychiatr Clin North Am. 2011;34:335–355. vii–iii. doi: 10.1016/j.psc.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruce ML, Ten Have TR, Reynolds CF, 3rd, et al. Reducing suicidal ideation and depressive symptoms in depressed older primary care patients: a randomized controlled trial. JAMA. 2004;291:1081–1091. doi: 10.1001/jama.291.9.1081. [DOI] [PubMed] [Google Scholar]
- 25.Unutzer J, Katon W, Callahan CM, et al. Collaborative care management of late-life depression in the primary care setting: a randomized controlled trial. JAMA. 2002;288:2836–2845. doi: 10.1001/jama.288.22.2836. [DOI] [PubMed] [Google Scholar]
- 26.Whyte EM, Dew MA, Gildengers A, et al. Time course of response to antidepressants in late-life major depression: therapeutic implications. Drugs Aging. 2004;21:531–554. doi: 10.2165/00002512-200421080-00004. [DOI] [PubMed] [Google Scholar]
- 27.Driscoll HC, Karp JF, Dew MA, et al. Getting better, getting well: understanding and managing partial and non-response to pharmacological treatment of non-psychotic major depression in old age. Drugs Aging. 2007;24:801–814. doi: 10.2165/00002512-200724100-00002. [DOI] [PubMed] [Google Scholar]
- 28.Alexopoulos GS, Meyers BS, Young RC, et al. ‘Vascular depression’ hypothesis. Arch Gen Psychiatry. 1997;54:915–922. doi: 10.1001/archpsyc.1997.01830220033006. [DOI] [PubMed] [Google Scholar]
- 29.Alexopoulos GS, Morimoto SS. The inflammation hypothesis in geriatric depression. Int J Geriatr Psychiatry. 2011 doi: 10.1002/gps.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Byers AL, Yaffe K. Depression and risk of developing dementia. Nat Rev Neurol. 2011;7:323–331. doi: 10.1038/nrneurol.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaupp R, Berrios GE, Pomarol-Clotet E. Depressive states in old age. (Classic Text No. 42). Hist Psychiatry. 2000;11:213–225. doi: 10.1177/0957154X0001104205. [DOI] [PubMed] [Google Scholar]
- 32.Firbank MJ, Lloyd AJ, Ferrier N, et al. A volumetric study of MRI signal hyperintensities in late-life depression. Am J Geriatr Psychiatry. 2004;12:606–612. doi: 10.1176/appi.ajgp.12.6.606. [DOI] [PubMed] [Google Scholar]
- 33.Krishnan KR, McDonald WM, Doraiswamy PM, et al. Neuroanatomical substrates of depression in the elderly. Eur Arch Psychiatry Clin Neurosci. 1993;243:41–46. doi: 10.1007/BF02191522. [DOI] [PubMed] [Google Scholar]
- 34.Aizenstein HJ, Andreescu C, Edelman KL, et al. fMRI Correlates of White Matter Hyperintensities in Late-Life Depression. Am J Psychiatry. 2011 doi: 10.1176/appi.ajp.2011.10060853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sparkman NL, Johnson RW. Neuroinflammation associated with aging sensitizes the brain to the effects of infection or stress. Neuroimmunomodulation. 2008;15:323–330. doi: 10.1159/000156474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Godbout JP, Johnson RW. Age and neuroinflammation: a lifetime of psychoneuroimmune consequences. Neurol Clin. 2006;24:521–538. doi: 10.1016/j.ncl.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 37.Harrison NA, Brydon L, Walker C, et al. Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry. 2009;66:407–414. doi: 10.1016/j.biopsych.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Basterzi AD, Aydemir C, Kisa C, et al. IL-6 levels decrease with SSRI treatment in patients with major depression. Hum Psychopharmacol. 2005;20:473–476. doi: 10.1002/hup.717. [DOI] [PubMed] [Google Scholar]
- 39.Maes M, Bosmans E, De Jongh R, et al. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. 1997;9:853–858. doi: 10.1006/cyto.1997.0238. [DOI] [PubMed] [Google Scholar]
- 40.Kim YK, Na KS, Shin KH, et al. Cytokine imbalance in the pathophysiology of major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1044–1053. doi: 10.1016/j.pnpbp.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 41.Collantes-Estevez E, Munoz-Villanueva MC, Canete-Crespillo JD, et al. Infliximab in refractory spondyloarthropathies: a multicentre 38 week open study. Ann Rheum Dis. 2003;62:1239–1240. doi: 10.1136/ard.2002.004879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Molina-Hernandez M, Tellez-Alcantara NP, Perez-Garcia J, et al. Antidepressant-like actions of minocycline combined with several glutamate antagonists. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:380–386. doi: 10.1016/j.pnpbp.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 43.Tyring S, Gottlieb A, Papp K, et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet. 2006;367:29–35. doi: 10.1016/S0140-6736(05)67763-X. [DOI] [PubMed] [Google Scholar]
- 44.Devanand DP, Sano M, Tang MX, et al. Depressed mood and the incidence of Alzheimer's disease in the elderly living in the community. Arch Gen Psychiatry. 1996;53:175–182. doi: 10.1001/archpsyc.1996.01830020093011. [DOI] [PubMed] [Google Scholar]
- 45.Steffens DC, Plassman BL, Helms MJ, et al. A twin study of late-onset depression and apolipoprotein E epsilon 4 as risk factors for Alzheimer's disease. Biol Psychiatry. 1997;41:851–856. doi: 10.1016/S0006-3223(96)00247-8. [DOI] [PubMed] [Google Scholar]
- 46.Butters MA, Young JB, Lopez O, et al. Pathways linking late-life depression to persistent cognitive impairment and dementia. Dialogues Clin Neurosci. 2008;10:345–357. doi: 10.31887/DCNS.2008.10.3/mabutters. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bremner JD, Narayan M, Anderson ER, et al. Hippocampal volume reduction in major depression. Am J Psychiatry. 2000;157:115–118. doi: 10.1176/ajp.157.1.115. [DOI] [PubMed] [Google Scholar]
- 48.Sheline YI. 3D MRI studies of neuroanatomic changes in unipolar major depression: the role of stress and medical comorbidity. Biol Psychiatry. 2000;48:791–800. doi: 10.1016/s0006-3223(00)00994-x. [DOI] [PubMed] [Google Scholar]
- 49.Rapp MA, Schnaider-Beeri M, Purohit DP, et al. Increased neurofibrillary tangles in patients with Alzheimer disease with comorbid depression. Am J Geriatr Psychiatry. 2008;16:168–174. doi: 10.1097/JGP.0b013e31816029ec. [DOI] [PubMed] [Google Scholar]
- 50.Rapp MA, Schnaider-Beeri M, Grossman HT, et al. Increased hippocampal plaques and tangles in patients with Alzheimer disease with a lifetime history of major depression. Arch Gen Psychiatry. 2006;63:161–167. doi: 10.1001/archpsyc.63.2.161. [DOI] [PubMed] [Google Scholar]
- 51.MacQueen GM, Campbell S, McEwen BS, et al. Course of illness, hippocampal function, and hippocampal volume in major depression. Proc Natl Acad Sci U S A. 2003;100:1387–1392. doi: 10.1073/pnas.0337481100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller DB, O'Callaghan JP. Effects of aging and stress on hippocampal structure and function. Metabolism. 2003;52:17–21. doi: 10.1016/s0026-0495(03)00296-8. [DOI] [PubMed] [Google Scholar]
- 53.Sapolsky RM. Glucocorticoids, stress, and their adverse neurological effects: relevance to aging. Exp Gerontol. 1999;34:721–732. doi: 10.1016/s0531-5565(99)00047-9. [DOI] [PubMed] [Google Scholar]
- 54.Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev. 1986;7:284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- 55.Wolkowitz OM, Epel ES, Reus VI, et al. Depression gets old fast: do stress and depression accelerate cell aging? Depress Anxiety. 2010;27:327–338. doi: 10.1002/da.20686. [DOI] [PubMed] [Google Scholar]
- 56.McEwen BS. Glucocorticoids, depression, and mood disorders: structural remodeling in the brain. Metabolism. 2005;54:20–23. doi: 10.1016/j.metabol.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 57.Glorioso C, Oh S, Douillard GG, et al. Brain molecular aging, promotion of neurological disease and modulation by Sirtuin5 longevity gene polymorphism. Neurobiol Dis. 2011;41:279–290. doi: 10.1016/j.nbd.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glorioso C, Sibille E. Between destiny and disease: genetics and molecular pathways of human central nervous system aging. Prog Neurobiol. 2011;93:165–181. doi: 10.1016/j.pneurobio.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Finch CE, Tanzi RE. Genetics of aging. Science. 1997;278:407–411. doi: 10.1126/science.278.5337.407. [DOI] [PubMed] [Google Scholar]
- 60.Lee CK, Weindruch R, Prolla TA. Gene-expression profile of the ageing brain in mice. Nat Genet. 2000;25:294–297. doi: 10.1038/77046. [DOI] [PubMed] [Google Scholar]
- 61.Lee CK, Klopp RG, Weindruch R, et al. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- 62.Blalock EM, Chen KC, Sharrow K, et al. Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J Neurosci. 2003;23:3807–3819. doi: 10.1523/JNEUROSCI.23-09-03807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang CH, Tsien JZ, Schultz PG, et al. The effects of aging on gene expression in the hypothalamus and cortex of mice. Proc Natl Acad Sci U S A. 2001;98:1930–1934. doi: 10.1073/pnas.98.4.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lu T, Pan Y, Kao SY, et al. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 65.Avramopoulos D, Szymanski M, Wang R, et al. Gene expression reveals overlap between normal aging and Alzheimer's disease genes. Neurobiol Aging. 2011;32:2319, e2327–2334. doi: 10.1016/j.neurobiolaging.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Erraji-Benchekroun L, Underwood MD, Arango V, et al. Molecular aging in human prefrontal cortex is selective and continuous throughout adult life. Biol Psychiatry. 2005;57:549–558. doi: 10.1016/j.biopsych.2004.10.034. [DOI] [PubMed] [Google Scholar]
- 67.Blalock EM, Geddes JW, Chen KC, et al. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miller JA, Horvath S, Geschwind DH. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc Natl Acad Sci U S A. 2010;107:12698–12703. doi: 10.1073/pnas.0914257107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tripp A, Kota RS, Lewis DA, et al. Reduced somatostatin in subgenual anterior cingulate cortex in major depression. Neurobiol Dis. 2011;42:116–124. doi: 10.1016/j.nbd.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morris HM, Hashimoto T, Lewis DA. Alterations in somatostatin mRNA expression in the dorsolateral prefrontal cortex of subjects with schizophrenia or schizoaffective disorder. Cereb Cortex. 2008;18:1575–1587. doi: 10.1093/cercor/bhm186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Douillard-Guilloux G, Guilloux JP, Lewis DA, et al. Anticipated brain molecular aging in Major Depression. American Journal of Geriatric Psychiatry. 2012 doi: 10.1016/j.jagp.2013.01.040. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- 73.Yeoman MS, Faragher RG. Ageing and the nervous system: insights from studies on invertebrates. Biogerontology. 2001;2:85–97. doi: 10.1023/a:1011597420036. [DOI] [PubMed] [Google Scholar]
- 74.Guarente L. Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N Engl J Med. 2011;364:2235–2244. doi: 10.1056/NEJMra1100831. [DOI] [PubMed] [Google Scholar]
- 75.Choi SW, Bhang S, Ahn JH. Diurnal variation and gender differences of plasma brain-derived neurotrophic factor in healthy human subjects. Psychiat Res. 2011;186:427–430. doi: 10.1016/j.psychres.2010.07.028. [DOI] [PubMed] [Google Scholar]
- 76.Castren E, Rantamaki T. The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Dev Neurobiol. 2010;70:289–297. doi: 10.1002/dneu.20758. [DOI] [PubMed] [Google Scholar]
- 77.Guilloux JP, Douillard-Guilloux G, Kota R, et al. Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry. 2011 doi: 10.1038/mp.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leppanen JM. Emotional information processing in mood disorders: a review of behavioral and neuroimaging findings. Curr Opin Psychiatry. 2006;19:34–39. doi: 10.1097/01.yco.0000191500.46411.00. [DOI] [PubMed] [Google Scholar]
- 79.Gaiteri C, Guilloux JP, Lewis DA, et al. Altered gene synchrony suggests a combined hormone-mediated dysregulated state in major depression. Plos One. 2010;5:e9970. doi: 10.1371/journal.pone.0009970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Waddington CH. Organizers and genes. Cambridge University Press; Cambridge: 1940. [Google Scholar]
- 81.Waddington CH. The strategy of the genes. Allen and Unwin; London: 1957. [Google Scholar]
- 82.Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nat Rev Neurosci. 2006;7:278–294. doi: 10.1038/nrn1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yankner BA, Lu T, Loerch P. The aging brain. Annu Rev Pathol. 2008;3:41–66. doi: 10.1146/annurev.pathmechdis.2.010506.092044. [DOI] [PubMed] [Google Scholar]
- 84.Wolkowitz OM, Mellon SH, Epel ES, et al. Leukocyte telomere length in major depression: correlations with chronicity, inflammation and oxidative stress--preliminary findings. PLoS One. 2011;6:e17837. doi: 10.1371/journal.pone.0017837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wolkowitz OM, Mellon SH, Epel ES, et al. Resting leukocyte telomerase activity is elevated in major depression and predicts treatment response. Mol Psychiatry. 2011 doi: 10.1038/mp.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]