

Abstract
Endocannabinoids are released ‘on-demand’ on the basis of physiological need, and can be pharmacologically augmented by inhibiting their catabolic degradation. The endocannabinoid anandamide is degraded by the catabolic enzyme fatty acid amide hydrolase (FAAH). Anandamide is implicated in the mediation of fear behaviors, including fear extinction, suggesting that selectively elevating brain anandamide could modulate plastic changes in fear. Here we first tested this hypothesis with preclinical experiments employing a novel, potent and selective FAAH inhibitor, AM3506 (5-(4-hydroxyphenyl)pentanesulfonyl fluoride). Systemic AM3506 administration before extinction decreased fear during a retrieval test in a mouse model of impaired extinction. AM3506 had no effects on fear in the absence of extinction training, or on various non-fear-related measures. Anandamide levels in the basolateral amygdala were increased by extinction training and augmented by systemic AM3506, whereas application of AM3506 to amygdala slices promoted long-term depression of inhibitory transmission, a form of synaptic plasticity linked to extinction. Further supporting the amygdala as effect-locus, the fear-reducing effects of systemic AM3506 were blocked by intra-amygdala infusion of a CB1 receptor antagonist and were fully recapitulated by intra-amygdala infusion of AM3506. On the basis of these preclinical findings, we hypothesized that variation in the human FAAH gene would predict individual differences in amygdala threat-processing and stress-coping traits. Consistent with this, carriers of a low-expressing FAAH variant (385A allele; rs324420) exhibited quicker habituation of amygdala reactivity to threat, and had lower scores on the personality trait of stress-reactivity. Our findings show that augmenting amygdala anandamide enables extinction-driven reductions in fear in mouse and may promote stress-coping in humans.
Keywords: amygdala, anxiety, cannabinoid, fear, gene, stress
Introduction
Fear extinction, the learned inhibition of a fear response, is readily quantifiable in laboratory rodents and provides an important behavioral assay for translational studies of anxiety disorders, which often entail impaired extinction.1,2 Implicating the endocannabinoid system in fear extinction, brain-wide deletion of the endocannabinoid CB1 receptor (CB1R) in mice results in profoundly impaired extinction and adaptation.3,4 However, because CB1Rs are ubiquitously expressed in the brain5 and mediate manifold functions in the brain and periphery, global activation of CB1R would produce widespread effects,6,7 some clinically undesirable.
Endocannabinoids are synthesized and released ‘on-demand’ on the basis of physiological need.8 Augmenting released endocannabinoids can be achieved pharmacologically by blocking their reuptake from the extracellular space9 or interfering with their catabolic degradation by inhibiting the activity of endocannabinoid-degrading enzymes. The endocannabinoids anandamide and 2-arachidonoylglycerol are predominantly degraded by the respective catabolic enzymes fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), and produce distinct behavioral effects.10,11 However, chronic inhibition of MAGL causes physical dependence, impaired endocannabinoid-mediated synaptic plasticity and CB1R desensitization, potentially limiting its therapeutic potential.12 These effects do not appear to be produced by chronic inhibition of FAAH.12
A number of prior findings suggest a potential link between FAAH, anandamide and extinction. First, extinction increases anandamide, but not 2-arachidonoylglycerol, levels in the mouse basolateral amygdala (BLA),3 a brain region critical for extinction.13,14 Second, constitutive deletion of the mouse FAAH gene promotes extinction of a spatial reference memory.15 Third, systemic administration of the endocannabinoid reuptake blocker AM404, which nonspecifically increases both anandamide and 2-arachidonoylglycerol,16 increases fear memory and/or extinction in rats.17,18
Taken together, these prior studies raise the possibility that inhibiting FAAH to selectively boost endogenously recruited anandamide in corticolimbic circuits could drive long-term reductions in fear following extinction. Furthermore, such effects would be predicted to occur in the absence of concomitant alterations in cannabinoid-mediated central nervous system and peripheral functions produced by nonspecifically augmenting anandamide and 2-arachidonoylglycerol levels, or by indiscriminate CB1R activation. This functional selectivity is of critical relevance to the side-effect burden and potential clinical utility of FAAH inhibitors for anxiety disorders. In fact, even prototypical FAAH inhibitors (for example, URB597) can produce unwanted peripheral effects, including hyperglycemia and insulin resistance.19 This study therefore employed a novel compound, AM3506 (5-(4-hydroxyphenyl)pentanesulfonyl fluoride), that is a highly potent and selective FAAH inhibitor,20 but devoid of undesirable hepatic effects.19
Our findings provide the first evidence that selectively augmenting the endocannabinoid anandamide decreases fear after extinction in a mouse model, and establish the neural locus underlying this behavioral action. In addition, we provide translational evidence from functional neuroimaging and genetic association studies in human subjects, supporting the therapeutic value of FAAH as a target for anxiety disorders.
Materials and Methods
Drugs and chemicals
Unless specified, all chemicals were obtained from Sigma-Aldrich (St Louis, MO, USA) or Fisher Chemicals (Fair Lawn, NJ, USA). AM3506 was synthesized at Northeastern University as described.19,20 SR141716 (Rimonabant) was provided by the National Institute on Drug Abuse Drug Supply program. Both drugs were dissolved in dimethylsulfoxide and aliquoted for storage at −80 °C. For in vivo administration, AM3506 was suspended in a 9:1 saline/dimethylsulfoxide solution and SR141716 was suspended in an 18:1:1 saline/Tween/dimethylsulfoxide solution. For systemic treatment, drugs were injected intraperitoneally in a volume of 10 ml/kg body weight. For intra-amygdala microinfusions, drugs were suspended as described above and infused, via bilateral 33-G injectors (Plastics One, Roanoke, VA, USA) projecting 1 mm past the guide cannula, in a volume of 0.5 μl per hemisphere over 2 min using a syringe pump (Harvard Apparatus PHD 22/2000, Holliston, MA, USA). Injectors were left in place for a further 3 min to allow diffusion into the tissue.
FAAH and MAGL activity time course
129S1/Sv1mJ mice (The Jackson Laboratory, Bar Harbor, ME, USA) were injected with 1.0 mg/kg AM3506. Mice were killed via cervical dislocation and rapid decapitation immediately or 1 h, 1 day, 3 days or 10 days after injection. Brains were removed and the forebrain and cerebellum tissue quickly dissected on ice. Tissue was homogenized in 10 mm (pH 7.6) Tris-HCl buffer containing 1 mm ethylenediaminetetraacetic acid and centrifuged at 1000g to remove cell debris. Tissue was assayed for FAAH activity via measurement of the release of [3H]ethanolamine from [3H]anandamide labeled on the ethanolamine moiety.21 Tissue was also assayed for MAGL activity by analyzing the release of [3H]glycerol from 2-oleoyl-[3H]glycerol in the presence of 1 mm of the FAAH blocker URB597 (Cayman Chemical Company, Ann Arbor, MI, USA).22
For the assays, the tissue homogenate (175 μg) was mixed with radiolabeled [3H]anandamide (specific activity 60 Ci/mmol) or 2-oleoyl-[3H]glycerol (specific activity 60 Ci/mmol) (containing 10 mg/ml fatty acid free bovine serum albumin) and unlabeled anandamide and 2-oleoyl-[3H]glycerol to produce samples of 200 μl total volume. Labeled and unlabeled chemicals were obtained from American Radiolabeled Chemicals (St Louis) and Cayman Chemical Company, respectively. Samples were incubated at 37 °C with continuous shaking in a water bath. ‘Blank’ samples that contained assay buffer, instead of the homogenate, were incubated in the same manner. Incubation was stopped after 15 min by placing the tubes on ice, and 400 μl of 1:1 chloroform/methanol was added to the samples. Samples were then vortexed three times, followed by centrifugation to produce phase separation. A measure of 200 μl of the upper aqueous phase was removed and analyzed for radioactivity by liquid scintillation counting. The blank sample values were subtracted from each count. Data were expressed as the percent of the 0 time point value (for 1 mg/kg AM3506) and the effect of time point analyzed using repeated measures analysis of variance (ANOVA).
Fear conditioning and extinction
Experimental procedures were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and approved by the local Animal Care and Use Committee.
Subjects
Subjects were male 8- to 12-week-old 129S1/Sv1mJ (S1) mice obtained from The Jackson Laboratory. This inbred strain exhibits normal fear, but is impaired in fear extinction learning and retrieval, which is rescued by various pharmacological interventions (for example, fluoxetine, yohimbine, zinc depletion).23–25 Mice were housed 2 per cage in a temperature- and humidity-controlled vivarium under a 12 h light/dark cycle (lights on 0600 h). The number of mice used in each experiment is indicated in the figure legends.
General procedure
Conditioning
Mice were fear conditioned as described previously.26,27 The conditioning context (‘context A’) was a 27 × 27 × 11 cm3 chamber with a metal-rod floor, cleaned with a 79.5% water/19.5% ethanol/1% vanilla-extract solution. After a 180 s acclimation period, there were 3 × pairings (60–120s inter-pairing interval) of the conditioned stimulus (CS; 30s, 80 dB, 3 kHz tone) and the unconditioned stimulus (US; 2 s, 0.6 mA scrambled foot shock), in which the US was presented during the last 2 s of the CS. The session ended 120 s after the final CS–US pairing. Stimulus presentation was controlled by the Med Associates VideoFreeze system (Med Associates, Burlington, VT, USA). Freezing (no visible movement except respiration) was scored every 5 s by an observer blind to condition/treatment and converted to a percentage ((freezing observations/total observations) × 100). Freezing during extinction was averaged to 5-trial blocks for analysis.
Extinction training
One day after conditioning, extinction training was conducted as described previously.28 Testing was conducted in a novel context (‘context B’) (cylinders with black/white-chequered walls and a solid Plexiglas opaque floor cleaned with a 1% acetic acid/99% water solution) housed in a different room from conditioning. After a 180s acclimation period, there were 50 × CS presentations (5-s inter-CS interval).
Extinction retrieval
Extinction retrieval was tested in context B 10 days after extinction training. After a 180s acclimation period, there were 3 × CS presentations (5-s inter-CS interval).
Effects of systemic AM3506 treatment
Fear conditioning and extinction testing was conducted as above, unless otherwise specified. Naïve cohorts of mice were used for each experiment. For a schematic summary of the experimental procedures, see Figure 1c.
Figure 1.
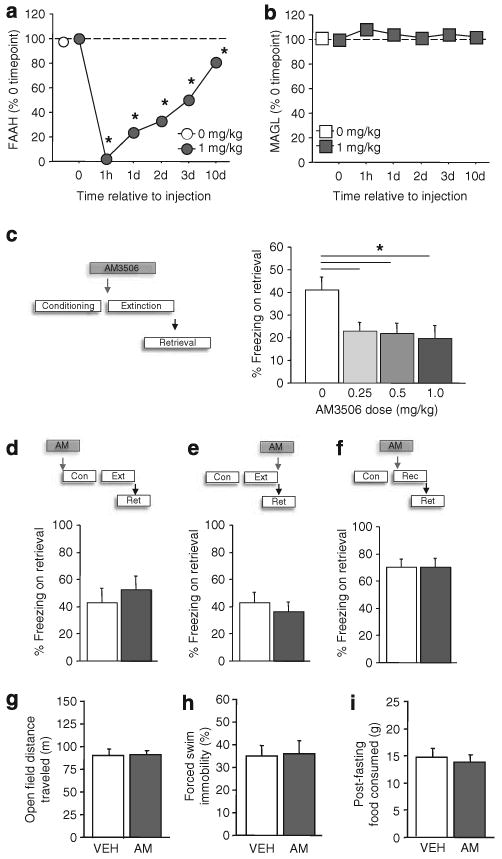
The fatty acid amide hydrolase (FAAH) inhibitor AM3506 (5-(4-hydroxyphenyl)pentanesulfonyl fluoride) facilitates fear extinction. Systemic treatment with AM3506 (n = 3 per time point) produced profound and lasting brain FAAH inhibition, as measured by [3H]ethanolamine activity (a), but not monoacylglycerol lipase, as assayed via [3H]glycerol activity (b) (*P<0.05 vs 1 mg/kg/time point 0). (c) Systemic AM3506 treatment before extinction training reduced fear on a retrieval test (n = 10–11 per treatment). Systemic AM3506 did not affect fear during a retrieval test when administered before conditioning (n = 8 per treatment) (d), after extinction training (n = 12 per treatment) (e) or before fear memory reactivation (n = 8 per treatment) (f). Systemic AM3506 did not affect fear open field locomotor activity (n = 8 per treatment) (g), forced swim ‘depression-related’ behavior (n = 8 per treatment) (h) or fasting-induced feeding (n = 8 per treatment) (i). Cond = conditioning; Ext = extinction; Rect = reactivation; Ret = retrieval. *P<0.05. Data are means±s.e.m.
Pre-extinction treatment
AM3506 was administered at a dose of 0,0.25, 0.5 or 1.0 mg/kg 60 min before extinction training. The purpose of this experiment was to test whether AM3506 facilitated extinction.
Pre-conditioning treatment
AM3506 was administered at a dose of 0 or 1.0 mg/kg 60 min before conditioning. The purpose was to test whether AM3506 affected fear memory learning.
Post-extinction treatment
AM3506 was administered at a dose of 0 or 1.0 mg/kg immediately after extinction training. The purpose was to test whether AM3506 facilitated post-training extinction consolidation.
Pre-reactivation treatment
Here, the extinction session was replaced with a 3 × CS session. AM3506 was administered at a dose of 0 or 1.0 mg/kg 60 min before reactivation. The purpose was to test whether AM3506 reduced long-term fear by impairing reconsolidation of the fear memory, rather than by facilitating extinction.
Pre-context-only treatment
Here, the extinction session was replaced with an equivalent length session in which there were no CS presentations in context B. AM3506 was administered at a dose of 0 or 1.0 mg/kg 60 min before context exposure. The purpose was to test whether AM3506 reduced long-term fear in the absence of extinction training.
Pre-extinction treatment in unconditioned mice
Here, there was no US presentation during conditioning. AM3506 was administered at a dose of 0 or 1.0 mg/kg 60 min before extinction training. The purpose was to test whether AM3506 produced nonspecific effects on unconditioned fear.
Pre-extinction treatment with SR141716 before AM3506
The purpose of this experiment was to test whether the extinction-facilitating effect of AM3506 was dependent on the action at CB1R. SR141716 (Rimonabant; 1.0 mg/kg) was administered 50 min before 0 or 1.0 mg/kg AM3506, and extinction training tested 60 min later. A control group of mice treated with vehicle instead of each drug was also included and tested in the same manner.
Statistical analysis
The effects of treatment and trial-block on freezing during extinction training were analyzed using two-factor ANOVA, with repeated measures for trial-block. The effect of drug treatment on freezing during retrieval or reactivation was statistically analyzed using ANOVA (>2 treatment conditions) or Student's t-test (2 treatment conditions).
Effects of intra-amygdala infusions
Stereotaxic surgery
Under isoflurane anesthesia, 26-G bilateral guide cannulae (Plastics One, Roanoke, VA, USA) were targeted to BLA (−1.40 mm anterior–posterior, ±3.30 mm mediolateral, −3.90 mm ventral to Bregma) and held in place with dental cement. Mice were singly housed and given a 7-day post-surgery recovery period during which dummy cannulae were replaced daily to habituate mice to handling and prevent cannulae blocking. On completion of testing, mice were perfused with 4% paraformaldehyde. Fixed brains were sectioned (50-μm thickness) on a vibrating microtome and stained with cresyl violet to verify the localization of the cannulae placements with the aid of a microscope.
Systemic AM3506 treatment + amygdala infusion of SR141716
AM3506 was administered at a dose of 0 or 1.0 mg/kg 60 min before extinction training. SR141716 (0 or 2 μg/μl) was bilaterally infused into the BLA 5 min before systemic treatment with 1.0 mg/kg AM3506. A control group of mice treated with vehicle instead of each drug was also included and tested in the same manner. The purpose of this experiment was to test whether the extinction-facilitating effect of AM3506 was dependent on the action at CB1R in the BLA. The effect of drug treatment on freezing during retrieval was statistically analyzed using ANOVA, followed by Newman–Keuls post hoc tests.
Amygdala infusion of AM3506
AM3506 (0 or 0.1 μg/μl) was bilaterally infused into the BLA 30 min before extinction training. The purpose of this experiment was to test whether the application of AM3506 to the amygdala was sufficient to produce an extinction-facilitating effect. The effect of drug treatment on freezing during retrieval was statistically analyzed using Student's t-test.
Non-fear-related behaviours
Open field locomotor activity
The effects of systemic AM3506 treatment on locomotor activity were tested in an open field test. The open field was in a 40 × 40 × 35 cm3 square arena (60 lx) constructed of white Plexiglas, as described previously.29 The mouse was placed in the perimeter and allowed to explore the apparatus for 30 min. Total distance traveled and time spent in the center square (20 × 20 cm2) was measured by the Ethovision videotracking system (Noldus Information Technology, Leesburg, VA, USA). AM3506 was administered at a dose of 0 or 1.0 mg/kg 60 min before testing. The effect of drug treatment was statistically analyzed using Student's t-test.
Forced swim test
The effects of systemic AM3506 treatment were tested in the forced swim test, a simple screen for antidepressant-related activity.30 The mouse was gently lowered into a 20-cm-diameter cylinder filled with 24±1.0°C water for a 6 min test, as described previously.31 Immobility (cessation of limb movements except minor movement necessary to keep the mouse afloat) was manually scored every 5 s during 3–6 min. AM3506 was administered at a dose of 0 or 1.0 mg/kg 60 min before testing. The effect of drug treatment was statistically analyzed using Student's t-test.
Fasting-induced feeding
The effects of systemic AM3506 treatment on fasting-induced feeding was tested using methods described previously.32 The mouse was food deprived for 16 h and presented with regular chow in the home cage. The amount of food consumed was measured over 1 h. AM3506 was administered at a dose of 0 or 1.0 mg/kg 60 min before testing. The effect of drug treatment was statistically analyzed using Student's t-test.
Glucose tolerance
The effects of systemic AM3506 treatment on anandamide-induced glucose intolerance were tested based on previous methods.19 Because we have shown that another FAAH inhibitor, URB597, produces glucose intolerance in obese mice,19 we examined the effects of URB597, for comparison with AM3506. The mouse was food deprived for 12–16 h and then administered saline vehicle, 1.0 mg/kg AM3506 or 1.0 mg/kg URB597. After 60 min, the mouse received 10 mg/kg anandamide and then, 10 min later, by 1.5 g/kg glucose. Blood glucose levels were measured from blood samples collected from the tail vein, 0,15,30,45, 60, 90 120, 150 and 180 min after glucose administration. The effect of drug treatment was statistically analyzed using a drug × time (repeated measures for time) ANOVA and, for data summarized as an area under the curve, Student's t-test.
Ex vivo endocannabinoid levels
Fear conditioning (referred to here as the ‘CS–US’ group) and extinction training was conducted as above, with the addition of an unconditioned group (as above for ‘Pre-extinction treatment in unconditioned mice’) that received CS-only trials during conditioning (referred to here as the ‘CS’ group). AM3506 was administered at a dose of 0 or 1.0 mg/kg 60 min before extinction training.
On completion of extinction training, mice were immediately killed via cervical dislocation and rapid decapitation. Brains were removed and the BLA, ventromedial prefrontal cortex and dorsal striatum were dissected on ice using 1- and 2-mm-diameter micropunches, respectively. Tissue was homogenized in 100 μl Tris (pH 8.0) buffer and protein concentrations determined using the Bradford assay with bovine serum albumin as a standard. Lipids were extracted and anandamide and 2-arachidonylglycerol levels quantified by liquid chromatography/tandem mass spectrometry, using multiple reactions monitoring, as described previously.33 The mass spectrometer was set for electrospray ionization operated in positive ion mode. The molecular ion and fragments for each compound measured were as follows: m/z 352.3 → 66.1 and 352.3 → 91 for [2H4] anandamide (CID energy: 12 and 56 V, respectively), m/z 348.3 → 62.1 and 348.3 → 91 for anandamide (CID energy: 12 and 48 V, respectively) and m/z 379.3 → 91 and 379.3 → 67.1 for 2-arachidonylglycerol (CID energy: 64 and 56 V, respectively). Analytes were quantified using MassHunter Workstation LC/QQQ Acquisition and MassHunter Workstation Quantitative Analysis (Agilent Technologies, Santa Clara, CA, USA). Levels of anandamide and 2-arachidonylglycerol in each brain region were determined against standard curves and expressed as fmol/mg or pmol/mg of protein. The effect of drug treatment and extinction training (CS–US vs CS) was statistically analyzed using ANOVA.
Slice electrophysiological recordings
Mice were killed by rapid cervical dislocation and decapitated and a 3-mm coronal block containing the amygdala was cut using a coronal brain matrix kept on ice. Coronal slices (300 μm) were made on a Leica VT1000S vibratome (Leica Microsystems, Bannockburn, IL, USA) in a 1−4 °C, oxygenated (95% O2, 5% CO2), low Na+ artificial cerebral spinal fluid (ACSF) containing (in mm): 194 sucrose, 20 NaCl, 2.5 KCl, 2 CaCl2, 1 MgSO4, 1.2 NaH2PO4, 10 glucose and 26 NaHCO3. Once cut, sections were transferred to a holding chamber containing oxygenated ACSF (in mm): 124 NaCl, 2.5 KCl, 2 CaCl2, 1.2 MgSO4, 1 NaH2PO4, 10 glucose and 26 NaHCO3 at 24 °C. After 1–4 h, sections were placed in the recording chamber superfused with oxygenated ACSF at a flow rate of 2 ml/min. To isolate pharmacologically IPSCs, ACSF was supplemented with a combination of 20 μm CNQX and 50 μm d/l AP-5. For field recordings, ACSF was supplemented with 25 μm picrotoxin. All experiments were carried out at 23–25 °C. ACSF was supplemented with 0.5 g/l fatty acid free bovine serum albumin. Slices were incubated in AM3506 (2 μm) or vehicle for at least 20–30 min before the start of recording.
For whole cell recordings, patch electrodes (2–3 Ωm) were filled with internal solution containing (in mm): CsMeSO3 119, TEA-Cl 10, NaCl 5, EGTA 2, HEPES 10, Na-ATP 4, Na-GTP 0.3, QX-314 5, with osmolarity adjusted to 275–285 mOsm (pH 7.25–7.35). Visually identified pyramidal neurons within the BLA were used for electrophysiological studies as described previously.34 Whole cell recordings were made using an Axopatch 700B amplifier (Molecular Devices, Sunnyvale, CA, USA). Voltage-clamp recordings were made at a holding potential of + 10 mV. Monosynaptically evoked inhibitory postsynaptic currents (eIPSCs) were elicited by constant current stimulation via a concentric bipolar stimulating electrode placed in the BLA just medial to the external capsule. eIPSC amplitudes were typically adjusted to 300–1200pA with stimulation intensities ranging from 10 to 40 μA. Test pulse simulations were elicited at 0.1 Hz. LTDi (long-term depression of inhibitory, GABAergic, transmission) was induced by eliciting 100 pulses at a frequency of 1 Hz (LFS1; low-frequency stimulation) at twice the stimulation intensity as the test pulse. Access resistance (Ra) was monitored online and cells showing changes in Ra of >20% or an Ra >20 mΩ upon break-in were excluded from the analysis. To determine the paired pulse ratio (PPR), paired stimulations were delivered at an interval of 100 ms. PPR was calculated as the peak amplitude of the second stimulation divided by the peak amplitude of the first stimulation. The percent increase in PPR between baseline and 35–40 min post-LFS were compared by paired t-test.
For field recordings, glass pipettes (1 ΩM) were placed in the lateral amygdala, and a bipolar stimulating electrode was positioned within the external capsule dorsal to the LA. Field potentials were recorded using an Axopatch 1D amplifier. Test stimuli were delivered at a frequency of 0.05 Hz, and LFS stimuli were delivered at 10 Hz for 10 min (LFS2), based on previous studies showing that this protocol induces endocannabinoid-mediated LTD in the cortex35 and in the nucleus accumbens.36 The amplitude of field excitatory postsynaptic potentials (fEPSPs) was calculated as a percent change from baseline.
The effect of drug and time relative to stimulation on the time course of eIPSC and fEPSP changes was statistically analyzed using ANOVA. Student's t-test was used to analyze the effect of drug on the overall magnitude of LTDi and PPR.
Imaging genetics
In all, 103 participants were recruited from the Adult Health and Behavior (AHAB) Study, which investigates a variety of behavioral and biological traits among non-patient, middle-aged community volunteers. All participants were in good general health and free of the following: (1) medical diagnoses of cancer, stroke, diabetes requiring insulin treatment, chronic kidney or liver disease, or a lifetime history of psychotic symptoms; (2) use of psychotropic, glucocorticoid or cardiovascular (for example, antihypertensive or antiarrhythmic) medication; (3) conditions that affect cerebral blood flow and metabolism (for example, hypertension); and (4) any current DSM-IV (Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition) axis I disorder as assessed by the non-patient version of the Structured Clinical Interview for DSM-IV.37 Written informed consent according to the guidelines of the University of Pittsburgh's Institutional Review Board was obtained from all participants upon their enrollment in the study. To minimize the risk of artifacts due to population stratification, we restricted analyses to Caucasian participants, after which both genotyping and functional magnetic resonance imaging data were available for 81 participants (43 women, mean age 44.70 ± 6.49 years).
Blood oxygen level-dependent functional magnetic resonance imaging paradigm
Participants completed an archival challenge paradigm, which robustly and consistently elicits threat-related reactivity of the amygdala.38–40 The paradigm consisted of four blocks of a face-processing task interleaved with five blocks of a sensorimotor control task. During the face-processing task, participants viewed a trio of faces (expressing either anger or fear) and selected one of two faces (bottom) that was identical to a target face (top). Each face-processing block consisted of six images, balanced for sex and target affect (angry or fearful), all of which were derived from a standard set of facial affect pictures.41 During the sensorimotor control blocks, participants viewed a trio of simple geometric shapes (circles and vertical and horizontal ellipses) and selected one of two shapes (bottom) that were identical to a target shape (top). Each sensorimotor control block consisted of six different shape trios. All blocks were preceded by a brief instruction (‘Match faces’ or ‘Match shapes’) that lasted 2 s. In the face-processing blocks, each of the six face trios was presented for 4 s with a variable inter-stimulus interval of 2–6 s (mean, 4 s), for a total block length of 48 s. In the sensorimotor control blocks, each of the six shape trios was presented for 4 s with a fixed inter-stimulus interval of 2 s, for a total block length of 36 s. Total task time was 390 s.
Functional magnetic resonance imaging acquisition parameters
Scans were acquired on a Siemens 3T MAGNETOM Allegra platform (Siemens AG, Erlangen, Germany). BOLD functional images were acquired with a gradient-echo echoplanar imaging sequence (TR = 2000 ms, TE = 25 ms, FOV = 20 cm, matrix 64×64), which covered 34 interleaved axial slices (3-mm slice thickness) aligned with the AC–PC plane and encompassing the entire cerebrum and most of the cerebellum. Before collecting functional magnetic resonance imaging data for each participant, we acquired a reference echoplanar imaging scan, which we visually inspected for artifacts (for example, ghosting) and good signal across the entire volume of acquisition, especially the amygdala. In addition, an autoshimming procedure was conducted before the acquisition of BOLD data in each participant to minimize field inhomogeneities.
BOLD functional magnetic resonance imaging data analysis
Whole-brain image analysis was completed using the general linear model of SPM2 (Wellcome Department of Imaging Neuroscience, London, UK). Images for each participant were realigned to the first volume in the time series to correct for head motion, spatially normalized into a standard stereotactic space (Montreal Neurological Institute template), using a 12-parameter affine model, and smoothed to minimize noise and residual difference in gyral anatomy with a Gaussian filter set at 6 mm full-width at half-maximum. Voxel-wise signal intensities were ratio-normalized to the whole-brain global mean. These pre-processed data sets were analyzed using second-level random-effects models that accounted for both scan-to-scan and participant-to-participant variability to determine task-specific regional responses.
The amygdala regions of interest were derived from the AAL atlas using the WFU PickAtlas Tool, version 1.04 (Wake Forest University School of Medicine, Winston-Salem, NC, USA). Amygdala habituation to threat-related stimuli was calculated as the linear decrease over successive face matching blocks (that is, block 1 > block 2 > block 3> block 4). Individual BOLD values from the functional amygdala clusters exhibiting habituation were extracted using the VOI tool in SPM2. Analyses involving FAAH genotype were conducted using these extracted values outside of SPM2, thereby eliminating any possibility of correlations that are artificially inflated because of extraction and correlation techniques that capitalize on the same data twice.42 In sum, our analytic method was quite conservative and yet still demonstrated robust effects as a function of genotype.
Genotyping
Participants were genotyped for a functional non-synonymous single-nucleotide polymorphism (C385A; rs324420), resulting in the conversion of a conserved proline residue to threonine (P129T) in the amino-acid sequence of FAAH.43 In human lymphocytes, FAAH 385A, with an allele frequency of ∼25% in populations of Western European ancestry, is associated with normal catalytic properties but reduced cellular expression of FAAH, possibly through enhanced sensitivity to proteolytic degradation43,44 The C385A is the most common polymorphism in FAAH.45 The 385A variant has previously been associated with obesity and reward-related pathologies, including street drug use and problem drug/alcohol abuse43,45 In addition, we previously linked the 385A allele in the current sample with relatively diminished threat-related reactivity of the amygdala.46
High-molecular-weight DNA was isolated from ethylenediaminetetraacetic acid anticoagulated whole blood samples obtained from all participants using the Puregene kit (Gentra Systems, Minneapolis, MN, USA). Samples were genotyped using the Illumina Human 610-Quad BeadChip; rs324420 data were extracted based on our a priori hypotheses, and no other polymorphisms were tested. Consistent with the manufacturer's protocol (Illumina, San Diego, CA, USA), approximately 200 ng of DNA was used to genotype each subject sample. A BeadArray scanner detected each specifically hybridized DNA that was fluorescently labeled by a single base extension reaction, which were then stained and imaged on an Illumina BeadArray Reader. Next, Illumina BeadStudio software produced SNP genotypes from fluorescent intensities using default cluster settings. The distribution of our observed genotypes (CC = 50, CA = 28 and AA = 3) did not deviate from Hardy–Weinberg equilibrium (χ2 = 0.145, P = 0.704).
Human genetic association study
Participants
Participants are members of the Dunedin Multidisciplinary Health and Development Study. Study members (N = 1037; 91% of eligible births; 52% male) were all individuals born between April 1972 and March 1973 in Dunedin, New Zealand, who were eligible for the longitudinal study based on residence in the province at age 3 years. The cohort represents the full range of socioeconomic status in the general population of New Zealand's South Island and is primarily white. Study members have been followed up repeatedly over their lives, with over 96% retention, and have given written informed consent before participating. The Otago Ethics Committee approved each phase of the study.
Personality assessment
Personality data were collected at age 26 years, from 975 Study members (96% of those still living). Participants completed the Multidimensional Personality Questionnaire,47 which provides, for each person, a profile of scores on 10 distinct personality traits that define three superfactors of personality: Negative Emotionality (indexed by three primary traits called Stress Reaction, Alienation and Aggression); Constraint vs Disinhibition (indexed by three primary traits called Traditionalism, Harm Avoidance and Self-Control); and Positive Emotionality (indexed by four primary traits called Social Potency, Achievement, Well Being and Social Closeness). In this study, we report about Negative Emotionality and its three primary traits. Internal reliability coefficients for all traits exceed 0.80.
Genotyping
DNA was isolated from either whole blood (93% of participants) or buccal swab (7%) samples. The FAAH C385A polymorphism (rs324420) was genotyped via Taqman allelic discrimination. Polymerase chain reactions contained 50 ng of genomic DNA, 1× quantitative polymerase chain reaction Rox Universal Genotyping master mix (Applied Biosystems, Carlsbad, CA, USA) and 1 × primer/probe mix (Applied Biosystems; Assay ID C189730610) made up to a total of 7 μl with distilled water. Thermocycling was performed using an Applied Biosystems 9700 PCR machine with the following parameters: 15 min at 95 °C, followed by 35 cycles of 95 °C for 15 s and 60 °C for 45 s. Genotypes were discerned via end-point fluorescence using an Applied Biosystems 7900HT machine set up in allelic discrimination mode and the SDS 2.1.1 software (Applied Biosystems).
Genotyping was performed blind to personality data, and the call rate was 99%. The distribution of our observed genotypes (CC = 539, AC = 295 and AA = 48) did not deviate from Hardy–Weinberg equilibrium (χ2 = 1.14, P = 0.56). To minimize the risk of artifacts due to population stratification, we restricted analyses to Caucasian participants, after which both genotyping and personality data were available for 881 individuals.
Statistical analysis
The association between rs324420 and personality traits was tested using ordinary least squares regression models (with sex as a covariate). Personality data were standardized to M = 0 and s.d. = 1. Regression coefficients (B with standard errors, t-tests and P-values) are reported comparing individuals carrying two copies of the A allele to C-carriers.
Results and Discussion
AM3506 selectively elevated brain anandamide
For this and subsequent experiments, we used the 129S1/SvImJ (S1) inbred mouse strain that provides a model of impaired fear extinction that can be rescued with pharmacological treatments.23,24
AM3506 has been shown to inhibit FAAH activity in recombinant human cells and increase anandamide in rat brain.19 Before behavioral experiments, we established the time course of FAAH inhibition by intraperitoneally injecting mice with vehicle or 1.0 mg/kg AM3506 and killing at time points from 1 h to 10 days post-injection. Forebrain tissue was extracted and processed for FAAH activity, as assayed by the release of [3H]ethanolamine from arachidonoyl-[3H]ethanolamine. AM3506 produced a near-complete inhibition of FAAH activity, relative to vehicle, within 1 h and produced >50% inhibition for up to 3 days before recovering to ∼80% by 10 days (ANOVA effect of time point: F5,12 = 410.34, P<0.01, followed by Newman–Keuls post hoc tests, n = 2–4) (Figure 1a). This long-lasting action of AM3506 is consistent with its profile as an irreversible FAAH inhibitor.19 FAAH inhibition was likely brain-wide, as there was a similarly potent and long-lasting inhibition of FAAH in cerebellar tissue extracts (Supplementary Figure S1). In the same samples, we also determined the activity of MAGL, by measuring the release of [3H]glycerol from 2-arachidonyl-[3H]glycerol. In contrast to FAAH inhibition, MAGL activity in the forebrain was unaltered by AM3506 treatment (n = 3–4) (Figure 1b). Thus, these data demonstrate that systemic administration of AM3506 produced a potent, lasting and preferential inhibition of FAAH in the brain.
AM3506 facilitates fear extinction
We next examined the effects of systemic AM3506 treatment on fear extinction. Mice were trained to fear an auditory CS via repeated pairings of the CS with footshock (US). A control group received CS-alone presentations. The day after conditioning, mice were intraperitoneally injected with AM3506 or vehicle 1 h before an extinction training session comprising 50× presentations of the CS alone.23 To evaluate the efficacy of extinction, a retrieval test was conducted 10 days later. The fear response to the CS during extinction and retrieval was measured by quantifying freezing behavior. This treatment and test design was meant to approximate the adjunctive drug treatment approach used in patients with anxiety disorders undergoing exposure therapy.48
Results showed that fear on the retrieval test was significantly lower in mice treated with any of the three doses of AM3506, relative to vehicle (ANOVA effect of treatment: F3,39 = 3.76, P<0.05, followed by post hoc tests, n = 10–11) (Figure 1c). This is consistent with significant facilitation of extinction by AM3506 treatment. It is notable that fear levels at the beginning of extinction were not affected by AM3506 treatment. This in line with prior studies showing that CB1R gene knockout or antagonism impairs fear extinction and adaptation without affecting fear expression.3,49 Interestingly, we also found that the long-term extinction-facilitating effects of AM3506 were independent of any demonstrable within-session extinction (Supplementary Figure S2a). This dissociation echoes the finding that pre-extinction treatment of S1 mice with the clinically efficacious anxiolytic, fluoxetine, also produces marked reductions in fear during retrieval without concomitant within-session reductions.25 It is also in line with the earlier observation that manipulations which facilitate long-term extinction in CB1R knockout mice (for example, spaced training) do not produce within-session extinction.50 On the other hand, there is evidence that nonspecific stimulation of the endocannabinoid system with the CB1 agonist WIN55212 or endocannabinoid reuptake blockade by AM404 treatment can facilitate within-session extinction in a strain of rats that, unlike S1, show intact trait extinction.51 The reasons for the absence of AM3056′s effects on extinction learning remain to be determined, but likely relate to either the specificity of the drug's actions on anandamide or the extinction-resistant model used, or a combination of the two.
AM3506 specifically affects fear extinction
To more fully elucidate the specificity of AM3506′s effects, we conducted a number of experiments in which 1.0 mg/kg AM3506 was systemically administered at different phases of conditioning, fear retrieval and extinction. We found that AM3506 given before conditioning did not affect fear or extinction the next day (Supplementary Figure S2b), or retrieval 10 days later (t-test: P>0.05, n = 8) (Figure 1d), indicating a lack of effect on fear acquisition and fear retrieval, as well as post-extinction fear. The latter negative effect is particularly interesting given that FAAH levels remain inhibited by ∼75% at the time of extinction training (that is, 1 day after pre-conditioning injection). This suggests that a certain threshold level of inhibition may be required at the time of extinction to enable long-term reductions in fear or, alternatively, that the timing of the injection, and the stress associated with it, may influence the efficacy of the drug's fear-reducing effects.52
Next, we showed that AM3506 given immediately after extinction training (Supplementary Figure S2c) did not reduce fear on the retrieval test 10 days later, indicating that augmenting anandamide during post-training consolidation is not sufficient to facilitate extinction (t-test: P>0.05, n = 12) (Figure 1e). An effect of AM3506 on reconsolidation was also ruled out by the finding that drug treatment before fear memory reactivation (and no extinction) (Supplementary Figure S2d) did not reduce fear on a later test (t-test: P>0.05, n = 8) (Figure 1f). Finally, we confirmed that AM3506 reduced fear on a retrieval test only in mice that had been extinguished, by showing that freezing was unaffected by AM3506 treatment before exposure to the extinction context per se (that is, no CS) (VEH = 74±5% freezing, AM = 69±8, t-test: P>0.05, n = 12), and only in mice that had been fear conditioned, by showing normal/negligible freezing in unconditioned mice given AM3506 before extinction (freezing <1% both groups, t-test: P>0.05, n = 6).
These results establish the specificity of AM3506′s effects on extinction and not other fear-related processes. However, previous rodent studies have shown that pharmacological manipulation of the endocannabinoid system has effects on locomotion, depression-related behavior and feeding.7 We therefore examined the effects of systemic AM3506 treatment, using the same 1 mg/kg dose, route and injection-test interval as we found facilitated extinction, on measures of these behaviors. Relative to vehicle, AM3506 treatment did not alter spontaneous locomotion (Figure 1g), ‘depression-related’ immobility in the forced swim test (Figure 1h) or food consumption after overnight fasting (Figure 1i) (all t-tests: P>0.05, n = 8). We also found that AM3506 did not promote glucose intolerance in anandamide-treated mice, an effect that contrasted with the pro-intolerance effect of URB597, replicating a dissociation between these compounds previously observed in diet-induced obese mice19 (Supplementary Figure S3). Taken together, these negative findings provide further support for the behavioral specificity of AM3506, although testing across a broad range of assays will be needed be provide a full pharmacodynamic profile of the drug.
AM3506 and extinction training boost amygdala anandamide
Our data suggest that FAAH inhibition targets endogenous anandamide signaling in extinction circuitry. To further interrogate this hypothesis, we assessed whether AM3506 treatment augmented anandamide levels in brain regions mediating fear extinction. Mice underwent conditioning and extinction training as above. AM3506 was injected intraperitoneally 1 h before extinction and mice were killed immediately after extinction. The tissue was extracted from two key nodes within the extinction circuit—the BLA and ventromedial prefrontal cortex,13 as well as the dorsal striatum (as a control) (Figure 2a). Anandamide and 2-arachidonoylglycerol levels were quantified using liquid chromatography tandem mass spectrometry.33
Figure 2.
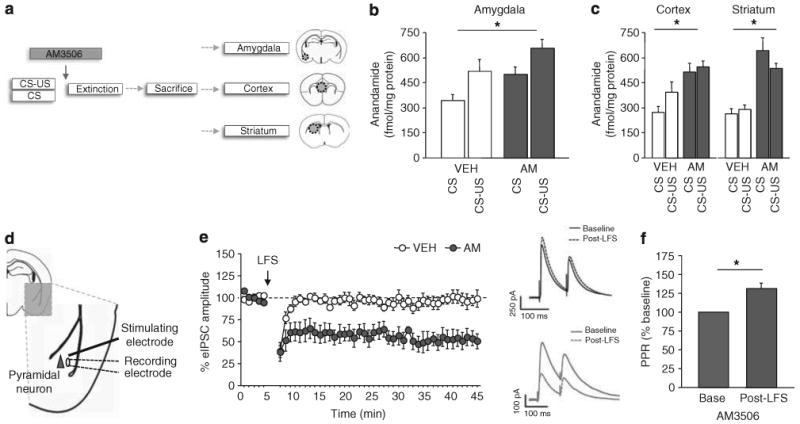
Extinction training and AM3506 (5-(4-hydroxyphenyl)pentanesulfonyl fluoride) increase anandamide levels in the basolateral amygdala. (a) Schematic of testing and tissue collection procedure. (b) Systemic AM3506 treatment and extinction training increased anandamide levels in the basolateral amygdala in fear-conditioned (CS–US) mice, as compared to non-conditioned (CS) controls (n = 10–11 per treatment). (c) Systemic AM3506 treatment and extinction training increased anandamide levels in the cortex and striatum, as compared to vehicle treated controls (n = 10–11 per treatment). (d) Low-frequency stimulation (LFS) of the external capsule was used to induce endocannabinoid-mediated long-term depression of inhibitory, GABAergic, transmission (LTDi) in the basolateral amygdala. (e) In slices treated with 2 μm AM3506, but not vehicle, there was a marked and lasting reduction in evoked inhibitory postsynaptic currents (eIPSCs) amplitude following LFS, relative to baseline (n = 7–8 per treatment). Example traces of eIPSC amplitudes are shown on the right. (f) Paired pulse ratio (PPR), measuring the peak change in eIPSC amplitude over two consecutive stimulations, was higher in AM3506-treated slices after stimulation, relative to pre-stimulation baseline. CS = conditioned stimulus; US = unconditioned stimulus. *P<0.05. Data are means±s.e.m.
Ex vivo levels of anandamide in the BLA were significantly elevated in mice that had received CS–US training, relative to CS-alone training (ANOVA effect of training: F1,38 = 7.03, P<0.05) (Figure 2b). In addition, AM3506 treatment increased anandamide levels in both training groups, relative to vehicle (ANOVA effect of treatment: F1,38 = 8.69, P<0.01, n = 10–11). AM3506-treated mice also increased anandamide levels in the ventromedial prefrontal cortex (ANOVA effect of treatment: F1,43 = 15.76, P<0.01, n = 10–13) and the dorsal striatum (ANOVA effect of treatment: F1,46 = 49.82, P<0.01, n = 11–15) (Figure 2c), relative to vehicle controls but, in contrast to BLA, neither region showed a significant increase as a result of CS–US training.
Our results are consistent with prior data showing that extinction training selectively increases anandamide levels in mouse BLA3 (but see Lin et al.53) and, furthermore, demonstrate that this increase in endogenous anandamide is augmented by AM3506 treatment. Examination of ex vivo 2-arachidonoylglycerol levels revealed only modest, statistically nonsignificant, increases in the BLA in CS–US, compared with CS-alone, trained mice. Moreover, AM3506 treatment had no effect on 2-arachidonoyl-glycerol levels either in the BLA or in the ventromedial prefrontal cortex or in the dorsal striatum (Supplementary Figure S4). These neurochemical data show that, by inhibiting FAAH, AM3506 boosts endogenous anandamide signaling in the BLA.
AM3506 enhances amygdala synaptic plasticity
To determine the consequences of these elevated anandamide levels for plasticity within amygdala circuits,54 we performed slice electrophysiological analysis of AM3506′s effects at the BLA synapses. LFS (100 stimuli at 1 Hz) of the external capsule has been shown to induce a form of endocannabinoid- and CB1R-mediated LTDi in the BLA.3,55,56 To test whether AM3506 enhanced this form of plasticity, 2 μm AM3506 or vehicle was applied to the amygdala slices. Monosynaptically eIPSCs were recorded from visually identified pyramidal neurons via stimulation by an electrode placed within the BLA just medial to the external capsule (Figure 2d). LTDi was induced by LFS at twice the stimulation intensity of a test pulse (paired pulses elicited at 0.1 Hz, with 100 ms inter-stimulus interval).
In slices treated with vehicle, this stimulation protocol did not produce a change in eIPSC amplitude from baseline, whereas there was a marked and lasting reduction in eIPSC amplitude in AM3506-pretreated slices (ANOVA effect of treatment: F1,44 = 469.30, P<0.01, n = 7–8) (Figure 2e). These data demonstrate that AM3506 enhances LTDi in the amygdala and echo the previous observation that a similar effect is produced by brain-wide gene deletion of faah in mice.56 PPR, which measures the peak change in eIPSC amplitude over two consecutive stimulations (for traces see Figure 2e), was increased in AM3506-treated slices after LFS, relative to baseline (t-test: t(4) = 4.33, P<0.05, n = 5) (Figure 2f, for traces see Figure 2e). Because alterations in PPR are typically associated with a change in presynaptic release probability, these data suggest that AM3506 promoted activity-dependent suppression of GABA release.
The effect of AM3506 on glutamatergic transmission in the BLA was tested by measuring fEPSP. Application of AM3506 produced a small, but significant, decrease in fEPSP amplitude (last 5-min time block vs baseline: t(5) = 3.06, P<0.05, n = 6) (Supplementary Figure S5a,b), consistent with a modest decrease in excitatory transmission. Next, we tested the ability of AM3506 pretreatment to gate the induction of excitatory LTD induced by low LFS2 (10 Hz for 10 min) using a protocol described previously.35,36 There was no change in fEPSPs, regardless of drug treatment (Supplementary Figure S5c,d), indicating this form of LTD was unaffected by AM3506 (n = 5–7) (Supplementary Figure S5c,d).
Collectively, these data show that AM3506-induced elevation of anandamide in the BLA is associated with an enhancement of LTDi, but less with other forms of synaptic plasticity, in this brain region. This profile is consistent with a lasting suppression of GABA release from amygdala interneurons, one consequence of which could be relief of a tonic inhibitory constraint on amygdala neurons engaged in the formation of extinction memories.14 Direct functional evidence in support of this mechanism will require additional study.
AM3506 facilitates fear extinction via the amygdala
The most likely receptor target of the effects of augmented anandamide in the BLA is CB1R, which is highly expressed in this region.5 To confirm that CB1R activation mediated the extinction-facilitating effect of AM3506, we repeated our experiment above, in which mice were systemically injected with AM3506 before extinction, but co-treated half of the AM3506-treated mice with 1 mg/kg of the CB1R antagonist SR141716 (Rimonabant). Neither SR141716 nor AM3506 altered baseline, pre-CS, freezing or CS-induced freezing during extinction training (ANOVA effect of treatment: P>0.05) (Supplementary Figure S6a). Replicating our earlier finding, fear measured at the 10-day retrieval test was significantly lower, relative to vehicle, in mice treated with AM3506 (ANOVA effect of treatment: F3,41 = 3.14, P<0.05, followed by post hoc tests, n = 9–12) (Figure 3a). By contrast, mice treated with SR141716+AM3506 were no different from vehicle controls, thereby indicating that systemic CB1R antagonism had blocked the extinction-facilitating effect of AM3506.
Figure 3.
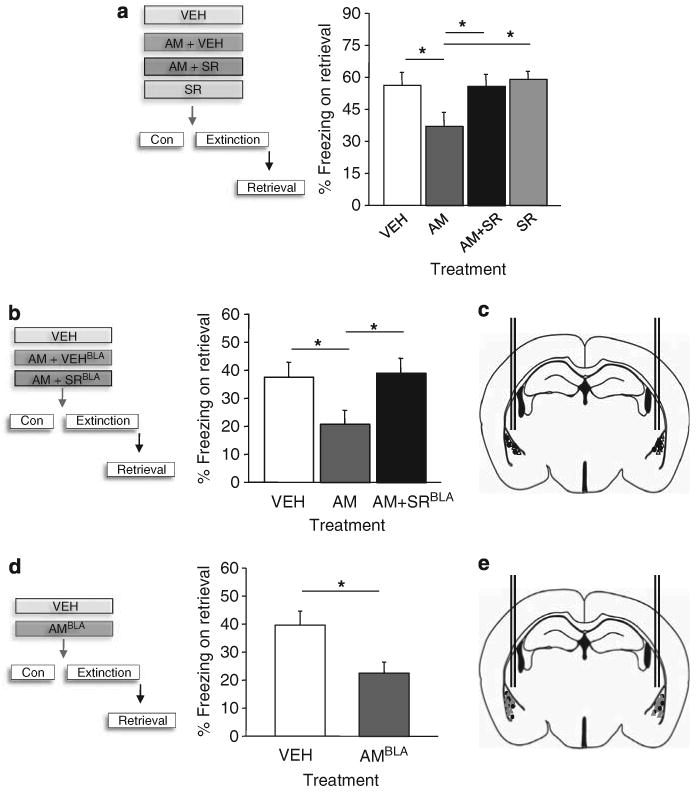
AM3506 (5-(4-hydroxyphenyl)pentanesulfonyl fluoride) facilitates fear extinction via CB1 receptor (CB1R) in the basolateral amygdala. (a) The ability of systemic AM3506 treatment before extinction to reduce fear on a retrieval test was blocked by systemic treatment with the CB1R antagonist SR141716 (n = 9–12 per treatment). (b) The ability of pre-extinction systemic AM3506 treatment to reduce fear on a retrieval test was blocked by intra-amygdala infusion of SR141716 (n = 7 per treatment). (c) Cannula tip localization. (d) Pre-extinction infusion of AM3506 into the amygdala was sufficient to reduce fear on a retrieval test (n = 8–11 per treatment). (e) Cannula tip localization. VEH = vehicle; SR = SR141716; AM = AM3506. *P<0.05. Data are means±s.e.m.
Next, to isolate the extinction-facilitating action of AM3506 to CB1Rs within the BLA, we stereotaxically implanted guide cannulae aimed bilaterally at the BLA. After recovering from surgery, mice underwent behavioral testing and pre-extinction systemic AM3506 treatment as above, with half receiving bilateral infusion of 2 μg/μl SR141716 (or equivalent volume of 0.5 μl per hemisphere vehicle) into the BLA 5 min before the systemic AM3506 treatment. Drug treatment did not affect baseline or CS-induced freezing during extinction training (ANOVA effect of treatment: P>0.05, n = 8–10) (Supplementary Figure S6b). Fear was again reduced in AM3506-treated mice, compared to vehicle, during the retrieval test (ANOVA effect of treatment: F2,19 = 3.54, P<0.05, followed by post hoc tests, n = 7–8). The key novel finding, however, was that this extinction-facilitating effect was absent in mice that had received BLA infusion of SR141716 (Figures 3b and c).
Our final step was to determine whether direct amygdala administration of AM3506 itself produced effects on extinction. We surgically prepared another set of mice and bilaterally infused 0.1 μg/μl AM3506 (or equivalent volume of 0.5 μl per hemisphere vehicle) into the amygdala 30 min before extinction training. Drug treatment did not affect baseline or CS-induced freezing during extinction training (ANOVA effect of treatment: P>0.05, n = 8–11) (Supplementary Figure S6c), but fear was reduced during retrieval test in amygdala-AM3506-treated mice, compared with vehicle-infused controls (t-test: t(17) = 2.77, P<0.05, n = 8–11) (Figures 3d and e). Thus, taken together this series of experiments demonstrate that augmenting endocannabinoid signaling at CB1R in the amygdala is both necessary and sufficient to facilitate extinction. It should be noted that this conclusion, and indeed all of the preclinical data in this study, is based on one compound and one model of impaired extinction, and it will be important to extend thesefindings to other rodent models, as well as other specific FAAH inhibitors.
FAAH gene variation modulates human amygdala habituation to threat
Our rodent experiments suggest that inhibiting FAAH, via AM3506 administration, facilitates extinction by augmenting endogenous anandamide signaling in the amygdala. By extension, this raises the possibility that genetic variation in endogenous anandamide levels could affect fear-circuit function and predisposition to fear-related disorders in humans. Thus, based on our preclinical data we generated the a priori hypothesis that human fear-related endophenotypes associate with a common functional FAAH gene variant (C385A; rs324420) that produces reduced FAAH expression in lymphocytes.43
In the first study, we used imaging genetics in a sample of middle-aged adults (N = 81) who performed a ‘threatening faces’ task that elicits robust amygdala reactivity on initial face-viewing, and subsequent amygdala habituation with repeated viewing.46 All subjects showed significant amygdala habituation to threatening faces, calculated as a linear decrease over successive viewing, in the right hemisphere dorsally (x = 24, y = −8, z = −11, z = 4.58, t-test: t(79) = 4.92, P<0.01) and ventrally (x = 32, y = −1, z = −20, z = 4.36, t-test: t(79) = 4.64, P<0.01), and left hemisphere (x = −26, y = −1, z = −13, z = 3.89, t-test: t(79) = 4.10, P<0.01). However, the rate of habituation differed significantly as a function of FAAH genotype (Figures 4a and b), with individuals carrying the lesser-expressing 385A variant demonstrating significantly greater habituation compared with C385 homozygotes in all three clusters (right dorsal: t-test: t(79) = 2.15, P<0.05; right ventral: t(79) = 3.15, P<0.01; left: t-test: t(79) = 2.26, P<0.05). While C385A genotype has previously been associated with human amygdala reactivity,46 these data show for the first time that FAAH variation modulates the ability of the amygdala to adapt to threat.
Figure 4.
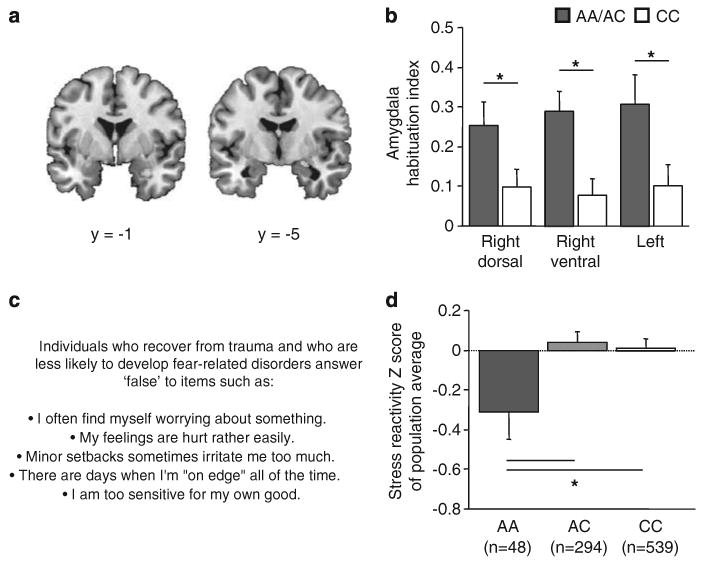
The lesser-expressing human fatty acid amide hydrolase (FAAH) gene 385A variant is associated with fear-related endophenotypes and traits. (a) Both the right and left amygdala exhibited significant habituation to repeated viewing of threatening faces. (b) Individuals carrying at least one lesser-expressing FAAH 385A variant showed greater amygdala-mediated habituation to threatening stimuli in all three clusters from (a). Given the low occurrence of homozygous AA carriers (n = 3) in this sample, A/A and C/A carriers (n = 31) were combined and compared with C/C homozygotes (n = 50). Data are means±s.e.m. (c, d) AA homozygotes scored significantly lower on a personality endophenotype of Stress Reactivity, measured by the Multidimensional Personality Questionnaire.47 *P<0.05. Data are means±s.e.m.
FAAH gene variation associates with low human stress reactivity We next tested the influence of FAAH C385A on fear-related traits via personality testing in a population-representative birth cohort of young adults (N = 881).57 On the basis of our preclinical and imaging genetics findings, we focused our analysis specifically on Negative Emotionality as the salient superfactor within the Multidimensional Personality Questionnaire47 because this is a premorbid personality trait that predisposes individuals who are exposed to trauma to develop fear-related disorders.58 Cohort members carrying two copies of the lesser-expressing 385A variant scored significantly lower for Negative Emotionality (β = −0.08, t = 2.51, P<0.05). Negative Emotionality comprises three specific facets: Stress Reactivity, Aggression and Alienation. Specificity analyses revealed that, as predicted by the above findings, those homozygous for the lesser-expressing 385A variant were especially likely to exhibit low Stress Reactivity (β = −0.08, t = 239, P<0.05) (Figures 4c and d), but did not differ from other cohort members on Aggression (β = −0.04, t = 1.31, P>0.05) or Alienation (β = −0.05, t = 1.64, P>0.05). Thus, FAAH genetic variation was associated with individual differences in specific fear-related traits. While these human genetic associations of FAAH with stress-reactive personality and habituation of amygdala-mediated threat responses await replication, they provide clear convergence with our rodent data showing that FAAH inhibition facilitates amygdala-mediated fear extinction. Among the key avenues for future studies will be a test of whether FAAH variation associates specifically with human fear extinction and prevalence of extinction-impaired neuropsychiatric disorders such as post-traumatic stress disorder.
Conclusions
The endocannabinoid system has emerged as an important regulator of stress and emotional processing, but a potentially intractable target for neuropsychiatric medications due to widespread effects on a variety of behaviors. Our mouse data suggest that, by preventing FAAH-mediated degradation, augmenting anandamide in the basolateral amygdala may boost on-demand recruitment of endocannabinoids to facilitate the extinction of traumatic fear memories. Further we report an association between a putative loss-of-function human FAAH gene variant, an amygdala fear-plasticity endophenotype, and reduced trait stress reactivity. The collective results of our study support the endocannabinoid system and FAAH as a key signaling mechanism regulating fear plasticity that is conserved across mouse and man. More generally, our approach illustrates the broad potential of focused translational research bridging animal models and human studies. Further work will be needed to test the possibility of targeting of endocannabinoids, via FAAH inhibition, as a novel approach to treating fear-related disorders.
Supplementary Material
Acknowledgments
This work was supported by the NIAAA Intramural Research Program (OGC, KPM, RC, GK, AH), Department of Defense in the Center for Neuroscience and Regenerative Medicine (OGC, AH), NIA (AG032282 to TEM), MRC (G0100527 to TEM), NIDA (DA031579 to ARH) and NIMH (MH077874 to AC; MH072837 to ARH, MH090412 to SP). AC is a Royal Society Wolfson Merit Award holder. We thank Daniel Fisher and Caitlin Schaapveld for assistance with behavioral experiments.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
References
- 1.Cryan JF, Holmes A. The ascent of mouse: advances in modelling human depression and anxiety. Nat Rev Drug Discov. 2005;4:775–790. doi: 10.1038/nrd1825. [DOI] [PubMed] [Google Scholar]
- 2.Quirk GJ, Pare D, Richardson R, Herry C, Monfils MH, Schiller D, et al. Erasing fear memories with extinction training. J Neurosci. 2010;30:14993–14997. doi: 10.1523/JNEUROSCI.4268-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 4.Kamprath K, Marsicano G, Tang J, Monory K, Bisogno T, Di Marzo V, et al. Cannabinoid CB1 receptor mediates fear extinction via habituation-like processes. J Neurosci. 2006;26:6677–6686. doi: 10.1523/JNEUROSCI.0153-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, et al. Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 7.Marsicano G, Lutz B. Neuromodulatory functions of the endocannabinoid system. J Endocrinol Invest. 2006;29:27–46. [PubMed] [Google Scholar]
- 8.Cravatt BF, Lichtman AH. Fatty acid amide hydrolase: an emerging therapeutic target in the endocannabinoid system. Curr Opin Chem Biol. 2003;7:469–475. doi: 10.1016/s1367-5931(03)00079-6. [DOI] [PubMed] [Google Scholar]
- 9.de Oliveira Alvares L, Pasqualini Genro B, Diehl F, Molina VA, Quillfeldt JA. Opposite action of hippocampal CB1 receptors in memory reconsolidation and extinction. Neuroscience. 2008;154:1648–1655. doi: 10.1016/j.neuroscience.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Kinsey SG, Long JZ, Cravatt BF, Lichtman AH. Fatty acid amide hydrolase and monoacylglycerol lipase inhibitors produce anti-allodynic effects in mice through distinct cannabinoid receptor mechanisms. J Pain. 2010;11:1420–1428. doi: 10.1016/j.jpain.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spradley JM, Guindon J, Hohmann AG. Inhibitors of monoacylglycerol lipase, fatty-acid amide hydrolase and endocannabinoid transport differentially suppress capsaicin-induced behavioral sensitization through peripheral endocannabinoid mechanisms. Pharmacol Res. 2010;62:249–258. doi: 10.1016/j.phrs.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci. 2010;13:1113–1119. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herry C, Ferraguti F, Singewald N, Letzkus JJ, Ehrlich I, Luthi A. Neuronal circuits of fear extinction. Eur J Neurosci. 2010;31:599–612. doi: 10.1111/j.1460-9568.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- 15.Varvel SA, Wise LE, Niyuhire F, Cravatt BF, Lichtman AH. Inhibition of fatty-acid amide hydrolase accelerates acquisition and extinction rates in a spatial memory task. Neuropsychopharmacology. 2007;32:1032–1041. doi: 10.1038/sj.npp.1301224. [DOI] [PubMed] [Google Scholar]
- 16.Hajos N, Kathuria S, Dinh T, Piomelli D, Freund TF. Endocannabinoid transport tightly controls 2-arachidonoyl glycerol actions in the hippocampus: effects of low temperature and the transport inhibitor AM404. Eur J Neurosci. 2004;19:2991–2996. doi: 10.1111/j.0953-816X.2004.03433.x. [DOI] [PubMed] [Google Scholar]
- 17.Tan H, Lauzon NM, Bishop SF, Chi N, Bechard M, Laviolette SR. Cannabinoid transmission in the basolateral amygdala modulates fear memory formation via functional inputs to the prelimbic cortex. J Neurosci. 2011;31:5300–5312. doi: 10.1523/JNEUROSCI.4718-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chhatwal JP, Davis M, Maguschak KA, Ressler KJ. Enhancing cannabinoid neurotransmission augments the extinction of conditioned fear. Neuropsychopharmacology. 2005;30:516–524. doi: 10.1038/sj.npp.1300655. [DOI] [PubMed] [Google Scholar]
- 19.Godlewski G, Alapafuja SO, Batkai S, Nikas SP, Cinar R, Offertaler L, et al. Inhibitor of fatty acid amide hydrolase normalizes cardiovascular function in hypertension without adverse metabolic effects. Chem Biol. 2010;17:1256–1266. doi: 10.1016/j.chembiol.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bashashati M, Storr MA, Nikas SP, Wood JT, Godlewski G, Liu J, et al. Inhibiting fatty acid amide hydrolase normalizes endotoxin-induced enhanced gastrointestinal motility in mice. Br J Pharmacol. 2012;165:1556–1571. doi: 10.1111/j.1476-5381.2011.01644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fowler CJ, Tiger G, Stenstrom A. Ibuprofen inhibits rat brain deamidation of anandamide at pharmacologically relevant concentrations. Mode of inhibition and structure–activity relationship. J Pharmacol Exp Ther. 1997;283:729–734. [PubMed] [Google Scholar]
- 22.Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, et al. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hefner K, Whittle N, Juhasz J, Norcross M, Karlsson RM, Saksida LM, et al. Impaired fear extinction learning and cortico-amygdala circuit abnormalities in a common genetic mouse strain. J Neurosci. 2008;28:8074–8085. doi: 10.1523/JNEUROSCI.4904-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whittle N, Hauschild M, Lubec G, Holmes A, Singewald N. Rescue of impaired fear extinction and normalization of cortico-amygdala circuit dysfunction in a genetic mouse model by dietary zinc restriction. J Neurosci. 2010;30:13586–13596. doi: 10.1523/JNEUROSCI.0849-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Camp M, MacPherson KP, Lederle L, Graybeal C, Gaburro S, DeBrouse L, et al. Genetic strain differences in learned fear inhibition associate with variation in neuroendocrine, autonomic and amygdala dendritic phenotypes. Neuropsychopharmacology. 2012;37:1534–1547. doi: 10.1038/npp.2011.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brigman JL, Wright T, Talani G, Prasad-Mulcare S, Jinde S, Seabold GK, et al. Loss of GluN2B-containing NMDA receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J Neurosci. 2010;30:4590–4600. doi: 10.1523/JNEUROSCI.0640-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang RJ, Mozhui K, Karlsson RM, Cameron HA, Williams RW, Holmes A. Variation in mouse basolateral amygdala volume is associated with differences in stress reactivity and fear learning. Neuropsychopharmacology. 2008;33:2595–2604. doi: 10.1038/sj.npp.1301665. [DOI] [PubMed] [Google Scholar]
- 28.Wellman CL, Izquierdo A, Garret JE, Martin KP, Carroll J, Millstein R, et al. Impaired stress-coping and fear extinction and abnormal corticolimbic morphology in serotonin transporter knock-out mice. J Neurosci. 2007;27:684–691. doi: 10.1523/JNEUROSCI.4595-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiedholz LM, Owens WA, Horton RE, Feyder M, Karlsson RM, Hefner K, et al. Mice lacking the AMPA GluR1 receptor exhibit striatal hyperdopaminergia and ‘schizophrenia-related’ behaviors. Mol Psychiatry. 2008;13:631–640. doi: 10.1038/sj.mp.4002056. [DOI] [PubMed] [Google Scholar]
- 30.Cryan JF, Markou A, Lucki I. Assessing antidepressant activity in rodents: recent developments and future needs. Trends Pharmacol Sci. 2002;23:238–245. doi: 10.1016/s0165-6147(02)02017-5. [DOI] [PubMed] [Google Scholar]
- 31.Norcross M, Mathur P, Enoch AJ, Karlsson RM, Brigman JL, Cameron HA, et al. Effects of adolescent fluoxetine treatment on fear-, anxiety- or stress-related behaviors in C57BL/6J or BALB/cJ mice. Psychopharmacology (Berl) 2008;200:413–424. doi: 10.1007/s00213-008-1215-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bellocchio L, Lafenetre P, Cannich A, Cota D, Puente N, Grandes P, et al. Bimodal control of stimulated food intake by the endocannabinoid system. Nat Neurosci. 2010;13:281–283. doi: 10.1038/nn.2494. [DOI] [PubMed] [Google Scholar]
- 33.Mukhopadhyay B, Cinar R, Yin S, Liu J, Tam J, Godlewski G, et al. Hyperactivation of anandamide synthesis and regulation of cell-cycle progression via cannabinoid type 1 (CB1) receptors in the regenerating liver. Proc Natl Acad Sci USA. 2011;108:6323–6328. doi: 10.1073/pnas.1017689108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel S, Kingsley PJ, Mackie K, Marnett LJ, Winder DG. Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signaling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology. 2009;34:2699–2709. doi: 10.1038/npp.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lafourcade M, Elezgarai I, Mato S, Bakiri Y, Grandes P, Manzoni OJ. Molecular components and functions of the endocannabinoid system in mouse prefrontal cortex. PLoS One. 2007;2:e709. doi: 10.1371/journal.pone.0000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang W, Sun D, Pan B, Roberts CJ, Sun X, Hillard CJ, et al. Deficiency in endocannabinoid signaling in the nucleus accumbens induced by chronic unpredictable stress. Neuropsychopharmacology. 2010;35:2249–2261. doi: 10.1038/npp.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.First M, Spitzer R, Gibbon M, Williams J. Structured Clinical Interview for DSM-IV Axis I Disorders, Research Version, Non-Patient Edition. New York State Psychiatric Institute, Biometrics Research Department; New York: 1996. [Google Scholar]
- 38.Brown SM, Manuck SB, Flory JD, Hariri AR. Neural basis of individual differences in impulsivity: contributions of corticolimbic circuits for behavioral arousal and control. Emotion. 2006;6:239–245. doi: 10.1037/1528-3542.6.2.239. [DOI] [PubMed] [Google Scholar]
- 39.Brown SM, Peet E, Manuck SB, Williamson DE, Dahl RE, Ferrell RE, et al. A regulatory variant of the human tryptophan hydroxylase-2 gene biases amygdala reactivity. Mol Psychiatry. 2005;10:884–888. 805. doi: 10.1038/sj.mp.4001716. [DOI] [PubMed] [Google Scholar]
- 40.Manuck SB, Brown SM, Forbes EE, Hariri AR. Temporal stability of individual differences in amygdala reactivity. Am J Psychiatry. 2007;164:1613–1614. doi: 10.1176/appi.ajp.2007.07040609. [DOI] [PubMed] [Google Scholar]
- 41.Ekman P, Friesen W. Pictures of Facial Affect. Consulting Psychologists Press; Palo Alto, CA: 1976. [Google Scholar]
- 42.Viviani R. Unbiased ROI selection in neuroimaging studies of individual differences. NeuroImage. 2010;50:184–189. doi: 10.1016/j.neuroimage.2009.10.085. [DOI] [PubMed] [Google Scholar]
- 43.Sipe JC, Chiang K, Gerber AL, Beutler E, Cravatt BF. A missense mutation in human fatty acid amide hydrolase associated with problem drug use. Proc Natl Acad Sci USA. 2002;99:8394–8399. doi: 10.1073/pnas.082235799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiang KP, Gerber AL, Sipe JC, Cravatt BF. Reduced cellular expression and activity of the P129T mutant of human fatty acid amide hydrolase: evidence for a link between defects in the endocannabinoid system and problem drug use. Hum Mol Genet. 2004;13:2113–2119. doi: 10.1093/hmg/ddh216. [DOI] [PubMed] [Google Scholar]
- 45.Flanagan JM, Gerber AL, Cadet JL, Beutler E, Sipe JC. The fatty acid amide hydrolase 385 A/A (P129T) variant: haplotype analysis of an ancient missense mutation and validation of risk for drug addiction. Hum Genet. 2006;120:581–588. doi: 10.1007/s00439-006-0250-x. [DOI] [PubMed] [Google Scholar]
- 46.Hariri AR, Gorka A, Hyde LW, Kimak M, Halder I, Ducci F, et al. Divergent effects of genetic variation in endocannabinoid signaling on human threat- and reward-related brain function. Biol Psychiatry. 2009;66:9–16. doi: 10.1016/j.biopsych.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patrick CJ, Curtin JJ, Tellegen A. Development and validation of a brief form of the multidimensional personality questionnaire. Psychol Assess. 2002;14:150–163. doi: 10.1037//1040-3590.14.2.150. [DOI] [PubMed] [Google Scholar]
- 48.Holmes A, Quirk GJ. Pharmacological facilitation of fear extinction and the search for adjunct treatments for anxiety disorders—the case of yohimbine. Trends Pharmacol Sci. 2010;31:2–7. doi: 10.1016/j.tips.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kamprath K, Romo-Parra H, Haring M, Gaburro S, Doengi M, Lutz B, et al. Short-term adaptation of conditioned fear responses through endocannabinoid signaling in the central amygdala. Neuropsychopharmacology. 2011;36:652–663. doi: 10.1038/npp.2010.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Plendl W, Wotjak CT. Dissociation of within- and between-session extinction of conditioned fear. J Neurosci. 2010;30:4990–4998. doi: 10.1523/JNEUROSCI.6038-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pamplona FA, Bitencourt RM, Takahashi RN. Short- and long-term effects of cannabinoids on the extinction of contextual fear memory in rats. Neurobiol Learn Mem. 2008;90:290–293. doi: 10.1016/j.nlm.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 52.Riebe CJ, Pamplona F, Kamprath K, Wotjak CT. Fear relief-toward a new conceptual frame work and what endocannabinoids gotta do with it. Neuroscience. 2012;204:159–185. doi: 10.1016/j.neuroscience.2011.11.057. [DOI] [PubMed] [Google Scholar]
- 53.Lin HC, Mao SC, Su CL, Gean PW. The role of prefrontal cortex CB1 receptors in the modulation of fear memory. Cereb Cortex. 2009;19:165–175. doi: 10.1093/cercor/bhn075. [DOI] [PubMed] [Google Scholar]
- 54.Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Luthi A. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 55.Chevaleyre V, Heifets BD, Kaeser PS, Sudhof TC, Castillo PE. Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1alpha. Neuron. 2007;54:801–812. doi: 10.1016/j.neuron.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Azad SC, Monory K, Marsicano G, Cravatt BF, Lutz B, Zieglgansberger W, et al. Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J Neurosci. 2004;24:9953–9961. doi: 10.1523/JNEUROSCI.2134-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krueger RF, Caspi A, Moffitt TE. Epidemiological personology: the unifying role of personality in population-based research on problem behaviors. J Pers. 2000;68:967–998. doi: 10.1111/1467-6494.00123. [DOI] [PubMed] [Google Scholar]
- 58.Miller MW. Personality and the etiology and expression of PTSD. Clin Psychol Sci Pract. 2003;10:373–393. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.