

Abstract
BACKGROUND
Glutathione (GSH) is present in all mammalian tissues as the most abundant non-protein thiol that defends against oxidative stress. GSH is also a key determinant of redox signaling, vital in detoxification of xenobiotics, regulates cell proliferation, apoptosis, immune function, and fibrogenesis. Biosynthesis of GSH occurs in the cytosol in a tightly regulated manner. Key determinants of GSH synthesis are the availability of the sulfur amino acid precursor, cysteine, and the activity of the rate-limiting enzyme, glutamate cysteine ligase (GCL), which is composed of a catalytic (GCLC) and a modifier (GCLM) subunit. The second enzyme of GSH synthesis is GSH synthetase (GS).
SCOPE OF REVIEW
This review summarizes key functions of GSH and focuses on factors that regulate the biosynthesis of GSH, including pathological conditions where GSH synthesis is dysregulated.
MAJOR CONCLUSIONS
GCL subunits and GS are regulated at multiple levels and often in a coordinated manner. Key transcription factors that regulate the expression of these genes include NF-E2 related factor 2 (Nrf2) via the antioxidant response element (ARE), AP-1, and nuclear factor kappa B (NFκB). There is increasing evidence that dysregulation of GSH synthesis contributes to the pathogenesis of many pathological conditions. These include diabetes mellitus, pulmonary and liver fibrosis, alcoholic liver disease, cholestatic liver injury, endotoxemia and drug-resistant tumor cells.
GENERAL SIGNIFICANCE
GSH is a key antioxidant that also modulates diverse cellular processes. A better understanding of how its synthesis is regulated and dysregulated in disease states may lead to improvement in the treatment of these disorders.
Keywords: GSH, glutamate-cysteine ligase, GSH synthase, Nrf2, MafG, c-Maf, antioxidant response element
1. Introduction
Glutathione (GSH) is a tripeptide, γ-L-glutamyl-L-cysteinylglycine, present in all mammalian tissues at 1–10 mM concentrations (highest concentration in liver) as the most abundant non-protein thiol that defends against oxidative stress. GSH is also a key determinant of redox signaling, vital in detoxification of xenobiotics, modulates cell proliferation, apoptosis, immune function, and fibrogenesis. This review is focused on factors that determine GSH synthesis and pathologies where dysregulation in GSH synthesis may play an important role with emphasis on the liver. This is because the liver plays a central role in the interorgan GSH homeostasis [1].
2. Structure and functions of GSH
GSH exists in the thiol-reduced and disulfide-oxidized (GSSG) forms [2]. GSH is the predominant form and accounts for >98% of total GSH [3–5]. Eukaryotic cells have three major reservoirs of GSH. Most (80–85%) of the cellular GSH are in the cytosol, 10–15% is in the mitochondria and a small percentage is in the endoplasmic reticulum [6–8]. Rat liver cytosolic GSH turns over rapidly with a half-life of 2–3 hours. The structure of GSH is unique in that the peptide bond linking glutamate and cysteine of GSH is through the γ-carboxyl group of glutamate rather than the conventional α-carboxyl group. The only enzyme that can hydrolyze this unusual bond is γ-glutamyltranspeptidase (GGT), which is only present on the external surfaces of certain cell types [9]. As a consequence, GSH is resistant to intracelluar degradation and is only metabolized extracellularly by cells that express GGT. This allows for released GSH to be broken down and its constituent amino acids taken up by cells and reincorporated into GSH (so called γ-glutamyl cycle, see below). The bulk of plasma GSH originates from the liver, which plays a central role in the interorgan homeostasis of GSH by exporting nearly all of the GSH it synthesizes into plasma and bile [1,10,11]. Thus, dysregulation of hepatic GSH synthesis has impact on GSH homeostasis systemically.
GSH serves several vital functions including antioxidant defense, detoxification of xenobiotics and/or their metabolites, regulation of cell cycle progression and apoptosis, storage of cysteine, maintenance of redox potential, modulation of immune function and fibrogenesis [4, 5,9,12–15]. Some of these key functions, namely antioxidant defense, redox signaling, storage of cysteine via the γ-glutamyl cycle, regulation of growth and death are described in more detail below.
2.1 Antioxidant function of GSH
The antioxidant function of GSH is accomplished largely by GSH peroxidase (GPx)-catalyzed reactions, which reduce hydrogen peroxide and lipid peroxide as GSH is oxidized to GSSG. GSSG in turn is reduced back to GSH by GSSG reductase at the expense of NADPH, forming a redox cycle [13]. Organic peroxides can also be reduced by GPx and GSH S-transferase. Catalase can also reduce hydrogen peroxide but it is present only in peroxisome. This makes GSH particularly important in the mitochondria in defending against both physiologically and pathologically generated oxidative stress [16,17]. As GSH to GSSG ratio largely determines the intracellular redox potential (proportional to the log of [GSH]2/[GSSG]) [5], to prevent a major shift in the redox equilibrium when oxidative stress overcomes the ability of the cell to reduce GSSG to GSH, GSSG can be actively exported out of the cell or react with a protein sulfhydryl group leading to the formation of a mixed disulfide. Thus, severe oxidative stress depletes cellular GSH [13] (Figure 2).
Fig. 2. Antioxidant function of GSH.
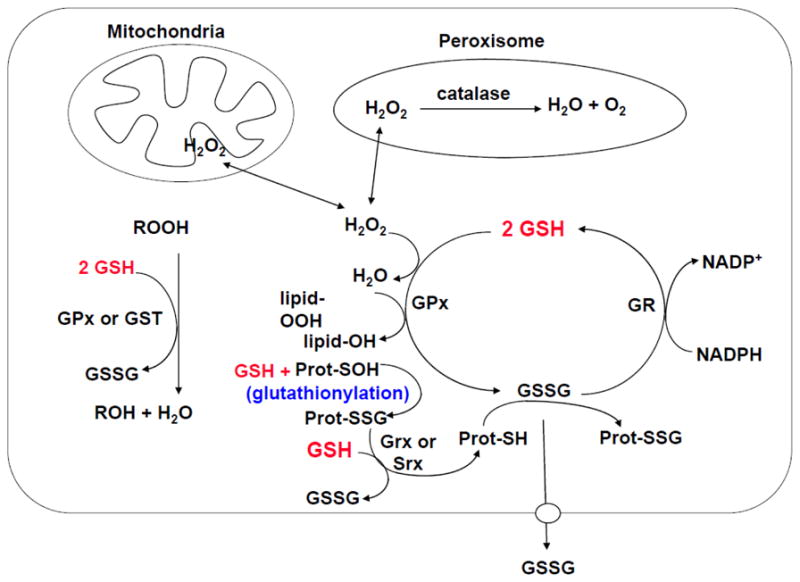
Aerobic metabolism generates hydrogen peroxide (H2O2), which can be metabolized by GSH peroxidase (GPx) in the cytosol and mitochondria, and by catalase in the peroxisome. GSSG can be reduced back to GSH by GSSG reductase (GR) at the expense of NADPH, thereby forming a redox cycle. Organic peroxides (ROOH) can be reduced by either GPx or GSH S-transferase (GST). GSH also plays a key role in protein redox signaling. During oxidative stress, protein cysteine residues can be oxidized to sulfenic acid (Prot-SOH), which can react with GSH to form protein mixed disulfides Prot-SSG (glutathionylation), which in turn can be reduced back to Prot-SH via glutaredoxin (Grx) or sulfiredoxin (Srx). This is a mechanism to protect sensitive protein thiols from irreversible oxidation and may also serve to prevent loss of GSH under oxidative conditions. The ability of the cell to reduce GSSG to GSH may be overcome during severe oxidative injury, leading to an accumulation of GSSG. To prevent a shift in the redox equilibrium, GSSG can either be actively transported out of the cell or react with a protein sulfhydryl (Prot-SH) to form a mixed disulfide (Prot-SSG).
2.2 GSH in redox signaling
GSH regulates redox-dependent cell signaling. This is largely accomplished by modifying the oxidation state of critical protein cysteine residues [5,18]. GSH can be reversibly bound to the –SH of protein cysteinyl residues (Prot-SH) by a process called glutathionylation, generating glutathionylated proteins (Prot-SSG), which can either activate or inactivate the protein [18]. This is a mechanism to protect sensitive protein thiols from irreversible oxidation and may also serve to prevent loss of GSH under oxidative conditions (Figure 2). Deglutathionylation can then occur through glutaredoxin and sulfiredoxin-catalyzed reactions using GSH as a reductant [15]. Many transcription factors and signaling molecules have critical cysteine residues that can be oxidized and this is an important mechanism whereby reactive oxygen and nitrogen species (ROS and RNS) regulate protein function and cell signaling that can be modulated by GSH [13,15].
2.3 GSH and the γ-glutamyl cycle
Alton Meister first described the γ-glutamyl cycle in the early 1970’s, which allows GSH to serve as a continuous source of cysteine [19] (Figure 3). This is an important function as cysteine is extremely unstable and rapidly auto-oxidizes to cystine extracellularly, which can generate potentially toxic oxygen free radicals [19]. In the γ-glutamyl cycle, GSH is released from the cell and the ecto-enzyme GGT transfers the γ-glutamyl moiety of GSH to an amino acid (the best acceptor being cystine), forming γ-glutamyl amino acid and cysteinylglycine. The γ-glutamyl amino acid can be transported back into the cell and once inside, the γ-glutamyl amino acid can be further metabolized to release the amino acid and 5-oxoproline, which can be converted to glutamate and used for GSH synthesis. Cysteinylglycine is broken down by dipeptidase to generate cysteine and glycine. Most cells readily take up cysteine. Once taken up, the majority of cysteine is incorporated into GSH, some is incorporated into protein, and some is degraded into sulfate and taurine [19].
Fig. 3. GSH is a continuous source of cysteine via the γ-glutamyl cycle.
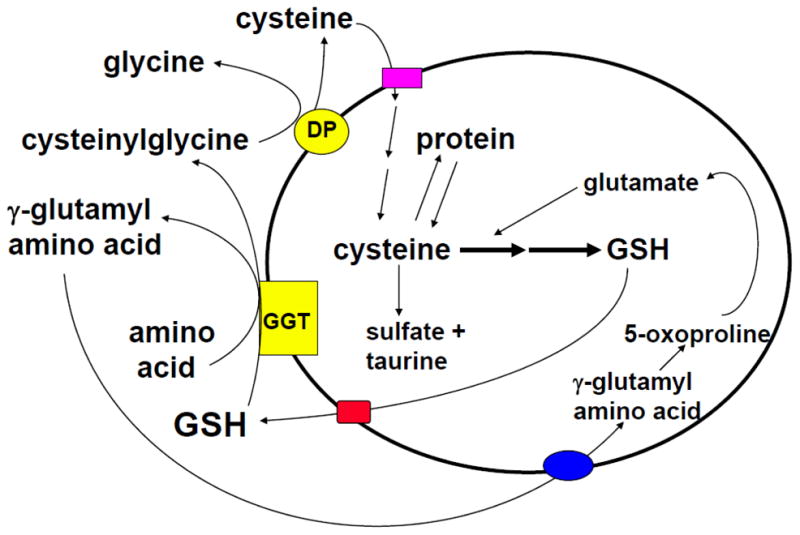
GSH is transported out of the cell where the ecto-enzyme γ-glutamylpeptidase (GGT) transfers the γ-glutamyl moiety of GSH to an amino acid (the best acceptor being cystine), forming γ-glutamyl amino acid and cysteinylglycine. The γ-glutamyl amino acid can then be transported back into the cell and once inside, the γ-glutamyl amino acid can be further metabolized to release the amino acid and 5-oxoproline, which can be converted to glutamate and reincorporated into GSH. Cysteinylglycine is broken down by dipeptidase (DP) to generate cysteine and glycine, which are also transported back into the cell to be reincorporated into GSH. Most of the cysteine taken up is incorporated into GSH while the rest is incorporated into newly synthesized proteins and/or broken down into sulfate and taurine.
2.4 GSH regulates growth and death
In many normal and malignant cell types, increased GSH level is associated with a proliferative response and is essential for cell cycle progression [20–26]. In normal hepatocytes, GSH level increases when cells shift from G0 to G1 phase of the cell cycle in vitro [25], and after 2/3 partial hepatectomy prior to the onset of increased DNA synthesis [27]. If this increase in GSH was blocked, DNA synthesis following partial hepatectomy was reduced by 33% [26]. In liver cancer and metastatic melanoma cells, GSH status also correlated with growth [26,28]. Interestingly, hepatocyte growth factor (HGF) induces the expression of GSH synthetic enzymes and acts as a mitogen in liver cancer cells only under subconfluent cell density condition and the mitogenic effect required increased GSH level [29]. A key mechanism for GSH’s role in DNA synthesis relates to maintenance of reduced glutaredoxin or thioredoxin, which are required for the activity of ribonucleotide reductase, the rate-limiting enzyme in DNA synthesis [30]. In addition, the GSH redox status can affect the expression and activity of many factors important for cell cycle progression. Of particular interest is the finding that GSH co-localizes to the nucleus at the onset of proliferation, which through redox changes can affect the activity of many nuclear proteins including histones [14,31]. These recent studies show that a reducing condition in the nucleus is necessary for cell cycle progression [14].
GSH also modulates cell death. Apoptosis, characterized by chromatin condensation, fragmentation and internucleosomal DNA cleavage, and necrosis, characterized by rupture or fragmentation of the plasma membrane and ATP depletion [32] can coexist and share common pathways, such as involvement of the mitochondria [33]. GSH modulates both types of cell death. GSH levels influence the expression/activity of caspases and other signaling molecules important in cell death [4,32]. GSH levels fall during apoptosis in many different cell types, due to ROS, enhanced GSH efflux, and decreased GCL activity (see section on post-translational regulation of GCLC) [34,35]. Although GSH efflux may be a mechanism to circumvent the normally protective role of GSH, it appears essential for apoptosis to occur in many cell types [4, 36]. However, profound GSH depletion can convert apoptotic to necrotic cell death [34], suggesting very high levels of ROS may overwhelm the apoptotic machinery. Consistently, severe mitochondrial GSH depletion leads to increased levels of ROS and RNS, mitochondrial dysfunction and ATP depletion, converting apoptotic to necrotic cell death [32].
3. Synthesis of GSH
The synthesis of GSH from its constituent amino acids involves two ATP-requiring enzymatic steps: formation of γ-glutamylcysteine from glutamate and cysteine and formation of GSH from γ-glutamylcysteine and glycine (Figure 1). The first step of GSH biosynthesis is rate limiting and catalyzed by GCL (EC 6.3.2.2; formerly γ-glutamylcysteine synthetase), which is composed of a heavy or catalytic (GCLC, Mr ~ 73 kDa) and a light or modifier (GCLM, Mr ~ 31 kDa) subunit, which are encoded by different genes in fruit flies, rodents and humans [37–41]. In contrast, GCL in yeast and bacteria have only a single polypeptide [41]. GCLC exhibits all of the catalytic activity of the isolated enzyme and feedback inhibition by GSH [42]. GCLM is enzymatically inactive but plays an important regulatory function by lowering the Km of GCL for glutamate and raising the Ki for GSH [38,43]. Thus, the holoenzyme is catalytically more efficient and less subject to inhibition by GSH than GCLC. However, GCLC alone does have enzymatic activity as Gclm knockout mice are viable but have markedly reduced tissue GSH levels (reduced by about 85 to 90%) [44]. Redox status can influence GCL activity via formation of the holoenzyme [45]. Most of the GCL holoenzyme can be reversibly dissociated by treatment with dithiothreitol [42], while oxidative stress may enhance holoenzyme formation as it increases GCL activity in the absence of any change in the expression of GCL subunits [45].
Fig. 1. GSH synthesis.
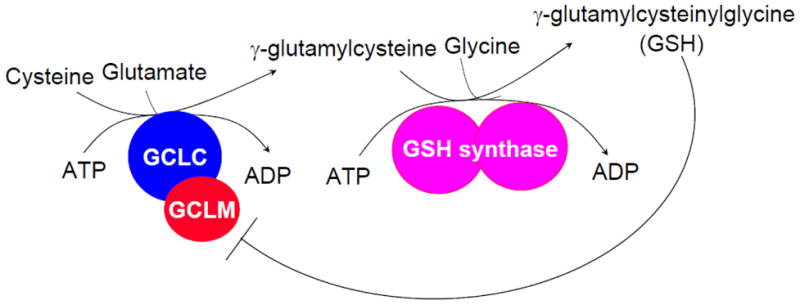
Synthesis of GSH occurs via a two-step ATP-requiring enzymatic process. The first step is catalyzed by glutamate-cysteine ligase (GCL), which is composed of catalytic and modifier subunits (GCLC and GCLM). This step conjugates cysteine with glutamate, generating γ-glutamylcysteine. The second step is catalyzed by GSH synthase, which adds glycine to γ-glutamylcysteine to form γ-glutamylcysteinylglycine or GSH. GSH exerts a negative feedback inhibition on GCL.
Under physiological conditions GCL is regulated by: (a) nonallosteric feedback competitive inhibition (with glutamate) by GSH (Ki=2.3mM) [46] and (b) availability of L-cysteine [9]. The Km values of GCL for glutamate and cysteine are 1.8 and 0.1–0.3 mM, respectively [46]. The intracellular glutamate concentration is 10-fold higher than the Km value but cysteine concentration approximates the apparent Km value [47].
The second step in GSH synthesis is catalyzed by GSH synthetase (GS, EC 6.3.2.3, also known as GSH synthase). GS is composed of two identical subunits (Mr ~ 118 kDa) and is not subject to feedback inhibition by GSH [48]. Since the product of GCL, γ-glutamylcysteine, is present at exceedingly low concentrations when GS is present, GCL is considered rate limiting [41]. In support of this is the finding that overexpression of GS failed to increase GSH level whereas overexpression of GCL increased GSH level [49]. Although GS is generally thought not to be important in the regulation of GSH synthesis, there is accumulating evidence that GS is important in determining overall GSH synthetic capacity in certain tissues and/or under stressful conditions [13]. Surgical trauma decreased GSH levels and GS activity in skeletal muscle while GCL activity was unchanged [50]. In rat hepatocytes, increased GS expression further enhanced GSH synthesis above that observed with increased GCLC expression alone [51].
3.1 Factors that determine cysteine availability
Cysteine is derived normally from the diet, protein breakdown and in the liver, from methionine via transsulfuration (see below). Cysteine is unstable extracellularly where it readily autoxidizes to cystine, which is taken up by some cells and is rapidly reduced to cysteine intracellularly [47]. In hepatocytes, the key factors that regulate cysteine availability include membrane transport of cysteine (via the ASC system), cystine (via the Xc- system which is induced under oxidative stress), methionine (via the L system) and the activity of the transsulfuration pathway [47,52,53].
3.2 Transsulfuration pathway
The transsulfuration pathway (also called the cystathionine pathway) allows the utilization of methionine for GSH synthesis [54] (Figure 4). Liver plays a central role in methionine metabolism as up to half of the daily intake of methionine is catabolized in the liver. The first step in methionine catabolism is the generation of S-adenosylmethionine (SAMe), the principal biological methyl donor, in a reaction catalyzed by methionine adenosyltransferase (MAT) [55]. Under normal conditions, most of the SAMe generated is used in transmethylation reactions [56]. SAMe donates its methyl group to a large variety of acceptor molecules in reactions catalyzed by methyltransferases (MTs), generating S-adenosylhomocysteine (SAH), which is in turn hydrolyzed to homocysteine (Hcy) and adenosine through a reversible reaction catalyzed by SAH hydrolase. SAH is a potent competitive inhibitor of methylation reactions so that prompt removal of Hcy and adenosine is required to prevent SAH accumulation. In hepatocytes, Hcy can be remethylated to generate methionine by methionine synthase (MS), which requires normal levels of folate and vitamin B12, and betaine homocysteine methyltransferase (BHMT), which requires betaine. Hcy can also be converted to cysteine via the transsulfuration pathway by a two-enzyme process. First, Hcy condenses with serine to form cystathionine in a reaction catalyzed by cystathionine β synthase (CBS), which requires vitamin B6. The second step cleaves cystathionine, catalyzed by another vitamin B6-dependent enzyme γ-cystathionase, and releases free cysteine for GSH synthesis [56]. The transsulfuration pathway is particularly active in hepatocytes but outside of the liver, it is either absent or present at very low levels [56]. The hepatic transsulfuration pathway activity is markedly impaired or absent in the fetus and newborn infant and in cirrhotic patients [13]. Part of the mechanism relates to decreased cofactor availability (such as B vitamins). In addition, cirrhotic patients also have decreased MAT activity and diminished SAMe biosynthesis, which further contribute to decreased GSH levels [55].
Fig. 4. Hepatic methionine metabolism.
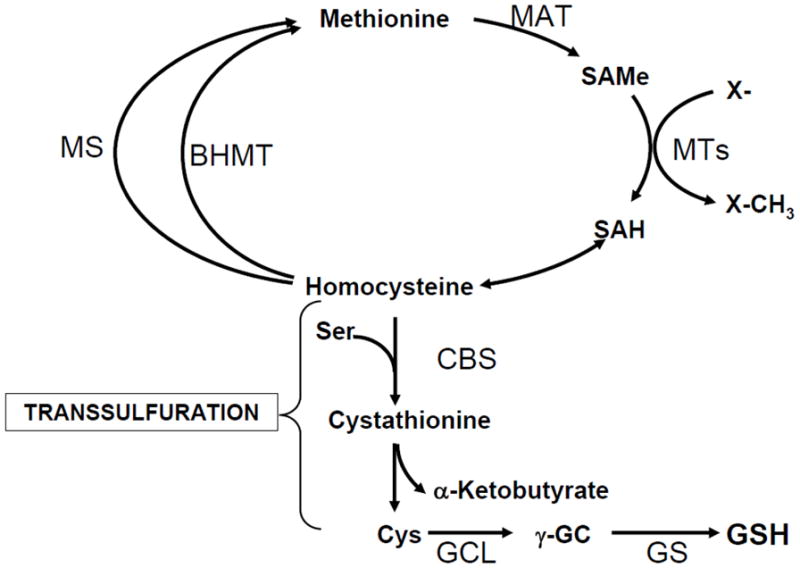
Liver plays a central role in methionine catabolism as up to half of the daily intake of methionine is catabolized to S-adenosylmethionine (SAMe) in the liver in a reaction catalyzed by methionine adenosyltransferase (MAT). SAMe is the principal biological methyl donor and donates its methyl group to a large variety of acceptor molecules in reactions catalyzed by methyltransferases (MTs). S-adenosylhomocysteine (SAH), generated as a result of transmethylation, is a potent inhibitor of all transmethylation reactions. To prevent SAH accumulation, it is hydrolyzed to homocysteine and adenosine is through a reversible reaction catalyzed by SAH hydrolase, whose thermodynamics favors biosynthesis rather than hydrolysis. Prompt removal of homocysteine and adenosine ensures SAH is hydrolyzed. Homcysteine can be remethylated to form methionine via methionine synthase (MS), which requires folate and vitamin B12 and betaine homocysteine methyltransferase (BHMT), which requires betaine. In hepatocytes, homocysteine can also undergo conversion to cysteine (Cys) via the transsulfuration pathway, a two-step enzymatic process catalyzed by cystathionine β-synthase (CBS) and cystathionase, both requiring vitamin B6. Liver has the highest activity of transsulfuration, which allows methionine and SAMe to be effectively utilized as GSH precursor.
4. Regulation of glutamate-cysteine ligase (GCL)
Changes in GCL activity can result from regulation at multiple levels affecting only GCLC or both GCLC and GCLM.
4.1 GCLC pre-translational regulation
Many conditions are known to affect GCLC pre-translationally. Drug-resistant tumor cell lines and oxidative stress are associated with increased cell GSH levels, GCL activity, GCLC mRNA levels and GCLC gene transcription [57–63]. While many of these treatments induced both GCLC and GCLM expression, selective transcriptional induction of only GCLC occurred when cultured rat hepatocytes were treated with insulin or hydrocortisone [64–66]. The physiologic significance of the hormonal effect was confirmed using insulin-deficient diabetic or adrenalectomized rats. Both exhibited lower hepatic GSH levels and GCL activity, which were prevented with hormone replacement [64]. Importantly, lower levels of GSH in the erythrocytes of diabetic patients and increased susceptibility to oxidative stress of these cells have been reported [67]. Kim et al reported that the effect of insulin on GSH levels and GCLC expression in rat hepatocytes involve PI3K/Akt/p70S6K but not ERK, JNK and p38 MAPK [68]. However, Li et al reported that insulin’s effect on GSH synthesis in cardiac myocytes required PI3K, MEK and p38 MAPK [69]. More recently, Langston et al reported that in human brain endothelial cell line, insulin activated GCLC promoter activity under altered glycemic condition (both low and high) that required PI3K/Akt/mTOR signaling [70]. Thus, while the PI3K signaling pathway appears to be a central player in mediating insulin’s effect in many cell types, other signaling pathways activated by insulin may act more in a tissue-specific manner in the up-regulation of GCLC expression and GSH level.
Another condition where GCLC is induced transcriptionally while GCLM is unchanged is during rapid liver growth. Plating hepatocytes under low-density and liver regeneration following partial hepatectomy are such examples [25,27,65]. Increased GSH levels and GCLC transcription and mRNA levels (GCLM expression was unchanged) also occur in human hepatocellular carcinoma (HCC) [26]. Thus, hormones and increased growth selectively regulate hepatic GCLC expression in most circumstances. Since an increase in GCLC expression alone led to an increase in GSH level, we speculated that GCLC might be limiting in hepatocytes. However, this point remains controversial, as others have reported the opposite, namely GCLM is limiting [13,45].
Transforming growth factor-β1 (TGF-β1), a pleiotropic cytokine implicated in the pathogenesis of idiopathic pulmonary fibrosis and in liver fibrosis, has also been shown to regulate GSH synthesis at the level of GCLC [71–73]. In type II alveolar epithelial cells, TGF-β1 lowered the transcriptional activity of GCLC [72]. Similarly, in rat hepatic stellate cells (HSCs) TGF-β1 suppressed the expression of GCLC (no effect on GCLM) and lowered GSH levels [73]. This was a key mechanism for TGF-β1-mediated profibrogenic effect in HSCs that is targeted by (−)-epigallocatechin-3-gallate, the major constituent of green tea that exerts antioxidant effect [73].
Other conditions known to influence expression of GCLC at the transcriptional level include treatments with antioxidants such as butylated hydroxyanisole [74,75], 5,10-dihydroindeno [1,2-b]indole and tert-butyl hydroquinone (TBH) [44,76,77], inducers of Phase II detoxifying enzymes such as β-naphthoflavone (β-NF) [78], formation of Michael reaction acceptors (containing an electrophilic electron-deficient center that is susceptible to nucleophilic attack) by treatment with diethyl maleate (DEM) to produce GSH conjugates [75], heat shock [79], zinc [80], melatonin [81], curcumin [82], and lipid peroxidation products such as 4-hydroxynonenal (4-HNE), 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) [83–86], trivalent arsenite (As3+) [87], and ajoene, a major compound in garlic extracts [88]. 4-HNE is a major end product of lipid peroxidation that is present in normal human plasma at concentrations ranging from 0.1μM to 1.4μM and this can increase more than 10 folds during oxidative stress in vivo [84]. 4-HNE was shown to induce GCLC with concentrations found in human plasma, suggesting that the “basal” GCLC expression is under regulation by products of lipid peroxidation [84]. In endothelial cells, nitric oxide (NO) was found to protect against H2O2-induced toxicity via induction of GCLC that required the participation of zinc and Nrf2 [89]. Thus, GCLC gene expression is up-regulated when increased cellular defense is needed. However, if the toxic or injurious insult persists, GCLC expression may become dysregulated. An example is treatment with the toxic bile acid lithocholic acid, which caused initial GCLC induction, followed later by suppression at the transcriptional level in hepatocytes [90]. A similar pattern also occurred during bile duct ligation (BDL) [90] (see below under GSH synthesis dysregulation during cholestatic liver injury).
There have been numerous studies that examined the molecular mechanism(s) of GCLC transcriptional regulation [13]. Rodents and human GCLC promoter regions share similar regulatory mechanisms. The promoter region of GCL subunits of human, rat, and mouse has been cloned [78,91–97]. Consensus NFκB, Sp-1, activator protein-1 (AP-1), AP-2, metal response (MRE), and ARE/EpRE elements have been identified in the human GCLC promoter. A proximal AP-1 element (−263 to −269) was found to be critical in mediating the effect of oxidative stress-induced increase in human GCLC transcription [98–103]. However, a distal ARE element located ~ 3.1kb upstream of the transcriptional start site of human GCLC was found to mediate constitutive and β-NF inducible expression in HepG2 cells (a human hepatoma cell line) [78]. The transcription factor Nrf2, possibly in complexes with other Jun or Maf proteins, was found to be responsible for trans-activating the human GCLC promoter via binding to ARE4 in response to numerous treatments including β-NF, pyrrolidine dithiocarbamate, TBH [104,105], and insulin [106]. The ARE4 is also the site of TGF-β1 action in type II alveolar cells, as TGF-β1 induced the binding of Fra-1/c-Jun dimer to the internal AP-1 sequence of the ARE4 site that led to suppression of GCLC transcriptional activity [72]. Thus, depending on the make up of the transcription factors bound to the ARE, opposite effects may occur. Indeed, this is further illustrated in cholestatic liver injury (see below). Thus, ARE appears to be the key element for various inducers of GCLC. However, As3+, despite being an inducer of oxidative stress, induced GCLC transcriptional activity by a mechanism that is independent of Nrf1 or Nrf2 [87]. The transcription factor(s) responsible for As3+-mediated GCLC induction is unknown.
Nrf2 is the key transcription factor required for activation of ARE. Nrf1 and Nrf2 are members of the cap ‘n’ collar-basic leucine zipper proteins (CNC-bZIP) and both can trans-activate ARE [107–109]. Under non-stressful physiological condition, Nrf2 is kept in the cytosol by Keap1, a component of an E3 ubiquitin ligase complex that targets Nrf2 for proteasomal degradation [110,111]. Oxidative stress and treatment with many agents that activate Nrf2 act by modifying either Keap1 or Nrf2 posttranslationally to cause their dissociation [88,110]. Once Nrf2 is released from Keap1, it escapes proteasomal degradation and translocates to the nucleus to induce genes involved in antioxidant defense [110]. NO’s activation of Nrf2 in endothelial cells required an increase in free zinc level intracellularly but how zinc activates Nrf2 is not clear [89]. Once Nrf2 translocates to the nucleus, it forms heterodimers with small Maf (MafG, MafK and MafF) and Jun (c-Jun, Jun-D, and Jun-B) proteins to bind to ARE [108]. Nrf2/MafG heterodimer generally activates ARE-dependent gene transcription and has also been reported to enhance Nrf2 nuclear retention [108,112]. Another key mechanism that controls Nrf2 nuclear level is importin α7-mediated Keap1 nuclear import [111]. Thus, Keap1 shuttles between the nucleus and the cytoplasm and the nuclear import requires the interaction of importin α7 with the C-terminal Kelch domain of Keap1 [111]. Keap1 contains a strong nuclear export signal, which facilitates the nuclear export of Keap1-Nrf2 complex to keep Nrf2 activation and ARE-dependent gene induction under control [111]. Keap1 does not control Nrf1’s activity; instead Nrf1 is primarily localized to the membrane of the endoplasmic reticulum and is released and translocates to the nucleus during endoplasmic reticulum stress [113]. Nrf1 knockout mice die in utero but fetal hepatocytes and embryonal fibroblasts have lower GSH levels and are more susceptible to oxidative stress [107,114]. Nrf2 knockout mice also exhibit lower GSH levels and are more susceptible to acetaminophen-induced liver injury [115]. Both Nrf1 and Nrf2 knockout mice have lower GCLC expression [114,115] and overexpression of Nrf1 and Nrf2 can induce the human GCLC promoter activity [104,116]. Nrf1 and Nrf2 may induce GCLC promoter directly and indirectly. Although the rat GCLC promoter also contain a distal ARE 4 kb upstream [117], the 1.8 kb 5′-flanking region of the rat GCLC does not contain any consensus ARE element (5′-G/ATG/TAG/CNNNGCA/G-3′) [105] and yet TBH treatment can induce the reporter activity driven by this 1.8 kb construct [118]. The explanation lies in cross talks between Nrf1/Nrf2 and AP-1 and NF B family members. The basal expression and nuclear binding activities of c-Jun, c-Fos, p50 and p65 are lower in fibroblast cells lacking Nrf1 or Nrf2. Other AP-1 and NF B family members are either unaffected (JunB, JunD) or increased (Fra-1, JAB1, and c-Rel). Overexpression of Nrf1 and Nrf2 restored the rat 1.8 kb-GCLC promoter activity and response to TBH by enhancing the expression of key NFκB and AP-1 family members [118]. However, this was blocked if the AP-1 and NFκB binding sites were mutated, proving the importance of these cis-acting elements at least in the rat GCLC. Interactions between NF B and AP-1 also occur [97]. Tumor necrosis factor α (TNFα) induces NFκB and AP-1 nuclear-binding activities and both are required for normal expression of both GCL subunits and GS in rat. While all three genes have multiple AP-1-binding sites, only GCLC has an NFκB-binding site. The explanation for the ability of NFκB to induce rat GCLM and GS promoter activity is that NFκB can increase AP-1 expression and nuclear binding activity. Thus, both c-Jun and NFκB are required for basal and TNFα-mediated induction of GSH synthetic enzymes in H4IIE cells (a rat hepatoma cell line). While NFκB may exert a direct effect on the GCLC promoter, it induces the GCLM and GS promoters indirectly via c-Jun. These findings further illustrate the complex cross talks among the different families of transcription factors. In mouse, both GCLC and GCLM have NFκB binding sites and the basal expression of these two genes requires NFκB [119].
In addition to ARE/EpRE, AP-1 and NFκB, c-Myc has been identified to also contribute to the basal expression and induction of human GCL under oxidative stress [120]. Thus, down-regulation of c-Myc lowered GSH while overexpression of c-Myc increased GSH. Two noncanonical c-Myc binding sites (CACATG, E box) are present in the human GCLC promoter at −559/−554 and −500/−495 and together with ARE4, are responsible for H2O2-induced GCLC promoter activity [120]. The signaling pathway activated by H2O2 involves ERK-mediated phosphorylation and activation of c-Myc [120].
Recently, c-AMP-response element binding protein (CREB) was found to be the key transcription factor rather than Nrf1 or Nrf2, in binding to ARE4 and induction of GCLC expression in response to anthocyanin (a flavonoid antioxidant) treatment in HepG2 cells [121]. The induction occurred when Nrf1 or Nrf2 was silenced using siRNA but whether CREB binds to the ARE4 alone or requires the participation of other transcription factors is unknown.
In addition to increased gene transcription, DEM and 4-HNE have also been found to stabilize the GCLC mRNA [102,122]. However, the mechanism of this effect remains unknown.
4.2 GCLC post-translational regulation
GCL is also regulated post-translationally. GSH synthesis was inhibited by hormone-mediated activation of various signal transduction pathways [123,124]. These hormones are secreted under stressful conditions, many of which have associated lower hepatic GSH levels [10,13]. The fall in hepatic GSH level occurs by both an increase in sinusoidal GSH efflux [125,126] and an inhibition of GSH synthesis [123]. This may represent the hepatic stress response by increasing the systemic delivery of GSH and cysteine and channeling cysteine to synthesis of stress proteins [127]. We showed that GCLC is phosphorylated directly by activation of protein kinase A (PKA), protein kinase C (PKC) or Ca2+-calmodulin kinase II (CMK) [128]. Cultured hepatocytes exhibit basal GCLC phosphorylation, which increased when treated with DBcAMP or phenylephrine, suggesting GCLC may be under a basal inhibitory tone [128]. Thus, phosphorylation-dephosphorylation may be an important physiologic regulator of GCL. Since many pathologic and toxic conditions can lead to activation of CMK and phosphorylation of GCLC, inhibition of GCL may further contribute to toxicity. Consistent with this notion is the report that toxic doses of acetaminophen suppressed hepatic GSH synthesis in rats [129].
GCLC can be cleaved by a caspase 3-dependent mechanism from the full-length 73 kDa to a 60 kDa form during apoptosis induced by TGF-β1, TNFα and α-Fas [45,130]. Cleavage of GCLC occurs at Asp499 within the sequence AVVD499G, which is located upstream of Cys553 thought to be important for disulfide bond formation with GCLM [35,45]. Theoretically, this would result in decreased GCL activity but this was not observed during apoptotic cell death [45]. Cleavage of GCLC at Asp499 generates a 13 kDa-C-terminal fragment with a N-terminal glycine residue that is predicted to be a myristoylation site [45]. While myristoylation was demonstrated when a GCLC fragment was overexpressed [131], whether this actually occurs during apoptotic cell death remains unclear.
Recently the lipid peroxidation product 4-HNE was shown to directly adduct GCLC Cys553 (and GCLM Cys35) in vitro [132]. Formation of 4-HNE GCLC adduct increased the activity of monomeric GCLC but inhibited formation of GCL holoenzyme and lowered GCL holoenzyme activity [132]. In cells where GCLC predominates, this mechanism may allow increased GSH synthesis by increasing the enzymatic activity of monomeric GCLC. However, whether this occurs in vivo remains to be examined.
4.3 Regulation of GCLM
GCLM plays a critical regulatory role on the overall function of GCL [38,43]. The two subunits of GCL are often coordinately induced by oxidative stress but as described above, hormones (insulin, hydrocortisone, TGF-β1) and rapid growth induce GCLC selectively in the liver. The exception is HGF, which induced the expression of both GCL subunits in hepatocytes under low-density condition [29]. GCLM is induced by xenobiotics such as β-NF and TBH [92,93,104,105,133,134]. Similar to the human GCLC, up-regulation of the human GCLM by β-NF involved binding of transcription factor Nrf2 (possibly in complexes with other Jun or Maf proteins as in GCLC) to a functional ARE/EpRE site located at −302 of the human GCLM [104,134]. Also, there is a critical c-Myc-binding E-box at −1609/−1604 of the human GCLM promoter that in conjunction with the proximal ARE, mediate the full induction of the GCLM promoter under H2O2-induced stress [120]. Nrf2 is also required for GCLM induction by 15d-PGJ2 and physiological 4-HNE concentrations [84–85]. Transcription factors and cis-acting elements important for mouse and rat GCLM genes are similar to the human gene. Fibroblast cells derived from Nrf1 and Nrf2 knockout mice have lower GSH levels and reduced basal expression of GCLM [107,118]. The rat GCLM promoter also has a functional ARE element (−295 to −285) [97]. This ARE element is important for basal expression and TNFα-mediated induction of rat GCLM [97]. AP-1, NFκB and Nrf2 are positive regulators of the rat GCLM gene and are induced by TNFα treatment [97]. While AP-1 and Nrf2 have direct effects on the rat GCLM promoter, NFκB activates it indirectly via AP-1 [97]. Not all inducers of oxidative stress induce GCLM. Ethanol and TGF-β1 treatments do not affect rat GCLM expression [73,135]. The reason for this discordance is not clear. Finally, GCLM expression also changes in a biphasic manner during BDL and when hepatocytes are treated with lithocholic acid [90,136 - see below under cholestatic liver injury].
Post-transcriptional regulation of GCLM also occurs with 4-HNE treatment, which increased the stability of GCLM mRNA by an unknown mechanism that required de novo protein synthesis [122]. This is in contrast to the effect of 4-HNE on GCLC mRNA stability, which did not require de novo protein synthesis. As3+ increased GCLM mRNA stability in addition to GCLM transcription but the mechanism is unclear [87].
5. Regulation of GSH synthase (GS)
GS has received relatively little attention in the field of GSH biosynthesis. GS is composed of two identical subunits and is not subject to feedback inhibition by GSH [48]. GS deficiency in humans can result in dramatic metabolic consequences because the accumulated γ-glutamylcysteine is converted to 5-oxoproline, which can cause severe metabolic acidosis, hemolytic anemia and central nervous system damage [137,138]. Choi et al described decreased hepatic GSH levels, which correlated with reduced GS activity in Tat transgenic mice [139]. A decrease in GS activity alone without a change in GCL and a fall in GSH levels occurred after surgical trauma in human skeletal muscle [50]. These findings seem to contradict the notion that GCL is rate-limiting. Although the specific activity of GS is normally 2 to 4 times that of GCL activity in normal liver [64,140], this may not be the case in other tissues and under stressful conditions. In fact, in normal human skeletal muscle, the specific activity of GS is only 36% higher than that of GCL [50]. Surgical trauma selectively reduced GS activity, which probably became rate limiting [50]. Recently, all-trans retinoic acid (ATRA), was shown to induce the expression of GS selectively (no effect on GCLC or GCLM) and GSH levels in myeloid-derived suppressor cells [141]. Taken together, these results suggest regulation of GS may also be important in determining the overall GSH synthetic capacity under certain conditions and especially in non-hepatic tissues.
We found treatments that increase the expression of both GCL subunits, such as DEM, buthionine sulfoximine (BSO), TBH, TNFα and HGF treatment of cultured rat hepatocytes and thioacetamide (TAA) treatment of rats, also increased the expression of GS [26,29,97]. In contrast, treatments that increase the expression of GCLC alone such as insulin, hydrocortisone in cultured hepatocytes and ethanol feeding in vivo, had no influence on GS expression. There are exceptions, one is liver regeneration after partial hepatectomy, another is HCC, and a third is liver-specific retinoid X receptor α (RXRα) knockout mice. GS mRNA levels changed in parallel to that of GCLC in all three conditions while GCLM mRNA levels were unchanged [26,51,142]. We speculated that when GCL is induced tremendously, the step catalyzed by GS might become limiting. A coordinated induction in the activity of both enzymes would further enhance GSH synthesis capacity. Consistent with this hypothesis, treatments that induced only GCLC increased the GSH synthesis capacity by 50 to 100% [64,135], whereas treatments that induced both GCLC and GS expression increased the GSH synthesis capacity by 161–200% [27,140].
Given the coordinated regulation of GCL and GS, it is not surprising that transcriptional regulation of these genes is quite similar. For the rat GS promoter, AP-1 serves as an enhancer directly while NF-1 acts as a repressor [143]. NFκB can also activate the rat GS promoter, albeit indirectly via AP-1 [97]. Both Nrf1 and Nrf2 overexpression induced the human GS promoter activity [144]. The human GS promoter contains two regions with homology to the NFE2 (nuclear factor erythroid 2) motif that are required for basal activity [144]. ATRA, which works via RXR, induced the expression of GS selectively and GSH levels in myeloid-derived suppressor cells [141]. ATRA treatment for 48 hours also increased GSH levels in mononuclear cells isolated from patients with metastatic renal cell carcinoma, but GS expression was not examined [141]. The mechanism of ATRA’s inductive effect on GS expression required ERK1/2 signaling but the mechanism is not clear, as ATRA treatment did not influence the expression of Nrf2 or NFκB and it had no effect on the GS promoter activity [141]. This suggests the possibility of post-transcriptional regulation of GS by ATRA but this remains to be examined. Post-translational regulation of GS has not been reported.
6. Dysregulation of GSH synthesis
There is accumulating data that reduced GSH levels occur in many human diseases and they contribute to worsening of the condition [4]. While oxidative injury plays a dominant role in GSH depletion in many of these disorders, some are causally related to reduced expression of GSH synthetic enzymes [13]. In the most severe cases, polymorphisms of GCLC and/or GCLM that result in significantly reduced GCL expression and activity can present with severe phenotype including hemolytic anemia, aminoaciduria and spinocerebellar degeneration [reviewed in 45]. GCLC and GCLM polymorphisms have been reported in many disorders, including schizophrenia, cardiovascular diseases, stroke, and asthma [45,145,146]. Outside of polymorphism, decreased GSH synthesis occurs during aging, diabetes mellitus, fibrotic diseases (including cystic fibrosis and pulmonary fibrosis), endotoxemia, and several hepatic disorders such as cholestatic and alcoholic liver injury [13,15]. The opposite situation, namely increased GSH synthesis, plays an important role in conferring drug and/or radiation resistance to many different cancers [13]. Targeting this increase in GCL activity using BSO (the irreversible GCL inhibitor) is now often used as an adjuvant chemotherapeutic agent in cancer treatment [13]. Given the central role of hepatic GSH in systemic GSH homeostasis, four liver disorders (cholestasis, endotoxemia, alcohol and fibrosis) where decreased GSH synthesis may participate in the pathogenesis of liver injury are described in more detail.
6.1. Cholestasis
Cholestasis is the underlying mechanism for many chronic liver diseases. The underlying mechanism for cell toxicity is thought to be retention of toxic bile acids, which can cause oxidative stress, apoptosis, fibrosis leading to cirrhosis [147,148]. Recently we used the BDL model in mice and showed that hepatic expression of GSH synthetic enzymes increased early on likely as an adaptive response to oxidative stress but decreased markedly along with GSH levels during later stages of BDL [136]. A key observation from this study was the fall in Nrf2 nuclear binding to the ARE two weeks after BDL. A similar pattern of early induction followed by fall in GSH synthetic enzymes also occurred when Huh-7 cells (a human hepatoma cell line) was treated with lithocholic acid [90]. In both BDL and lithocholic acid-treated Huh-7 cells, the fall in expression of GSH synthetic enzymes coincided with an increase in the expression of several Maf proteins (c-Maf, MafG and MafK) as well as increased c-Maf and MafG nuclear binding to ARE. MafG and MafK are small Mafs that have been reported to heterodimerize with Nrf2 to either activate or repress ARE-dependent genes [108,149]. Small Mafs lack transcriptional activation domain and can form homodimers to repress ARE-mediated gene expression [150]. In addition, large Maf protein such as c-Maf can bind to ARE as homodimers and heterodimers with small Mafs (but not Nrfs) to repress ARE-mediated gene expression [151]. Given these known effects of small Mafs and c-Maf, we speculated that the induction in Mafs and displacement of Nrf2 from nuclear binding to ARE during cholestasis might have caused the fall in the expression of GSH synthetic enzymes. Consistent with this, blocking either c-Maf or MafG induction during BDL protected against the fall in expression of GSH synthetic enzymes, GSH levels and BDL-induced liver injury [90]. Interestingly, ursodeoxycholic acid (UDCA), the only medication approved by the FDA for the treatment of primary biliary cirrhosis [152], a chronic cholestatic disorder, and SAMe were able to raise nuclear Nrf2 level, block the increase in MafG and c-Maf expression, protect against the fall in expression of GSH synthetic enzymes and GSH levels in these models [90,136]. Combining UDCA and SAMe exerted additional benefit, suggesting they have different mechanisms. Murine cholestatic liver injury is the first example that illustrates the importance of Maf proteins on ARE-dependent gene expression in liver pathology.
6.2 Endotoxemia
Lipopolysaccharide (LPS, synonymous as endotoxin) is a major constituent of the outer cell wall of all gram-negative bacteria that can trigger the synthesis and release of pro-inflammatory cytokines and inducible nitric oxide synthase (iNOS) [153,154]. Liver clears gut-derived LPS [154]. This explains why endotoxemia occurs in cirrhotic patients and the degree of endotoxemia correlates with the degree of liver failure [154]. Endotoxemia also participates in worsening of alcoholic liver disease and non-alcoholic steatohepatitis [154,155]. Endotoxemia lowers GSH levels in the liver [156,157], peritoneal macrophages and lymphocytes [158]. Septic patients have lower blood GSH:GSSG ratios [159]. Exogenous GSH treatment suppressed LPS-induced systemic inflammatory response and reduced mortality [160]. GSH level is an important variable that determines susceptibility to LPS-induced injury in multiple tissues [157,160,161]. This may be related to GSH’s ability to influence toll like receptor 4 (TLR4) signaling. Specifically, LPS-induced mortality and TNFα secretion were higher when GSH level was reduced [162]. The fall in GSH is multifactorial. In liver, increased GSH efflux and increased oxidative stress both contribute [153,156]. One study showed that a major mechanism of hepatic GSH depletion during endotoxemia is a fall in GCLC mRNA level and GCL activity [157]. Consistent with this, we found that hepatic GSH level fell more than 50% following LPS, coinciding with a comparable fall in the mRNA and protein levels of GCLC and GCLM (50–60%) [163]. GS expression fell to a lesser extent (40% fall). SAMe pretreatment protected against liver injury and prevented the fall in GCLC and GCLM expression and GSH level. Nearly maximum inhibition in the expression of GSH synthetic enzymes occurred as early as six hours after LPS administration [163]. The molecular mechanisms remain to be elucidated.
6.3 Alcohol
Alcoholic liver disease patients have low hepatic and plasma GSH levels due to multiple mechanisms such as oxidative stress, nutritional deficiency and abnormalities in the methionine metabolic pathway that impairs cysteine availability [164–166]. In addition, we found a 50% fall in the mRNA levels of GCLC and GS (GCLM was unchanged) in patients hospitalized for alcoholic hepatitis [166]. The mechanism for this is unclear but these abnormalities may contribute to the high morbidity and mortality associated with this disorder.
6.4 Fibrogenesis
HSCs are the key effectors in hepatic fibrogenesis [167]. HSCs reside in the space of Disse and in normal liver are the major storage sites of vitamin A. Following chronic liver injury, HSCs proliferate, lose their vitamin A and undergo a major phenotypical transformation to α-smooth muscle actin (α-SMA) positive activated HSCs, which produce a wide variety of collagenous and non-collagenous extracellular matrix (ECM) proteins [168]. The profibrogenic potential of activated HSCs is due to their capacity to synthesize fibrotic matrix proteins and components that inhibit fibrosis degradation. Pro-fibrogenic factors include TGF-β [169], connective tissue growth factor [170], leptin [171] and platelet derived growth factor [172]. Activation of HSC is mediated by various cytokines and ROS released from damaged hepatocytes and activated Kupffer cells [173]. Hence, inhibition of HSC activation and its related events such as ECM formation and cellular proliferation are important targets for therapeutic intervention. In both hepatic and pulmonary fibrosis, TGF-β1 has been shown to target GSH synthesis (see above under GCLC regulation) [15,73,82]. EGCG and curcumin, two agents that exert anti-fibrotic effect in hepatic HSCs, require de novo GSH synthesis to exert this effect [73,82]. In BDL, preventing the fall in hepatic GSH also resulted in amelioration of hepatic fibrosis [90]. Consistent with the importance of GSH in hepatic fibrogenesis, we found that a lower hepatic GSH level greatly potentiated BDL-induced fibrosis and if induction in GCLC expression was blocked (by using RNAi), the therapeutic efficacy of UDCA and SAMe was nearly lost [174]. We also established that GCLC expression is a critical factor in determining the phenotype of rat HSCs (GCLM and GS were unchanged at the protein level) [174]. Specifically, GCLC expression fell during HSC activation and increased as activated HSCs revert to quiescence. Blocking the increase in GCLC expression kept HSCs in an activated state. Although activated HSCs have increased nuclear MafG level, formation of Nrf2/MafG heterodimer and binding to ARE is greatly diminished. In contrast, quiescent HSCs have markedly lower total nuclear MafG level but increased Nrf2/MafG heterodimerization and binding to ARE. This is due to enhanced sumoylation of Nrf2 and MafG by SUMO-1 in the quiescent state, which facilitated heterodimerization and binding to ARE [174]. Thus, a key mechanism that controls Nrf2/MafG trans-activation of GCLC ARE is sumoylation by SUMO-1 in HSCs. Taken together, a fall in GSH facilitates activation of HSCs and fibrosis to proceed. Targeting this is an attractive therapeutic strategy that yielded promising results in animal models of pulmonary and hepatic fibrosis [15, 90] but human trials have not been as positive [15].
7. Concluding remarks
Up until recently, most of the literature on GSH synthesis has focused on understanding how the enzymes are regulated transcriptionally and post-transcriptionally. There are now increasing evidence that show dysregulation of GSH synthesis in multiple conditions, such as aging, diabetes, pulmonary and hepatic fibrosis, alcoholic and cholestatic liver injuries. Some of these have been confirmed to occur also in humans. GCLC and GCLM polymorphisms have also gained attention as another determinant of chronic oxidative injury to various organs. While the status of screening for these polymorphisms remains to be established, uncovering the molecular mechanisms responsible for the dysregulation in GSH synthesis may provide novel therapeutic approaches.
Highlights.
GSH regulates antioxidant defense, growth, death, immune function, and fibrogenesis.
GSH is synthesized via two enzymatic steps that are regulated at multiple levels.
GSH synthesis is dysregulated in multiple human diseases.
Acknowledgments
This work was supported by NIH grant R01DK092407
Abbreviations (in alphabetical order)
- 4-HNE
4-hydroxynonenal
- 15d-PGJ2
15-deoxy-Δ12,14-prostaglandin J2
- AP-1
activator protein-1
- As3+
trivalent arsenite
- α-SMA
a-smooth muscle actin
- ARE
antioxidant response element
- ATRA
all-trans retinoic acid
- BDL
bile duct ligation
- BHMT
betaine homocysteine methyltransferase
- β-NF
β-naphthoflavone
- BSO
buthionine sulfoximine
- CBS
cystathionine β synthase
- CMK
Ca2+-calmodulin kinase II
- CNC-bZIP
cap ‘n’ collar-basic leucine zipper proteins
- CREB
c-AMP-response element binding protein
- DEM
diethyl maleate
- ECM
extracellular matrix
- EpRE
electrophile response element
- GCL
glutamate-cysteine ligase
- GCLC
GCL-catalytic subunit
- GCLM
GCL-modifier subunit
- GGT
γ-glutamyltranspeptidase
- GPx
GSH peroxidase
- GS
GSH synthase
- GSH
glutathione
- GSSG
oxidized GSH
- HCC
hepatocellular carcinoma
- Hcy
homocysteine
- HGF
hepatocyte growth factor
- HSC
hepatic stellate cell
- iNOS
inducible nitric oxide synthase
- LPS
lipopolysaccharide
- MAT
methionine adenosyltransferase
- MRE
metal response element
- MS
methionine synthase
- MT
methyltransferase
- NFE2
nuclear factor erythroid 2
- NO
nitric oxide
- Nrf2
nuclear factor-erythroid 2 related factor 2
- PKA
protein kinase A
- PKC
protein kinase C
- RNS
reactive nitrogen species
- RXRα
retinoid X receptor α
- ROS
reactive oxygen species
- SAH
S-adenosylhomocysteine
- SAMe
S-adenosylmethionine
- TAA
thioacetamide
- TBH
tert-butyl hydroquinone
- TGF-β1
transforming growth factor-β1
- TLR4
toll like receptor 4
- TNFα
tumor necrosis factor α
- UDCA
ursodeoxycholic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ookhtens M, Kaplowitz N. Role of the liver in interorgan homeostasis of glutathione and cyst(e)ine. Sem Liv Dis. 1998;18:313–329. doi: 10.1055/s-2007-1007167. [DOI] [PubMed] [Google Scholar]
- 2.Kaplowitz N, Aw TY, Ookhtens M. The regulation of hepatic GSH. Ann Rev Pharm Toxicol. 1985;25:714–744. doi: 10.1146/annurev.pa.25.040185.003435. [DOI] [PubMed] [Google Scholar]
- 3.Akerboom TPM, Bilizer M, Sies H. The relationship of biliary GSSG efflux and intracellular GSSG content in perfused rat liver. J Biol Chem. 1982;257:4248–4252. [PubMed] [Google Scholar]
- 4.Ballatori N, Krance SM, Notenboom S, Shi S, Tieu K, Hammond CL. Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem. 2009;390:191–214. doi: 10.1515/BC.2009.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forman HJ, Zhang H, Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Asp Med. 2009;30:1–12. doi: 10.1016/j.mam.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meredith MJ, Reed DJ. Status of the mitochondrial pool of glutathione in the isolated hepatocyte. J Biol Chem. 1982;257:3747–3753. [PubMed] [Google Scholar]
- 7.Hwang C, Sinsky AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 8.Yuan L, Kaplowitz N. Glutathione in liver diseases and hepatotoxicity. Mol Asp Med. 2009;30:29–41. doi: 10.1016/j.mam.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 9.Meister, Anderson ME. Glutathione. Ann Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 10.Lauterburg BH, Adams JD, Mitchell JR. Hepatic glutathione homeostasis in the rat: Efflux accounts for glutathione turnover. Hepatology. 1984;4:586–590. doi: 10.1002/hep.1840040402. [DOI] [PubMed] [Google Scholar]
- 11.Fernández-Checa J, Lu SC, Ookhtens M, DeLeve L, Runnegar M, Yoshida H, Saiki H, Kannan R, Garcia-Ruiz C, Kuhlenkamp JF, Kaplowitz N. The Regulation of Hepatic Glutathione. In: Tavoloni N, Berk PD, editors. Hepatic Anion Transport and Bile Secretion: Physiology and Pathophysiology. Marcel Dekker; New York: 1992. pp. 363–395. [Google Scholar]
- 12.Suthanthiran M, Anderson ME, Sharma VK, Meister A. Glutathione regulates activation-dependent DNA synthesis in highly purified normal human T lymphocytes stimulated via the CD2 and CD3 antigens. Proc Natl Acad Sci USA. 1990;87:3343–3347. doi: 10.1073/pnas.87.9.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu SC. Regulation of glutathione synthesis. Mol Asp Med. 2009;30:42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pallardó FV, Markovic J, García JL, Viña J. Role of nuclear glutathione as a key regulator of cell proliferation. Mol Asp Med. 2009;30:77–85. doi: 10.1016/j.mam.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Liu RM, Gaston Pravia KA. Oxidative stress and glutathione in TGF-β-mediated fibrogenesis. Free Rad Biol Med. 2010:1–15. doi: 10.1016/j.freeradbiomed.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernández-Checa J, Kaplowitz N, Garcia-Ruiz C, Colell A, Miranda M, Mai M, Ardite E, Morales A. GSH transport in mitochondria: defense against TNF-induced oxidative stress and alcohol-induced defect. Am J Physiol. 1997;273:G7–G17. doi: 10.1152/ajpgi.1997.273.1.G7. [DOI] [PubMed] [Google Scholar]
- 17.Garcia-Ruiz C, Fernández-Checa JC. Mitochondrial glutathione: hepatocellular survival-death switch. J Gastroenterol Hepatol. 2006;21:S3–6. doi: 10.1111/j.1440-1746.2006.04570.x. [DOI] [PubMed] [Google Scholar]
- 18.Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Meister A. Glutathione. In: Arias IM, Jakoby WB, Popper H, Schachter D, Shafritz DA, editors. The Liver: Biology and Pathobiology. 2. Raven Press; New York: 1988. pp. 401–417. [Google Scholar]
- 20.Shaw JP, Chou I. Elevation of intracellular glutathione content associated with mitogenic stimulation of quiescent fibroblasts. J Cell Physiol. 1986;129:193–198. doi: 10.1002/jcp.1041290210. [DOI] [PubMed] [Google Scholar]
- 21.Messina JP, Lawrence DA. Cell cycle progression of glutathione-depleted human peripheral blood mononuclear cells is inhibited at S phase. J Immunol. 1989;143:1974–1981. [PubMed] [Google Scholar]
- 22.Hamilos DL, Zelarney P, Mascali JJ. Lymphocyte proliferation in glutathione-depleted lymphocytes: direct relationship between glutathione availability and the proliferative response. Immunopharmacology. 1989;18:223–235. doi: 10.1016/0162-3109(89)90020-9. [DOI] [PubMed] [Google Scholar]
- 23.Iwata S, Hori T, Sato N, Ueda-Taniguchi Y, Yamabe T, Nakamura H, Masutani H, Yodoi J. Thiol-mediated redox regulation of lymphocyte proliferation. Possible involvement of adult T cell leukemia-derived factor and glutathione in transferrin receptor expression. J Immunol. 1994;152:5633–5642. [PubMed] [Google Scholar]
- 24.Poot M, Teubert H, Rabinovitch PS, Kavanagh TJ. De novo synthesis of glutathione is required for both entry into and progression through the cell cycle. J Cell Physiol. 1995;163:555–560. doi: 10.1002/jcp.1041630316. [DOI] [PubMed] [Google Scholar]
- 25.Lu SC, Ge J. Loss of suppression of GSH synthesis under low cell density in primary cultures of rat hepatocytes. Am J Physiol. 1992;263:C1181–1189. doi: 10.1152/ajpcell.1992.263.6.C1181. [DOI] [PubMed] [Google Scholar]
- 26.Huang ZZ, Chen CJ, Zeng ZH, Yang HP, Oh J, Chen LX, Lu SC. Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. FASEB J. 2000 doi: 10.1096/fj.00–0445fje. [DOI] [PubMed] [Google Scholar]
- 27.Huang ZZ, Li H, Cai J, Kuhlenkamp J, Kaplowitz N, Lu SC. Changes in glutathione homeostasis during liver regeneration in the rat. Hepatology. 1998;27:147–153. doi: 10.1002/hep.510270123. [DOI] [PubMed] [Google Scholar]
- 28.Carretero J, Obrador E, Anasagasti MJ, Martin JJ, Vidal-Vanaclocha F, Estrela JM. Growth-associated changes in glutathione content correlate with liver metastatic activity of B16 melanoma cells. Clin Exp Metastasis. 1999;17:567–574. doi: 10.1023/a:1006725226078. [DOI] [PubMed] [Google Scholar]
- 29.Yang HP, Magilnick N, Xia M, Lu SC. Effects of hepatocyte growth factor on glutathione synthesis, growth, and apoptosis is cell density-dependent. Exp Cell Res. 2008;314:398–412. doi: 10.1016/j.yexcr.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holmgren A. Regulation of ribonucleotide reductase. Current Topics in Cell Reg. 1981;19:47–76. doi: 10.1016/b978-0-12-152819-5.50019-1. [DOI] [PubMed] [Google Scholar]
- 31.Vivancos PD, Wolff T, Markovic J, Pallardó FV, Foyer CH. A nuclear glutathione cycle within the cell cycle. Biochem J. 2010;431:169–178. doi: 10.1042/BJ20100409. [DOI] [PubMed] [Google Scholar]
- 32.Garcia-Ruiz C, Fernández-Checa JC. Redox regulation of hepatocyte apoptosis. J Gastroenterol Hepatol. 2007;22:S38–42. doi: 10.1111/j.1440-1746.2006.04644.x. [DOI] [PubMed] [Google Scholar]
- 33.Lemasters JJ. Dying a thousand deaths: redundant pathways from different organelles to apoptosis and necrosis. Gastroenterology. 2005;129:351–360. doi: 10.1053/j.gastro.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Hall AG. The role of glutathione in the regulation of apoptosis. Eur J Clin Invest. 1999;29:238–245. doi: 10.1046/j.1365-2362.1999.00447.x. [DOI] [PubMed] [Google Scholar]
- 35.Franklin CC, Krejsa CM, Pierce RH, White CC, Fausto N, Kavanagh TJ. Caspase-3-dependent cleavage of the gluatamate-L-cysteine ligase catalytic subunit during apoptotic cell death. Am J Pathol. 2002;160:1887–1894. doi: 10.1016/S0002-9440(10)61135-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghibelli L, Fanelli C, Rotilio G, Lafavia E, Coppola S, Colussi C, Civitareale P, Ciriolo MR. Rescue of cells from apoptosis by inhibition of active GSH extrusion. FASEB J. 1998;12:479–486. doi: 10.1096/fasebj.12.6.479. [DOI] [PubMed] [Google Scholar]
- 37.Yan N, Meister A. Amino acid sequence of rat kidney g-glutamylcysteine synthetase. J Biol Chem. 1990;265:1588–1593. [PubMed] [Google Scholar]
- 38.Huang C, Anderson ME, Meister A. Amino acid sequence and function of the light subunit of rat kidney γ-glutamylcysteine synthetase. J Biol Chem. 1993;268:20578–20583. [PubMed] [Google Scholar]
- 39.Gipp JJ, Chang C, Mulcahy RT. Cloning and nucleotide sequence of a full-length cDNA for human liver γ-glutamylcysteine synthetase. Biochem Biophys Res Comm. 1992;185:29–35. doi: 10.1016/s0006-291x(05)80950-7. [DOI] [PubMed] [Google Scholar]
- 40.Gipp JJ, Bailey HH, Mulcahy RT. Cloning and sequence of the cDNA for the light subunit of human liver γ-glutamylcysteine synthetase and relative mRNA levels for heavy and light subunits in human normal tissues. Biochem Biophys Res Comm. 1995;206:584–589. doi: 10.1006/bbrc.1995.1083. [DOI] [PubMed] [Google Scholar]
- 41.Dalton TP, Chen Y, Schneider SN, Nebert DW, Shertzer HG. Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Rad Biol Med. 2004;37:1511–1526. doi: 10.1016/j.freeradbiomed.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 42.Seelig GF, Simondsen RP, Meister A. Reversible dissociation of γ-glutamylcysteine synthetase into two subunits. J Biol Chem. 1984;259:9345–9347. [PubMed] [Google Scholar]
- 43.Huang C, Chang L, Anderson ME, Meister A. Catalytic and regulatory properties of the heavy subunit of rat kidney γ-glutamylcysteine synthetase. J Biol Chem. 1993;268:19675–19680. [PubMed] [Google Scholar]
- 44.Yang Y, Dieter MZ, Chen Y, Shertzer HG, Nebert DW, Dalton TP. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm (−/−) knockout mouse: novel model system for a severely compromised oxidative stress response. J Biol Chem. 2002;277:49446–49452. doi: 10.1074/jbc.M209372200. [DOI] [PubMed] [Google Scholar]
- 45.Franklin CC, Backos DS, Mohar I, White CC, Forman HJ, Kavanagh TJ. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate-cysteine ligase. Mol Asp Med. 2009:86–98. doi: 10.1016/j.mam.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Richman PG, Meister A. Regulation of γ-glutamylcysteine synthetase by nonallosteric feedback inhibition by glutathione. J Biol Chem. 1975;250:1422–1426. [PubMed] [Google Scholar]
- 47.Bannai S, Tateishi N. Role of membrane transport in metabolism and function of glutathione in mammals. J Membrane Biol. 1986;89:1–8. doi: 10.1007/BF01870891. [DOI] [PubMed] [Google Scholar]
- 48.Oppenheimer L, Wellner VP, Griffith OW, Meister A. Glutathione synthetase. Purification from rat kidney and mapping of the substrate binding sites. J Biol Chem. 1979;254:5184–90. [PubMed] [Google Scholar]
- 49.Grant CM, MacIver FH, Dawes IW. 1997. Glutathione synthetase is dispensable for growth under both normal and oxidative stress conditions in the yeast Saccharomyces cerevisiae due to an accumulation of the dipeptide γ-glutamylcysteine. Mol Bio Cell. 1997;8:1699–1707. doi: 10.1091/mbc.8.9.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo JL, Hammarqvist F, Andersson K, Wernerman J. Surgical trauma decreases glutathione synthetic capacity in human skeletal muscle tissue. Am J Physiol. 1998;275:E359–365. doi: 10.1152/ajpendo.1998.275.2.E359. [DOI] [PubMed] [Google Scholar]
- 51.Huang ZZ, Yang HP, Chen CJ, Zeng ZH, Lu SC. Inducers of γ-glutamylcysteine synthetase and their effects on glutathione synthetase expression. Biochim Biophys Acta. 2000;1493:48–55. doi: 10.1016/s0167-4781(00)00156-1. [DOI] [PubMed] [Google Scholar]
- 52.Kilberg MS. Amino acid transport in isolated rat hepatocytes. J Membrane Biol. 1982;69:1–12. doi: 10.1007/BF01871236. [DOI] [PubMed] [Google Scholar]
- 53.Takada A, Bannai S. Transport of cystine in isolated rat hepatocytes in primary culture. J Biol Chem. 1984;259:2441–2445. [PubMed] [Google Scholar]
- 54.Tarver H, Schmidt CLA. The conversion of methionine to cystine: Experiments with radioactive sulfur. J Biol Chem. 1939;130:67–80. [Google Scholar]
- 55.Lu SC, Mato JM. S-adenosylmethionine in liver health, injury and cancer. Physiol Rev. 2012 doi: 10.1152/physrev.00047.2011. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem. 1990;1:228–237. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- 57.Mulcahy RT, Bailey HH, Gipp JJ. Up-regulation of γ-glutamylcysteine synthetase activity in melphalan-resistant human multiple myeloma cells expressing increased glutathione levels. Cancer Chemother Pharmacol. 1994;34:67–71. doi: 10.1007/BF00686114. [DOI] [PubMed] [Google Scholar]
- 58.Mulcahy RT, Untawale S, Gipp JJ. Transcriptional up-regulation of g-glutamylcysteine synthetase gene expression in melphalan-resistant human prostate carcinoma cells. Mol Pharmacol. 1994;46:909–914. [PubMed] [Google Scholar]
- 59.Mulcahy RT, Bailey HH, Gipp JJ. Transfection of complementary DNAs for the heavy and light subunits of human γ-glutamylcysteine synthetase results in an elevation of intracellular glutathione and resistance to melphalan. Cancer Res. 1995;55:4771–4775. [PubMed] [Google Scholar]
- 60.Godwin AK, Meister A, O’Dwyer PJ, Huang CS, Hamilton TC, Anderson ME. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc Natl Acad Sci USA. 1992;89:3070–3074. doi: 10.1073/pnas.89.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woods JS, Davis HA, Baer RP. Enhancement of γ-glutamylcysteine synthetase mRNA in rat kidney by methyl mercury. Arch Biochem Biophys. 1992;296:350–353. doi: 10.1016/0003-9861(92)90583-i. [DOI] [PubMed] [Google Scholar]
- 62.Shi MM, Kugelman A, Iwamoto T, Tian L, Forman HJ. Quinone-induced oxidative stress elevates glutathione and induces γ-glutamylcysteine synthetase activity in rat lung epithelial L2 cells. J Biol Chem. 1994;269:26512–26317. [PubMed] [Google Scholar]
- 63.Yamane Y, Furuichi M, Song R, Van NT, Mulcahy RT, Ishikawa T, Kuo MT. Expression of multidrug resistance protein/GS-X pump and γ-glutamylcysteine synthetase genes is regulated by oxidative stress. J Biol Chem. 1998;273:31075–31085. doi: 10.1074/jbc.273.47.31075. [DOI] [PubMed] [Google Scholar]
- 64.Lu SC, Ge J, Kuhlenkamp J, Kaplowitz N. Insulin and glucocorticoid dependence of hepatic γ-glutamylcysteine synthetase and GSH synthesis in the rat: Studies in cultured hepatocytes and in vivo. J Clin Invest. 1992;90:524–532. doi: 10.1172/JCI115890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cai J, Sun WM, Lu SC. Hormonal and cell density regulation of hepatic γ-glutamylcysteine synthetase gene expression. Molecular Pharmacol. 1995;48:212–218. [PubMed] [Google Scholar]
- 66.Cai J, Huang ZZ, Lu SC. Differential regulation of γ-glutamylcysteine synthetase heavy and light subunit gene expression. Biochem J. 1997;326:167–172. doi: 10.1042/bj3260167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoshida K, Hirokawa J, Tagami S, Kawakami Y, Urata Y, Kondo T. Weakened cellular scavenging activity against oxidative stress in diabetes mellitus: regulation of glutathione synthesis and efflux. Diabetologia. 1995;38:201–210. doi: 10.1007/BF00400095. [DOI] [PubMed] [Google Scholar]
- 68.Kim SK, Woodcroft KJ, Khodadadeh SS, Novak RF. Insulin signaling regulates γ-glutamylcysteine ligase catalytic subunit expression in primary cultured rat hepatocytes. J Pharmacol Exp Therap. 2004;311:99–108. doi: 10.1124/jpet.104.070375. [DOI] [PubMed] [Google Scholar]
- 69.Li S, Li X, Rozanski GJ. Regulation of glutathione in cardiac myocytes. J Mol Cell Cardiol. 2003;35:1145–1152. doi: 10.1016/s0022-2828(03)00230-x. [DOI] [PubMed] [Google Scholar]
- 70.Langston JW, Circu ML, Aw TY. Insulin stimulation of γ-glutamylcysteine ligase catalytic subunit expression increases endothelial GSH during oxidative stress: Influence of low glucose. Free Radic Biol Med. 2008;45:1591–1599. doi: 10.1016/j.freeradbiomed.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arsalane K, Dubois CM, Muanza T, Begin R, Boudreau F, Asselin C, Cantin AM. Transforming growth factor-beta1 is a potent inhibitor of glutathione synthesis in the lung epithelial cell line A549: transcriptional effect on the GSH rate-limiting enzyme gamma-glutamylcysteine synthetase. Am J Respir Cell Mol Biol. 1997;17:599–607. doi: 10.1165/ajrcmb.17.5.2833. [DOI] [PubMed] [Google Scholar]
- 72.Jardine H, MacNee W, Donaldson K, Rahman I. Molecular mechanism of transforming growth factor (TGF)-β2-induced glutathione depletion in alveolar epithelial cells. J Biol Chem. 2002;277:21158–21166. doi: 10.1074/jbc.M112145200. [DOI] [PubMed] [Google Scholar]
- 73.Fu Y, Zhou YJ, Lu SC, Chen AP. Epigallocatechin-3-gallate inhibits growth of activated hepatic stellate cells by enhancing the capacity of glutathione synthesis. Mol Pharmacol. 2008;73:1465–73. doi: 10.1124/mol.107.040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eaton DL, Hamel DM. Increase in γ-glutamylcysteine synthetase activity as a mechanism for butylated hydroxyanisole-mediated elevation of hepatic glutathione. Toxicol Appl Pharmacol. 1994;126:145–149. doi: 10.1006/taap.1994.1100. [DOI] [PubMed] [Google Scholar]
- 75.Borroz KI, Buetler TM, Eaton DL. Modulation of γ-glutamylcysteine synthetase large subunit mRNA expression by butylated hydroxianisole. Toxicol Appl Pharmacol. 1994;126:150–155. doi: 10.1006/taap.1994.1101. [DOI] [PubMed] [Google Scholar]
- 76.Liu RM, Vasiliou V, Zhu H, Duh JL, Tabor MW, Puga A, Nebert DW, Sainsbury M, Shertzer HG. Regulation of [Ah] gene battery enzymes and glutathione levels by 5,10-dihyroindeno[1,2-b]indole in mouse hepatoma cell lines. Carcinogenesis. 1994;15:2347–2352. doi: 10.1093/carcin/15.10.2347. [DOI] [PubMed] [Google Scholar]
- 77.Liu RM, Hu H, Robison TW, Forman HJ. Differential enhancement of γ-glutamyl transpeptidase and γ-glutamylcysteine synthetase by tert-butylhydroquinone in rat lung epithelial L2 cells. Am J Respir Cell Mol Biol. 1996;14:186–191. doi: 10.1165/ajrcmb.14.2.8630269. [DOI] [PubMed] [Google Scholar]
- 78.Mulcahy RT, Wartman MA, Bailey HH, Gipp JJ. Constitutive and β-naphthoflavone-induced expression of the human γ-glutamylcysteine synthetase heavy subunit gene is regulated by a distal antioxidant response element/TRE sequence. J Biol Chem. 1997;272:7445–7454. doi: 10.1074/jbc.272.11.7445. [DOI] [PubMed] [Google Scholar]
- 79.Kondo T, Yoshida K, Urata Y, goto S, Gasa S, Taniguchi N. γ-Glutamylcysteine synthetase and active transport of glutathione S-conjugate are responsive to heat shock in K562 erythroid cells. J Biol Chem. 1993;268:20366–20372. [PubMed] [Google Scholar]
- 80.Ha KN, Chen Y, Cai J, Sternberg P., Jr Increased glutathione synthesis through an ARE-Nrf2-dependent pathway by zinc in the RPE: Implication for protection against oxidative stress. Invest Ophthalmol Vis Sci. 2006;47:2709–2715. doi: 10.1167/iovs.05-1322. [DOI] [PubMed] [Google Scholar]
- 81.Urata Y, Honma S, Goto S, Todoroki S, Iida T, Cho S, Honma K, Kondo T. Melatonin induces γ-glutamylcysteine synthetase mediated by activator protein-1 in human vascular endothelial cells. Free Rad Biol Med. 1999;27:838–847. doi: 10.1016/s0891-5849(99)00131-8. [DOI] [PubMed] [Google Scholar]
- 82.Zheng S, Yumei F, Chen A. De novo synthesis of glutathione is a prerequisite for curcumin to inhibit hepatic stellate cell (HSC) activation. Free Rad Biol Med. 2007;43:444–453. doi: 10.1016/j.freeradbiomed.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Iles KE, Liu RM. Mechanisms of glutamate cysteine ligase (GCL) induction by 4-hydroxynonenal. Free Rad Biol Med. 2005;38:547–556. doi: 10.1016/j.freeradbiomed.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 84.Zhang H, Court N, Forman HJ. Submicromolar concentrations of 4-hydroxynonenal induce glutamate cysteine ligase expression in HBE1 cells. Redox Report. 2007;12:101–106. doi: 10.1179/135100007X162266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen ZH, Yoshida Y, Saito Y, Sekine A, Noguchi N, Niki E. Induction of adaptive response and enhancement of PC12 cell tolerance by 7-hydroxycholesterol and 15-deoxy-delta(12,14)-prostaglandin J2 through up-regulation of cellular glutathione via different mechanisms. J Biol Chem. 2006;281:14440–14445. doi: 10.1074/jbc.M600260200. [DOI] [PubMed] [Google Scholar]
- 86.Lim SY, Jang JH, Lu SC, Rahman I, Surh YJ. 15-Deoxy12,14-Prostaglandin J2 protects against nitrosative neuronal PC12 cell death through up-regulation of intracellular glutathione synthesis. J Biol Chem. 2004;279:46263–46270. doi: 10.1074/jbc.M406555200. [DOI] [PubMed] [Google Scholar]
- 87.Thompson JA, White CC, Cox DP, Chan JY, Kavanagh TJ, Fausto N, Franklin CC. Distinct Nrf1/2-independent mechanisms mediate As 3+-induced glutamate-cysteine ligase subunit gene expression in murine hepatocytes. Free Radic Biol Med. 2009;46:1614–1625. doi: 10.1016/j.freeradbiomed.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kay HY, Won Yang J, Kim TH, Lee DY, Kang B, Ryu JH, Jeon R, Kim SG. Ajoene, a stable garlic by-product, has an antioxidant effect through Nrf2-mediated glutamate-cysteine ligase induction in HepG2 cells and primary hepatocytes. J Nutr. 2010;140:1211–1219. doi: 10.3945/jn.110.121277. [DOI] [PubMed] [Google Scholar]
- 89.Cortese-Krott MM, Suschek CV, Wetzel WW, Kröncke KD, Kolb-Bachofen V. Nitric oxide-mediated protection of endothelial cells from hydrogen peroxide is mediated by intracellular zinc and glutathione. Am J Physiol Cell Physiol. 2009;296:C811–820. doi: 10.1152/ajpcell.00643.2008. [DOI] [PubMed] [Google Scholar]
- 90.Yang HP, Ko K, Xia M, Li TWH, Oh P, Li J, Lu SC. Induction of avian musculoaponeurotic fibrosarcoma proteins by toxic bile acid inhibits expression of GSH synthetic enzymes and contributes to cholestatic liver injury in mice. Hepatology. 2010;51:1291–1301. doi: 10.1002/hep.23471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mulcahy RT, Gipp JJ. Identification of a putative antioxidant response element in the 5′-flanking region of the human γ-glutamylcysteine synthetase heavy subunit gene. Biochem Biophys Res Comm. 1995;209:227–233. doi: 10.1006/bbrc.1995.1493. [DOI] [PubMed] [Google Scholar]
- 92.Galloway DC, Blake DG, Shepherd AG, McLellan LI. Regulation of human γ-glutamylcysteine synthetase: co-ordinate induction of the catalytic and regulatory subunits in HepG2 cells. Biochem J. 1997;328:99–104. doi: 10.1042/bj3280099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Moinova HR, Mulcahy RT. An electrophile responsive element (EpRE) regulates β-naphthoflavone induction of the human γ-glutamylcsteine synthetase regulatory subunit gene. J Biol Chem. 1998;273:14683–14689. doi: 10.1074/jbc.273.24.14683. [DOI] [PubMed] [Google Scholar]
- 94.Hudson FN, Kavanagh TJ. Cloning and characterization of the proximal promoter region of the mouse glutamate-L-cysteine ligase regulatory subunit gene. Biochim Biophys Acta. 2000;1492:447–451. doi: 10.1016/s0167-4781(00)00128-7. [DOI] [PubMed] [Google Scholar]
- 95.Yang HP, Huang ZZ, Wang JH, Ou XP, Lu SC. Cloning and characterization of the 5′-flanking region of the rat glutamate-cysteine ligase catalytic subunit. Biochem J. 2001;357:447–455. doi: 10.1042/0264-6021:3570447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Solis WA, Dalton TP, Dieter MZ, Freshwater S, Harrer JM, He L, Shertzer HG, Nebert DW. Glutamate-cysteine ligase modifier subunit: mouse Gclm gene structure. p. 1754. [DOI] [PubMed] [Google Scholar]
- 97.Yang HP, Magilnick N, Ou XP, Lu SC. Tumor necrosis alpha induces coordinated activation of rat GSH synthetic enzymes via NFκB and AP-1. Biochem J. 2005;391:399–408. doi: 10.1042/BJ20050795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morales A, García-Ruiz C, Miranda M, Marí M, Colell A, Ardite E, Fernández-Checa JC. Tumor necrosis factor increases hepatocellular glutathione by transcriptional regulation of the heavy subunit chain of γ-glutamylcysteine synthetase. J Biol Chem. 1997;272:30371–30379. doi: 10.1074/jbc.272.48.30371. [DOI] [PubMed] [Google Scholar]
- 99.Morales A, Miranda M, Sanchez-Reyes A, Colell A, Biete A, Fernández-Checa JC. Transcriptional regulation of the heavy subunit chain of γ-glutamylcysteine synthetase by ionizing radiation. FEBS Lett. 1998;427:15–20. doi: 10.1016/s0014-5793(98)00381-0. [DOI] [PubMed] [Google Scholar]
- 100.Rahman, Smith CA, Lawson MF, Harrison DJ, MacNee W. Induction of γ-glutamylcysteine synthetase by cigarette smoke is associated with AP-1 in human alveolar epithelial cells. FEBS Lett. 1996;396:21–25. doi: 10.1016/0014-5793(96)01027-7. [DOI] [PubMed] [Google Scholar]
- 101.Rahman, Smith CA, Antonicelli F, MacNee W. Characterization of γ-glutamylcysteine synthetase-heavy subunit promoter: a critical role for AP-1. FEBS Lett. 1998;427:129–133. doi: 10.1016/s0014-5793(98)00410-4. [DOI] [PubMed] [Google Scholar]
- 102.Sekhar KR, Long M, Long J, Xu ZQ, Summar ML, Freeman ML. Alteration of transcriptional and post-transcriptional expression of gamma-glutamylcysteine synthetase by diethyl maleate. Radiation Res. 1997;147:592–297. [PubMed] [Google Scholar]
- 103.Sekhar KR, Meredith MJ, Kerr LD, Soltaninassab SR, Spitz DR, Xu ZQ, Freeman ML. Expression of glutathione and γ-glutamylcysteine synthetase mRNA is Jun dependent. Biochem Biophys Res Commun. 1997;234:588–593. doi: 10.1006/bbrc.1997.6697. [DOI] [PubMed] [Google Scholar]
- 104.Wild AC, Moinova HR, Mulcahy RT. Regulation of γ-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem. 1999;274:33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- 105.Erickson AM, Nevarea Z, Gipp JJ, Mulcahy RT. Identification of a variant antioxidant response element in the promoter of the human glutamate-cysteine ligase modifier subunit gene. J Biol Chem. 2002;277:30730–30737. doi: 10.1074/jbc.M205225200. [DOI] [PubMed] [Google Scholar]
- 106.Langston JW, Li W, Harrison L, Aw TY. Activation of promoter activity of the catalytic subunit of γ-glutamylcysteine ligase (GCL) in brain endothelial cells by insulin requires antioxidant response element 4 and altered glycemic status: implication for GCL expression and GSH synthesis. Free Radic Biol Med. 2011;51:1749–1757. doi: 10.1016/j.freeradbiomed.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kwong M, Kan YW, Chan JY. The CNC basic leucine zipper factor, Nrf1, is essential for cell survival in response to oxidative stress-inducing agents. J Biol Chem. 1999;274:37491–37498. doi: 10.1074/jbc.274.52.37491. [DOI] [PubMed] [Google Scholar]
- 108.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Review. Free Rad Biol Med. 2004;36:1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 109.Chan JY, Kwong M. Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim Biophys Acta. 2000;1517:19–26. doi: 10.1016/s0167-4781(00)00238-4. [DOI] [PubMed] [Google Scholar]
- 110.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol, Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 111.Sun Z, Wu T, Zhao F, Lau A, Birch CM, Zhang DD. KPNA6 (Importin α7)-mediated nuclear import of Keap1 represses the Nrf2-dependent antioxidant response. Mol Cell Biol. 2011;31:1800–1811. doi: 10.1128/MCB.05036-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li W, Yu S, Liu T, Kim JH, Blank V, Li H, Kong ANT. Heterodimerization with small Maf proteins enhances nuclear retention of Nrf2 via masking the NESzip motif. Biochim Biophys Acta. 2008;1783:1847–1856. doi: 10.1016/j.bbamcr.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang W, Chan JY. Nrf1 is targeted to the endoplasmic reticulum membrane by an N-terminal transmembrane domain. J Biol Chem. 2006;281:19676–19687. doi: 10.1074/jbc.M602802200. [DOI] [PubMed] [Google Scholar]
- 114.Chen L, Kwong M, Lu R, Ginzinger D, Lee C, Leung L, Chan JY. Nrf1 Is critical for redox balance and survival of liver cells during development. Mol Cell Biol. 2003;23:4673–4686. doi: 10.1128/MCB.23.13.4673-4686.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: Detoxification of acetaminophen. Proc Nat Acad Sci USA. 2001;98:4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Myhrstad MCW, Husberg C, Murphy P, Nordström O, Blomhoff R, Moskaug JØ, Kolstø AB. TCF11/Nrf1 overexpression increases the intracellular glutathione level and can transactivate the gamma-glutamylcysteine synthetase (GCS) heavy subunit promoter. Biochim Biophys Acta. 2001;1517:212–219. doi: 10.1016/s0167-4781(00)00276-1. [DOI] [PubMed] [Google Scholar]
- 117.Li M, Chiu JF, Kelsen A, Lu SC, Fukagawa NK. Identification and characterization of a Nrf2-mediated ARE upstream of the rat glutamate cysteine ligase catalytic subunit gene. J Cell Biochem. 2009;107:944–954. doi: 10.1002/jcb.22197. [DOI] [PubMed] [Google Scholar]
- 118.Yang HP, Magilnick N, Lee C, Kalmaz D, Ou XP, Chan JY, Lu SC. Nrf1 and Nrf2 regulate rat glutamate-cysteine ligase catalytic subunit transcription indirectly via AP-1 and NFκB. Mol Cell Biol. 2005;25:5933–5946. doi: 10.1128/MCB.25.14.5933-5946.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Peng Z, Geh E, Chen L, Meng Q, Fan Y, Sartor M, Shertzer HG, Liu ZG, Puga A, Xia Y. Inhibitor of κB kinase β regulates redox homeostasis by controlling the constitutive levels of glutathione. Mol Pharmacol. 2010;77:784–792. doi: 10.1124/mol.109.061424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Benassi A, Fanciulli M, Fiorentino F, Porrello A, Chiorino G, Loda M, Zupi G, Biroccio A. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol Cell. 2006;21:509–519. doi: 10.1016/j.molcel.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 121.Zhu W, Jia Q, Wang Y, Zhang Y, Xia M. The anthocyanin cyanidin-3-O-β-glucoside, a flavonoid, increases hepatic glutathione synthesis and protects hepatocytes against reactive oxygen species during hyperglycemia: Involvement of a cAMP-PKA-dependent signaling pathway. Free Radic Biol Med. 2012;52:314–327. doi: 10.1016/j.freeradbiomed.2011.10.483. [DOI] [PubMed] [Google Scholar]
- 122.Liu RM, Gao L, Choi J, Forman HJ. γ-Glutamylcysteine synthetase: mRNA stabilization and independent subunit transcription by 4-hydroxy-2-nonenal. Am J Physiol. 1998;275:L861–L869. doi: 10.1152/ajplung.1998.275.5.L861. [DOI] [PubMed] [Google Scholar]
- 123.Lu SC, Kuhlenkamp J, Garcia-Ruiz C, Kaplowitz NN. Hormone-mediated down-regulation of hepatic GSH synthesis in the rat. J Clin Invest. 1991;88:260–269. doi: 10.1172/JCI115286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Estrela JM, Gil F, Vila JM, Vina J. α-Adrenergic modulation of glutathione metabolism in isolated rat hepatocytes. Am J Physiol. 1988;255:E801–805. doi: 10.1152/ajpendo.1988.255.6.E801. [DOI] [PubMed] [Google Scholar]
- 125.Lu SC, Garcia-Ruiz C, Kuhlenkamp J, Ookhtens M, Salas-Prato M, Kaplowitz N. Hormonal regulation of GSH efflux. J Biol Chem. 1990;265:16088–16095. [PubMed] [Google Scholar]
- 126.Raiford DS, Sciuto AM, Mitchell MC. Effects of vasopressor hormones and modulators of protein kinase C on glutathione efflux from perfused rat liver. Am J Physiol. 1991;24:G578–584. doi: 10.1152/ajpgi.1991.261.4.G578. [DOI] [PubMed] [Google Scholar]
- 127.Hidalgo J, Garvey JS, Armario A. On the metallothionein, glutathione and cysteine relationship in rat liver. J Pharm Exp Therap. 1990;255:554–564. [PubMed] [Google Scholar]
- 128.Sun WM, Huang ZZ, Lu SC. Regulation of γ-glutamylcysteine synthetase by protein phosphorylation. Biochem J. 1996;320:321–328. doi: 10.1042/bj3200321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lauterburg BH, Mitchell JR. Toxic doses of acetaminophen suppress hepatic glutathione synthesis in rats. Hepatology. 1982;2:8–12. doi: 10.1002/hep.1840020103. [DOI] [PubMed] [Google Scholar]
- 130.Franklin CC, Rosenfled-Franklin ME, White C, Kavanagh TJ, Fausto N. TGFβ1-induced suppression of glutathione antioxidant defenses in hepatocytes: caspase-dependent posttranslational and caspase-independent transcriptional regulatory mechanisms. FASEB J. 2003 doi: 10.1096/fj.02–0867fje. [DOI] [PubMed] [Google Scholar]
- 131.Martin DD, Vilas GL, Prescher JA, Rajaiah G, Falck JR, Bertozzi CR, Berthiaume LG. Rapid detection, discovery, and identification of posttranslationally myristoylated proteins during apoptosis using a bio-orthogonal azidomyristate analog. FASEB J. 2008;22:797–806. doi: 10.1096/fj.07-9198com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Backos DS, Fritz KS, Roede JR, Petersen DR, Franklin CC. Posttranslational modification and regulation of glutamate-cysteine ligase by the α,β-unsaturated aldehyde 4-hydroxy-2-nonenal. Free Radic Biol Med. 2011;50:14–26. doi: 10.1016/j.freeradbiomed.2010.10.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Galloway DC, McLellan LI. Inducible expression of the γ-glutamylcysteine synthetase light subunit by t-butylhydroquinone in HepG2 cells is not dependent on an antioxidant-responsive element. Biochem J. 1998;336:535–539. doi: 10.1042/bj3360535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Moinova HR, Mulcahy RT. Up-regulation of the human γ-glutamylcysteine synthetase regulatory subunit gene involves binding of Nrf-2 to an electrophile responsive element. Biochem Biophys Res Commun. 1999;261:661–668. doi: 10.1006/bbrc.1999.1109. [DOI] [PubMed] [Google Scholar]
- 135.Lu SC, Huang ZZ, Yang J, Tsukamoto H. Effect of ethanol and high fat feeding on hepatic γ-glutamate-cysteine ligase subunit expression in the rat. Hepatology. 1999;30:209–214. doi: 10.1002/hep.510300134. [DOI] [PubMed] [Google Scholar]
- 136.Yang HP, Ramani K, Xia M, Ko KS, Li TWH, Oh P, Li J, Lu SC. Dysregulation of glutathione synthesis during cholestasis in mice: molecular mechanisms and therapeutic implications. Hepatology. 2009;49:1982–1991. doi: 10.1002/hep.22908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Huang A, He W, Meister A, Anderson ME. Amino acid sequence of rat kidney glutathione synthetase. Proc Natl Acad Sci USA. 1995;92:1232–1236. doi: 10.1073/pnas.92.4.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shi ZZ, Habib GM, Rhead WJ, Gahl WA, He X, Sazer S, Lieberman MW. Mutations in the glutathione synthetase gene cause 5-oxoprolinuria. Nature Genetics. 1996;14:361–365. doi: 10.1038/ng1196-361. [DOI] [PubMed] [Google Scholar]
- 139.Choi J, Liu RM, Kundu RK, Sangiorgi F, Wu W, Maxson R, Forman HJ. Molecular emchanism of decreased glutathione content in human immunodeficiency virus type 1 Tat-transgenic mice. J Biol Chem. 2000;275:3693–3698. doi: 10.1074/jbc.275.5.3693. [DOI] [PubMed] [Google Scholar]
- 140.Lu SC, Huang ZZ, Yang H, Tsukamoto H. Effect of thioacetamide on hepatic γ-glutamylcysteine synthetase subunit expression. Tox Appl Pharmacol. 1999;159:161–168. doi: 10.1006/taap.1999.8729. [DOI] [PubMed] [Google Scholar]
- 141.Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007;67:11021–11028. doi: 10.1158/0008-5472.CAN-07-2593. [DOI] [PubMed] [Google Scholar]
- 142.Wu Y, Zhang X, Robel RCV, Chen LX, Zeng Y, Aguilo J, Ao Y, Hwang K, French SW, Lu SC, Wan YJY. Retinoid X receptor a regulates amino acid metabolism, glutathione homeostasis and xenobiotic detoxification processes in mouse liver. Mol Pharmacol. 2004;65:550–557. doi: 10.1124/mol.65.3.550. [DOI] [PubMed] [Google Scholar]
- 143.Yang HP, Zeng Y, Lee TD, Yang T, Ou XP, Chen LX, Haque M, Rippe R, Lu SC. Role of AP-1 in the co-ordinate induction of rat glutamate-cysteine ligase and glutathione synthetase by tert-butylhydroquinone. J Biol Chem. 2002;277:35232–35239. doi: 10.1074/jbc.M203812200. [DOI] [PubMed] [Google Scholar]
- 144.Lee TD, Yang HP, Whang J, Lu SC. Cloning and characterization of the human glutathione synthetase 5′-flanking region. Biochem J. 2005;390:521–528. doi: 10.1042/BJ20050439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nakamura S, Kugiyama K, Sugiyama S, Miyamoto S, Koide S, Fukushima H, Honda O, Yoshimura M, Ogawa H. Polymorphism in the 5′-flanking region of human glutamate-cysteine ligase modifier subunit gene is associated with myocardial infarction. Circulation. 2002;105:2968–2973. doi: 10.1161/01.cir.0000019739.66514.1e. [DOI] [PubMed] [Google Scholar]
- 146.Nakamura S, Sugiyama S, Fujioka D, Kawabata K, Ogawa H, Kugiyama K. Polymorphism in glutamate-cysteine ligase modifier subunit gene is associated with impairment of nitric oxide-mediated coronary vasomotor function. Circulation. 2003;108:1425–7. doi: 10.1161/01.CIR.0000091255.63645.98. [DOI] [PubMed] [Google Scholar]
- 147.Neuschwander-Tetri BA, Nicholson C, Wells LD, Tracy TF., Jr Cholestatic liver injury down-regulates hepatic GSH synthesis. J Surg Res. 1996;63:447–451. doi: 10.1006/jsre.1996.0290. [DOI] [PubMed] [Google Scholar]
- 148.Tan KP, Yang M, Ito S. Activation of nuclear factor (erythroid-2 like) factor 2 by toxic bile acids provokes adaptive defense responses to enhance cell survival at the emergence of oxidative stress. Mol Pharmacol. 2007;72:1380–1390. doi: 10.1124/mol.107.039370. [DOI] [PubMed] [Google Scholar]
- 149.Nguyen T, Huang HC, Pickett CB. Transcriptional regulation of the antioxidant response element. J Biol Chem. 2000;275:15466–15473. doi: 10.1074/jbc.M000361200. [DOI] [PubMed] [Google Scholar]
- 150.Dhakshinamoorthy S, Jaiswal AK. Small Maf (MafG and MafK) proteins negatively regulate antioxidant response element-mediated expression and antioxidant induction of the NAD(P)H:Quinone oxidoreductase 1 gene. J Biol Chem. 2000;275:40134–40141. doi: 10.1074/jbc.M003531200. [DOI] [PubMed] [Google Scholar]
- 151.Dhakshinamoorthy S, Jaiswal AK. C-Maf negatively regulates ARE-mediated detoxifying enzyme genes expression and anti-oxidant induction. Oncogene. 2002;21:5301–5312. doi: 10.1038/sj.onc.1205642. [DOI] [PubMed] [Google Scholar]
- 152.Kumar A, Tandon RK. Use of ursodeoxycholic acid in liver disease. J Gastro and Hepatol. 2001;16:3–14. doi: 10.1046/j.1440-1746.2001.02376.x. [DOI] [PubMed] [Google Scholar]
- 153.Zhang C, Walker LM, Hinson JA, Mayeux PR. Oxidant stress in rat liver after lipopolysaccharide administration: effect of inducible nitric-oxide synthase inhibition. J Pharmacol, Exp Therap. 2000;293:968–972. [PubMed] [Google Scholar]
- 154.SuL G. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am J Physiol Gastrointest Liver Physiol. 2002;283:G256–265. doi: 10.1152/ajpgi.00550.2001. [DOI] [PubMed] [Google Scholar]
- 155.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA. 1997;94:2557–62. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jaeschke H. Enhanced sinusoidal glutathione efflux during endotoxin-induced oxidant stress in vivo. Am J Physiol Gastrointest Liver Physiol. 1992;263:G60–68. doi: 10.1152/ajpgi.1992.263.1.G60. [DOI] [PubMed] [Google Scholar]
- 157.Payabvash S, Ghahremani MH, Goliaei A, Mandegary A, Shafaroodi H, Amanlou M, Dehpour AR. Nitric oxide modulates glutathione synthesis during endotoxemia. Free Rad Biol Med. 2006;41:1817–1828. doi: 10.1016/j.freeradbiomed.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 158.Victor VM, De La Fuente M. Immune cells redox state from mice with endotoxin-induced oxidative stress. Involvement of NF-κB. Free Rad Res. 2003;37:19–27. doi: 10.1080/1071576021000038522. [DOI] [PubMed] [Google Scholar]
- 159.Németh, Boda D. Xanthine oxidase activity and blood glutathione redox ration in infants and children with septic shock syndrome. Int Care Med. 2001;27:216–221. doi: 10.1007/s001340000791. [DOI] [PubMed] [Google Scholar]
- 160.Sun S, Zhang H, Xue B, Wu Y, Wang J, Yin Z, Luo L. Protective effect of glutathione against lipopolysaccharide-induced inflammation and mortality in rats. Inflamm Res. 2006;55:504–510. doi: 10.1007/s00011-006-6037-7. [DOI] [PubMed] [Google Scholar]
- 161.Gould NS, Min E, Day BJ. Macropinocytosis of extracellular glutathione ameliorates tumor necrosis factor α release in activated macrophages. PLoS One. 2011;6:e25704. doi: 10.1371/journal.pone.0025704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ko KS, Yang HP, Noureddin M, Iglesia-Ara A, Xia M, Wagner C, Luka Z, Mato JM, Lu SC. Changes in S-adenosylmethionine and glutathione homeostasis during endotoxemia in mice. Lab Invest. 2008;88:1121–1129. doi: 10.1038/labinvest.2008.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Vendemiale G, Altomare E, Trizio T, Le Grazie C, Di Padova C, Salerna MT, Carrieri V, Albano O. Effect of oral S-adenosyl-L-methionine on hepatic glutathione in patients with liver disease. Scan J Gastroenterol. 1989;24:407–415. doi: 10.3109/00365528909093067. [DOI] [PubMed] [Google Scholar]
- 165.Tsukamoto HC, Lu SC. Current concepts in the pathogenesis of alcoholic liver injury. FASEB J. 2001;15:1335–1349. doi: 10.1096/fj.00-0650rev. [DOI] [PubMed] [Google Scholar]
- 166.Lee TD, Sadda ME, Mendler MH, Bottiglieri T, Kanel G, Mato JM, Lu SC. Abnormal hepatic methionine and GSH metabolism in patients with alcoholic hepatitis. Alcoholism. Clin Exp Res. 2004;28:173–181. doi: 10.1097/01.ALC.0000108654.77178.03. [DOI] [PubMed] [Google Scholar]
- 167.Friedman SL, Roll FJ, Boyles J, Arenson DM, Bissell DM. Maintenance of differentiated phenotype of cultured rat hepatic lipocytes by basement membrane matrix. J Biol Chem. 1989;264:10756–10762. [PubMed] [Google Scholar]
- 168.Burt AD. Cellular and molecular aspects of hepatic fibrosis. J Pathol. 1993;70:105–114. doi: 10.1002/path.1711700203. [DOI] [PubMed] [Google Scholar]
- 169.Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7:793–807. doi: 10.2741/A812. [DOI] [PubMed] [Google Scholar]
- 170.Paradis V, Dargere D, Bonvoust F, Vidaud M, Segarini P, Bedossa P. Effects and regulation of connective tissue growth factor on hepatic stellate cells. Lab Invest. 2002;82:767–774. doi: 10.1097/01.lab.0000017365.18894.d3. [DOI] [PubMed] [Google Scholar]
- 171.Saxena NK, Saliba G, Floyd JJ, Anania FA. Leptin induces increased alpha2(I) collagen gene expression in cultured rat hepatic stellate cells. J Cell Biochem. 2003;89:311–320. 2003. doi: 10.1002/jcb.10494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kamphorst BE, Stoll D, Gressner AM, Weiskirchen R. Inhibitory effect of soluble PDGF-beta receptor in culture-activated hepatic stellate cells. Biochem Biophys Res Commun. 2004;317:451–462. doi: 10.1016/j.bbrc.2004.03.064. [DOI] [PubMed] [Google Scholar]
- 173.Iredale JP. Hepatic stellate cell behavior during the resolution of liver injury. Sem Liver dis. 2001;21:427–436. doi: 10.1055/s-2001-17557. [DOI] [PubMed] [Google Scholar]
- 174.Ramani K, Tomasi ML, Yang H, Ko K, Lu SC. Mechanism and significance of changes in glutamate-cysteine ligase expression during hepatic fibrogenesis. J Biol Chem. 2012 doi: 10.1074/jbc.M112.370775. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]