

Abstract
Ras GTPases have been a subject of intense investigation since the early-80’s, when single point mutations in Ras were shown to cause deregulated cell growth control. Subsequently, Ras was identified as the most prevalent oncogene found in human cancer. Ras proteins regulate a host of pathways involved in cell growth, differentiation, and apoptosis by cycling between inactive GDP-bound and active GTP-bound states. Regulation of Ras activity is controlled by cellular factors that alter guanine nucleotide cycling. Oncogenic mutations prevent protein regulatory factors from down-regulating Ras activity, thereby maintaining Ras in a chronically activated state. The central dogma in the field is that protein modulatory factors are the primary regulators of Ras activity. Since the mid-90’s, however, evidence has accumulated that small molecule reactive nitrogen species (RNS) can also influence Ras guanine nucleotide cycling. Herein, we review the basic chemistry behind RNS formation and discuss the mechanism through which various RNS enhance nucleotide exchange in Ras proteins. In addition, we present studies that demonstrate the physiological relevance of RNS-mediated Ras activation within the context of immune system function, brain function, and cancer development. We also highlight future directions and experimental methods that may enhance our ability to detect RNS-mediated activation in cell cultures and in vivo. The development of such methods may ultimately pave new directions for detecting and elucidating how Ras proteins are regulated by redox species, as well as for targeting redox-activated Ras in cancer and other disease states.
Keywords: Ras GTPase, Ras activation, S-nitrosation, nitric oxide, nucleotide exchange, radical-mediated exchange, immuno-spin trapping
Introduction: Ras GTPases and Free Radical Oxidants
Ras superfamily GTPases are considered critical signaling proteins that regulate pathways involved in cell growth, differentiation, apoptosis, cell motility and vesicular transport [1]. This regulatory action stems from the cycling of Ras between an “active” GTP bound state and an “inactive” GDP bound state. Protein modulatory factors (GTPase Activating Proteins (GAPs) and Guanine Nucleotide Exchange Factors (GEFs) (Figure 1) have been shown to bind to Ras proteins and regulate their activity by modulating guanine nucleotide cycling. The binding of GEFs to Ras enhances the rate of GDP dissociation. Given the excess molar ratio of GTP/GDP in vivo (~ 10 : 1) [2], GEFs activate Ras by promoting exchange of GDP for GTP. Conversely, GAPs have the opposite function and cause inactivation of Ras by facilitating hydrolysis of GTP to GDP. While research on protein modulatory factors (i.e. GAPs and GEFs) has dominated the literature on Ras regulation, reactive small molecule oxidants have also been shown to regulate Ras activity [3–5]. Similar to the action of GEFs, reactive nitrogen species have been shown to facilitate Ras activation, both in vitro and in vivo, by enhancing the rate of guanine nucleotide dissociation. However, the mechanisms through which Ras is regulated by redox control remain less well understood.
Figure 1.

Guanine nucleotide exchange is commonly regulated by protein modulatory factors, such as GEFs and GAPs. GEFs facilitate exchange of GDP to GTP, while GAPs promote intrinsic hydrolysis of GTP to GDP. Recent evidence suggests small free radical oxidants, such as •NO2, also facilitate Ras guanine nucleotide exchange similar to the action of GEFs.
Numerous endogenous and exogenous sources of reactive nitrogen and oxygen species exist. Many reactive nitrogen and oxygen species (RNS and ROS, respectively) are found in the form of small free radicals. Some endogenous enzymes, such as nitric oxide synthases (NOSs) and NADPH oxidases which produce nitric oxide (•NO) and superoxide (O2•−), respectively, are common sources of free radicals in vivo. UV radiation and gas-phase smoke, from cigarettes or air pollution, highlight a small portion of exogenous sources. Free radicals, themselves, are generally short-lived molecules which possess one or more unpaired electrons. They can be highly reactive species or relatively non-reactive depending on their oxidation potential. Most free radicals are, however, particularly reactive toward other free radicals. Antioxidant enzymes, such Cu,Zn-superoxide dismutase (SOD), glutathione peroxidase and peroxiredoxins, act alongside many small molecule antioxidants, such as ascorbate, glutathione, and tocotrienols, to quench free radicals in the body. Nonetheless, free radicals can still react with lipids, DNA, or proteins such as Ras, and alter their biological function.
Herein, we review in vitro and in vivo studies that detail the effects of RNS on Ras proteins. For simplification, we limit our discussion of RNS to nitric oxide (•NO), nitrogen dioxide (•NO2), dinitrogen trioxide (N2O3), and to a lesser extent peroxynitrite anion (ONOO−). Initially, we present a brief introduction to the chemistry and kinetics governing the formation of these RNS species. Current theories on the mechanism of RNS-mediated Ras regulation are also presented alongside numerous studies that may demonstrate this event in cells. Finally, we discuss future directions which highlight new efforts to detect and better characterize Ras regulation by RNS within a cellular context.
Understanding the kinetics that dictate RNS formation is required before delving into the •NO-mediated processes that regulate Ras GTPase activity. Within the body, •NO is generated by three main NOS isoforms. •NOS isoforms produce •NO at different levels in response to numerous stimuli such as Ca+ and calmodulin, post-translational modifications, and inflammatory agents [6]. Nitric oxide, itself, is a very poor oxidant with a low standard oxidation potential (Em7 = −0.8 V vs. NHE) [7]. Even though •NO has a low oxidation potential, it remains the parent compound for a number of reactive nitrogen species. Some of the best studied interactions of •NO entail its reaction with O2 or superoxide (O2•−). Initially we examine the former reaction with O2.
Formation of Reactive Nitrogen Species
Autoxidation of •NO
One major route for RNS production begins with the reaction between nitric oxide and oxygen. Autoxidation of •NO occurs in aqueous solutions with a rate constant of ~ 2.9 × 106 M−2 s−1 and yields the moderate oxidant nitrogen dioxide, •NO2 (Reaction 1, Em7 = ~1 V vs. NHE) [8–11]. The autoxidation of •NO is reported to be over 30 times more rapid within the hydrophobic environment of biological membranes [12]. This is significant, as •NO2 is known to react and
| (1) |
modify a variety of biomolecules resulting in lipid peroxidation [13, 14], guanine base nitration [15, 16], tyrosine side chain nitration [17, 18], and oxidation of thiols to thiyl radicals [19]. Thiyl radical intermediates, whether protein or small molecule based (i.e., glutathione), act as a precursor for radical-based S-nitrosation events. This process is considered extremely important for activation of Ras GTPases and will be discussed in more detail later. In addition, •NO2 is known to react with unsaturated hydrocarbons by both reversible addition and irreversible H-abstraction mechanisms [20]. The biological importance of this reactivity is apparent from the reaction of •NO2 and arachidonic acid [21]. •NO2 has been shown to induce cis-trans isomerization of arachidonic acid leading to increased levels of trans-fatty acids in human plasma [21].
The reactivity of •NO2 toward other free radical species, through well-established radical recombination reactions, contrasts its reactions with small organics (lipids) and large macromolecules (protein thiols). •NO2 reacts with •NO near the diffusion limit (k = 1 × 1010 M−1 s−1) with a forward reaction rate of 1.1 × 109 M−1 s−1. This reaction produces non-radical dinitrogen trioxide, N2O3, (Reaction 2) [8, 22]. N2O3 is considered a potent S-nitrosation agent for small molecule thiols (i.e. glutathione, cysteine) and protein thiols (i.e. cysteine side chains)
| (2) |
| (3) |
[23, 24]. N2O3 readily hydrolyzes under aqueous conditions, however, with a rate of 4.75 × 104 s−1 (Reaction 3) [23, 25]. In addition to its reaction with •NO, •NO2 also dimerizes to form an additional oxidizing species, dinitrogen tetroxide (N2O4) (Reaction 4) [8, 26, 27]. The reported solubility of N2O4 is ~2 orders of magnitude higher than monomeric •NO2 (g) [8, 26, 27]. Levels of •NO2 in vivo are likely to be low (nM range) under typical cellular conditions [8, 28], with monomeric •NO2 being the predominant species in vivo as its dimerization rate depends on the square of •NO2 concentration [8, 28]. Both N2O3 and N2O4 exist in equilibrium with their precursor radicals (•NO2 and •NO for N2O3, •NO2 for N2O4) and have equilibrium constants of K = 2.6 × 102 M−1 and K = 7 × 104 M−1, respectively [8, 22, 26, 27]. Under aqueous conditions, N2O4 hydrolyzes to produce nitrite (NO2−) and nitrate (NO3−) with a reaction rate of ~ 1 × 103 s−1 (Reaction 5) [8, 29].
| (4) |
| (5) |
Peroxynitrite Formation
Another well-studied RNS is produced from the reaction of •NO with superoxide (O2•−). This radical recombination reaction yields the non-radical oxidant, peroxynitrite anion (ONOO−) [30]. The importance of this oxidizing species, within a biological context, was presented over two decades ago [31], and its significance has been the focus of several thorough reviews [32–35]. As shown in Reaction 6, •NO reacts with O2•− near the diffusion limit with a reaction rate between 4.3 and 19 × 109 M−1 s−1 [36, 37]. This rate is notably faster than the very efficient reaction of O2•− with superoxide dismutase (SOD, k = 2 × 109 M−1 s−1) [38]. SOD facilitates the
| (6) |
dismutation of O2•− to H2O2 (hydrogen peroxide) and is considered a major scavenger of superoxide in vivo. Peroxynitrite has a pKa of ~ 6.8 and readily diffuses through cell membranes in both anionic (through anion channels) and protonated peroxynitrous acid forms (via passive diffusion) [33, 39]. While peroxynitrite is considered a strong oxidant and nitrating agent, it is a poor nitrosating agent [34]. Given the pKa of peroxynitrite, it is estimated that ~ 80 % exists as ONOO− anion (peroxynitrite) and ~ 20 % as protonated HONOO (peroxynitrous acid) in the absence of carbon dioxide [34]. Peroxynitrous acid, if present at all under physiological conditions, may undergo O-O bound homolysis yielding both •NO2 and the omnipotent hydroxyl radical (•OH, Em7 = 2.3 V vs NHE) at a rate of k = 0.9 s−1 (Reaction 7) [31, 34, 40, 41]. The yield of these decomposition products has been intensely debated, but studies indicate ~ 10 % of
| (7) |
peroxynitrous acid decomposes to form hydroxyl radicals [41]. The remaining 90 % is believed to isomerize into nitric acid (HNO3) [41]. Only a small fraction of peroxynitrite/peroxynitrous acid likely proceeds to homolysis in vivo (~ 5 %) [34], as the oxidant is predominately consumed through other reactions, notably reactions involving carbon dioxide and hemoglobin.
As mentioned, one major target for peroxynitrite in vivo is carbon dioxide [33, 34, 42, 43]. Peroxynitrite reacts with CO2 with a pH independent reaction rate of 5.8 × 104 M−1 s−1 to yield a theoretical nitrosoperoxocarboxylate intermediate species (ONOOCO2−, Reaction 8) [42]. The nitrosoperoxocarboxylate intermediate is believed to rapidly decompose into a mixture
| (8) |
of ~35 % •NO2 + CO3•− and ~ 65 % NO3− + CO2 [42, 44]. The carbonate radical anion (CO3•−) produced during this process has a high oxidizing potential (Em7 = ~1.8 V) and is known to perform a one-electron oxidation of glutathione and cysteine to the thiyl radical with rates of 5.3 × 106 M−1 s−1 and 4.6 × 107 M−1 s−1, respectively [45, 46].
RNS and Ras GTPases
A correlation between RNS and Ras activation was first discovered when human Jurkat T cells were exposed to ~ 1 mM •NO (g). As a result of this exposure, the cells were found to have elevated levels of active, GTP-bound Ras [47]. Later studies revealed increased Ras activity was correlated to S-nitrosation of a single, solvent-exposed cysteine (Cys118) [48]. Subsequent mutation of this cysteine to a serine (C118S) prevented S-nitrosation and •NO-mediated guanine nucleotide exchange of Ras in vitro and Ras activation in cells [48]. As described in later sections, Lander et al. went on to show that RNS could not only promote Ras activation but also Ras signaling to downsteam targets [48]. Work from our laboratory (Williams et al.) subsequently probed the specific mechanism of •NO-mediated regulation of H-ras GTPase activity in vitro [49]. We found that stable S-nitrosation of H-ras at Cys118 actually had no effect on the structure or guanine exchange rate relative to non-nitrosated H-ras [49], and concluded an intermediate formed during the reaction rather than the endpoint of Ras S-nitrosation must be a causative factor for increased nucleotide exchange [49].
RNS can produce S-nitrosothiols through different mechanisms; a non-radical pathway that involves S-nitrosation via transnitrosation [50–53] or electrophilic N2O3 [24, 54, 55] and a radical pathway (by reaction of •NO2 and the thiol) which consists of a one-electron oxidation followed by radical recombination (i.e. RS• + •NO) [24, 55–57]. Formation of a S-nitrosothiol at Cys118 on Ras by N2O3 is considered unlikely, however. The physiological concentrations of Ras would never be high enough to permit sufficient S-nitrosation through this pathway, as the rate of N2O3 hydrolysis dominates the fate of this nitrosating species (Reaction 3) [23, 55]. With this in mind, we turn our focus to the free radical pathway involving an initial one-electron oxidation via •NO2.
Given that H-ras S-nitrosation does alter on guanine nucleotide exchange [49] we followed up with a series of studies that provided support for RNS-mediated generation of a Ras thiyl radical intermediate (a precursor of S-nitrosothiol formation) and its role in modulating Ras activity [4, 58, 59]. Results from these studies indicated that one-electron oxidation of Cys118 in Ras, using authentic •NO2 (g), stimulated enhanced guanine nucleotide exchange. The mechanism of free radical Ras activation was postulated to involve electron transfer between bound guanine nucleotide and the Cys118 thiyl radical (Figure 2) [4, 59]. According to the NMR structure of GDP-bound Ras (pdb 1CRR), the Cys118 sulfhydryl lies within 8 Å of the bound GDP guanine base [60]. Intramolecular electron transfer over such distances is commonly observed, particularly when a suitable pathway for the transfer is available. In some proteins, electron transfer pathways between donor and acceptor side chains have been cited at distances as large as 35 Å [61]. We have previously shown that treatment of Ras-GDP with •NO2 perturbs GDP binding due to accelerated release of GDP and formation of a 5-guanidino-4-nitroimidazole diphosphate product (NIm-DP) [59]. These results suggest dissociation, rearrangement and nitration of the GDP moiety when GDP-bounds Ras is exposed to •NO2 in vitro. The guanine base was not observed to be oxidized or nitrated when Cys118 was mutated to serine, indicating that it was Ras-bound GDP rather than GDP in the bulk solvent that became oxidized to NIm-DP. Does this mechanism occur in vivo? Quantifying GDP oxidation in vivo is non-trivial, given the large pool of GDP associated with various metabolities, nucleic acids and other GDP-binding proteins. Thus, there is currently no evidence that exposure of endogenous Ras to RNS in cells, results in guanine base oxidation. Collectively, the in vitro data suggest an altered oxidation potential for the GDP moiety when bound to Ras, as GDP cannot be oxidized by •NO2 alone in solution [59]. Nonetheless, if electron transfer occurs between the thiyl radical at Cys118 and bound GDP, a potential competition must exist between electron transfer and S-nitrosation via radical recombination (RS• + •NO) when •NO is available. This theory may also help explain why high levels of Ras activity were initially correlated with S-nitrosation of Cys118, as both S-nitrosation and radical-mediated exchange can be correlated to a precursor Cys118 thiyl radical species.
Figure 2.

In the presence of •NO2, Cys118 in Ras undergoes a one-electron oxidation. Subsequent electron transfer between the resulting Cys118 thiyl radical and bound guanine nucleotide causes oxidation of the guanine base and premature release guanine nucleotide from Ras. The closest distance between the sulfur of Cys118 and bound GDP is ~ 7.54 Å, as presented by the 1CRR NMR structure [71].
Detecting Free Radical Ras Activation in Cells
Detection of radical-mediated Ras activation in cells is an experimental challenge due to the transient nature of protein radicals. Immunoprecipitation of S-nitrosated Ras is not necessarily a good biomarker for free radical activation events. As mentioned, S-nitrosation can occur through many processes, including transnitrosation [50–53] and electrophilic N2O3 [23–25]. While detection of S-nitrosated Ras could indicate free radical activation, it should not be construed as a conclusive indicator. Detecting free radical induced Ras activation ultimately rests in our ability to detect the Ras thiyl radical intermediate, as this species is a direct precursor to radical-mediated activation (Figure 3).
Figure 3.
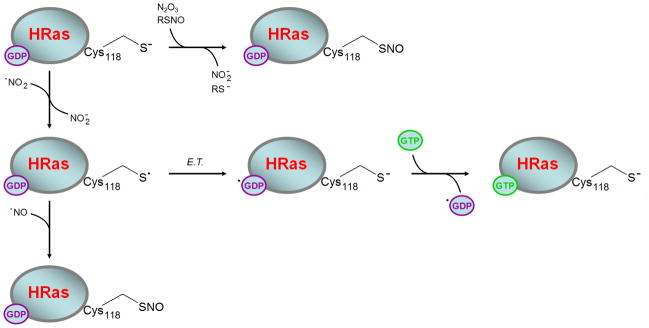
Transnitrosation or electrophilic N2O3 are believed to S-nitrosate Ras without promoting guanine nucleotide exchange. In the presence of •NO2, however, a transient Ras Cys118 thiyl radical is produced. Electron transfer (E.T.) between the Cys118 thiyl radical and bound GDP is believed to initiate release the nucleotide. In vivo this would result in population of the active GTP-bound Ras. Once formed, the transient Ras Cys118 thiyl radical can also undergo radical recombination with •NO to produce S-nitrosated Ras. For these reasons, S-nitrosation cannot be construed as a conclusive indicator of radical-mediated exchange. Detection of S-nitrosated Ras can, however, be useful as a biomarker for potential radical-mediated activation events.
One promising avenue for this may lie in a technique pioneered by the Mason lab at the National Institute of Environmental Health Sciences (NIEHS). The technique, immuno-spin trapping, offers an immunological-based approach for detecting protein radicals [62, 63]. Immuno-spin trapping (IST) consists of using the 5,5′-dimethyl-1-pyrroline N-oxide (DMPO) spin trap to isolate protein radical intermediates. DMPO will not react with either •NO or •NO2 alone. Once the Ras thiyl protein radical is trapped, recently developed anti-DMPO antibodies can be employed to detect the bound DMPO-nitrone adduct using highly sensitive enzyme-linked immunosorbent assays (ELISAs) or simple Western blotting [62, 63]. Adduction of DMPO to Ras also adds ~111 Da. to its molecular weight. Therefore, IST can be coupled with tandem mass spectrometry (MS/MS) approaches to unambiguously identify protein radical sites [64–68]. Perhaps the main advantage of immuno-spin trapping, however, lies in its suitability for detecting protein radical formation in living cells or animal models [69]. This suitability is mostly attributable to the overall redox stability of DMPO and its inert reactivity toward non-radical species [63].
Although IST offers a promising route for detecting radical-mediated activation, the technique has been limited in its applications and has been used almost exclusively to detect protein radicals in metalloproteins [62–68]. Detection of Ras protein radicals by IST in vitro is complex and depends on competing reaction pathways shown in Figure 4. Efforts to detect the Ras Cys118 thiyl radical in vitro and in cultured cells using IST-based approaches are currently underway in our laboratory. While IST may not be able to differentiate Ras radical formation via primary or secondary radicals formed in vivo, IST studies of cells expressing the Ras C118S variant, should allow discrimination of cellular redox events that require radical formation at Cys118. Moreover, the activation state of Ras should also be assessed so that IST detection of Ras Cys118 radical intermediates can be correlated with Ras activity. Successful detection of Ras protein radicals by IST, under physiological relevant conditions, may bring attention to this activation mechanism in cancers and other disease states where Ras activation plays a significant role.
Figure 4.

Detection of Ras DMPO-nitrone adducts by IST, upon treatment of Ras with •NO in vitro, involves a series of complex and competing reaction kinetics. Blue text highlights the pathway which results in observable Ras DMPO-nitrone adducts.
Biological Consequences of RNS-mediated Ras Activation
Previously, we highlighted mechanisms through which RNS are formed and discussed how these oxidants enhance Ras GTPase guanine nucleotide exchange in vitro. We now turn our focus to representative cell-based experiments that show a direct correlation between RNS and regulation of Ras activity. While this review focuses primarily on RNS and Ras activation, a brief discussion of other Ras regulation mechanisms is warranted.
Currently, guanine nucleotide exchange factors (GEFs) and oncogenic mutations remain the best understood mechanisms of Ras activation in cells. RasGEFs activate Ras by binding the GDP-bound form and catalyzing release of the nucleotide. The relative GTP to GDP ratio in human cells is ~ 10 : 1 [2]. This ratio dictates Ras will likely to bind GTP upon GDP dissociation, as affinity for the two nucleotides is nearly identical [70]. Eight well-established RasGEFs represent three distinct families: SOS (Son of Sevenless) 1 and 2, RasGRPs (Ras guanyl nucleotide-releasing proteins) 1–4, and RasGRFs (Ras guanine nucleotide releasing factors) 1 and 2. The large number of RasGEFs permit spatio-temporal activation of functionally distinct pools of Ras in response to different stimuli such as growth factors, calcium, and neurotransmitters [71–73]. Oncogenic mutation of Ras, most commonly somatic mutation of residues 12, 13 and 61, is another well-characterized activation mechanism. Mutation of Ras in cancer cells typically reduces the ability of RasGAPs to hydrolyze Ras-GTP, thereby chronically populating Ras in an active, GTP-bound state [74, 75].
While RasGEFs and oncogenic mutations represent well-established activation mechanisms, efforts of the past decade have greatly enhanced our understanding of how RNS contribute to Ras activity. In the following sections, we highlight several studies that provide strong evidence of Ras modification by RNS in cells. The specific methodologies of the highlighted investigations will be examined to better understand the strengths, weaknesses, and physiological relevance of data implicating RNS in Ras activation. As discussed earlier, no direct method exists for detecting radical-mediated Ras activation in cells. While immuno-spin trapping may offer a viable route, the technique has yet to be implemented for Ras GTPases in a cellular context. Therefore, we must look at simple Ras S-nitrosation as a biomarker for potential radical-mediated events. This has several drawbacks, as S-nitrosation can occur through non-radical processes, such as transnitrosation and electrophilic N2O3. In addition, other oxidative modifications (e.g. S-glutathiolation) may or may not reflect formation of a Ras radical species. Nonetheless, Ras S-nitrosation may be used as a compass to identify potential radical-mediated activation events [55]. Indeed, this notion has also been echoed in a recent publication by Lancaster et al [76]. Detection of S-nitrosated Ras may provide a useful initial screen for more specific targeting efforts in the future. Likewise, the use of Ras mutants which lack the reactive thiol have proven to be critical in a number of cellular studies linking RNS to altered Ras signaling processes.
Ras Activation and the Immune System
Ras and the downstream MAPK (Mitogen-Activated Protein Kinase) and PI3K (Phosphatidylinositol-3-Kinase) pathways have especially crucial functions in T cell activation from the T lymphocyte antigen receptor. Specifically, deregulation of Ras signaling in T cells has been implicated during development of the autoimmune disorder lupus [77]. Ras has interesting and complex properties in T cells. Ras is activated at various intracellular locations by distinct stimuli, which leads to differential downstream signaling and biological effects [78]. For example, K-ras is activated specifically on the plasma membrane, while N-ras is activated on the endoplasmic reticulum [79]. Although it was shown that the RasGEF, RasGRP1, was responsible for full Golgi-specific Ras activation [80], it is still unclear precisely how distinct pools of Ras are activated in a coordinated manner.
The significance of •NO-mediated Ras activation in T cells was first demonstrated by Lander and co-workers, who reported that addition of exogenous •NO (g) to Jurkat T cells resulted in increased GTP-bound Ras concentrations. Furthermore, addition of •NO gas to purified H-ras in vitro resulted in greater nucleotide exchange rates [47]. Another important observation from this study was that lower levels of exogenous •NO (.01 μM) activated Ras in T cells, while higher levels (> 0.1 μM) inactivated Ras. This is significant as the physiological concentration of •NO in cells is believed to range from 0.005 – 4 μM [32, 81]. The levels of •NO required for Ras activation in vitro were found to be much higher, however (~ 75 μM). This may be due to different reactions of •NO that occur in vitro versus in cells. These data suggest that •NO can be a double-edged sword; activating or inactivating Ras depending on the cellular context. In addition, •NO activated Ras in T cells only at early time points. Activation was reduced to baseline after a few hours, suggesting activation through •NO is both reversible and temporally regulated.
As highlighted in earlier sections, autoxidation of •NO within biological membranes is extremely fast (~ 30 times faster than its aqueous rate of k = 2.9 × 106 M−2 s−1) [12]. Therefore, direct addition of exogenous •NO (g) to cells likely results in a multitude of NO-oxides (i.e. •NO2, N2O3, N2O4). In fact, in this study Ras was exposed to •NO (g) that was not scrubbed for removal of these higher NO-oxides. This indicates that RNS, other than simple •NO may have been present. Evidence of this can be seen in other studies, which demonstrated specific S-nitrosation of Cys118 upon addition of •NO (g) [48]. This is significant, as •NO itself is incapable of producing S-nitrosothiols. Subsequent mutation of this cysteine (H-Ras C118S) rendered Ras insensitive to RNS/ROS modification. In addition, it also abolished the ability of •NO to enhance nucleotide exchange, both in vitro and in T cells [82]. A follow-up study, using exogenous NO-donating compounds, found that addition of •NO to T cells also activated the MAPK pathway, but not when a C118S Ras mutant was expressed [48]. Importantly, our in vitro studies of H-Ras C118S indicate mutation of the redox active cysteine to a serine does not affect the structure and biochemical activity of Ras [83].
A more recent study solidified •NO-mediated Ras regulation during T cell activation [84]. Ibiza et al. demonstrated that physiological stimulation of the T-cell receptor (TCR) by antigen causes S-nitrosation of wild-type but not C118S N-ras. Again, S-nitrosation of Ras was correlated with levels of active Ras. The authors importantly demonstrated that exogenous •NO S-nitrosated and activated both N-ras and K-ras, but TCR stimulation only caused S-nitrosation of N-ras. It is possible that the differential effects on the Ras isoforms may be due to differences in cellular •NO levels by exogenous versus endogenous sources. Nevertheless, this finding highlights potential isoform specificity of •NO-mediated regulation of Ras activity. Furthermore, the role of endogenous •NO, produced from active endothelial nitric oxide (eNOS) was also examined. Reduction of •NO levels, through the NOS inhibitor L-NAME (L-Nitro-Arginine-Methyl-Ester) in T cells, or in T cells from mice lacking the eNOS gene (eNOS −/−), was found to significantly reduce levels of activated and S-nitrosated Ras. The specific Ras isoforms which exhibited reduced activity were never determined. Overexpression of wild type but not C118S N-ras was also found to induce apoptosis upon TCR stimulation. This implies an important biological role for modification of the reactive cysteine (Cys118) in Ras, although which Ras effectors induce this differential effect remains unclear. Furthermore, the authors demonstrated that active (phosphorylated) eNOS co-localized with active N-ras on the Golgi upon TCR stimulation. This could signify spatio-temporal regulation of Ras by an endogenous nitric oxide source.
Although these studies described interesting cellular mechanisms for Ras activation by nitric oxide, many questions remain. The RasGEFs, SOS1 and RasGRP1, are known to be important for proper Ras activation during T cell activation and development [77]. Therefore, what is the interplay between eNOS-mediated and GEF-mediated Ras activation? Are they each responsible for activation of specific subcellular pools of Ras, or do they combine for a full activation of the same Ras pools? Is eNOS-mediated Ras activation important for a subset of Ras-mediated T-cell functions, such as clonal expansion or differentiation, and is this activation mechanism important in pathological settings such as lupus? A general model for •NO-mediated control of Ras in T cells is shown in Figure 5. Further studies as discussed in Future Directions will help provide the answers to these and other questions.
Figure 5.

Simplified model of Ras activation in T cells. The T-cell receptor is activated by antigen resulting in a variety of biological outcomes. K-ras activation at the plasma membrane, mediated by SOS1, contributes to proliferation and differentiation. Activation of N-ras at the Golgi is mediated by both eNOS and RasGEFs. Calcium activates RasGRP1 which subsequently activates N-ras and leads to integrin-mediated adhesion. Alternatively, activation of eNOS can activate N-ras in a •NO-dependent manner to promote apoptosis. This figure highlights how diverse stimuli can activate different pools of Ras to promote specific cellular outcomes. It also demonstrates how RasGEF-mediated activation of Ras can have a different functional outcome than •NO-mediated activation.
Ras Activation and Brain Function
The Ras family is highly expressed in both the developing and adult brain. Ras GTPases play crucial roles in neuronal survival, differentiation, and synaptic plasticity, in response to both growth factor and calcium signaling [85–87]. Like the immune system, Ras activation in neurons is complex. Activation of Ras involves growth factors, phospholipase isoforms, and calcium through the Ras GEFs, SOS, RasGRPs and RasGRFs [88]. In addition, studies have demonstrated a role for nitric oxide in the activation of Ras through various NOS isoforms.
In neurons, the glutamate-binding NMDA (N-methyl-D-aspartate) receptor is important for controlling synaptic plasticity and memory through regulation of calcium signaling [89]. Deregulation of NMDA receptor function contributes to seizure, depression, and neurodegenerative diseases [90]. As Ras and nitric oxide signaling are involved in similar neuronal processes, understanding if and how NMDA receptor and Ras signaling are related may provide novel avenues to treat these diseases. As an important step towards these connections, NMDA receptor stimulation has been correlated to activation of Ras in primary cortical neurons [91]. Furthermore, inhibition of eNOS with the NOS inhibitor L-NAME blocked NMDA receptor activation of Ras. Ras activation via NMDA receptor activation was also blocked in mice which lacked the eNOS gene. As an important control, brain-derived neutrophic factor could still activate Ras, demonstrating some aspects of Ras signaling were still functional. Mechanistically, L-NAME also blocked phosphorylation of the downstream Ras effector ERK (extracellular signal-related kinase) [91]. It remains unclear, however, if Ras is directly or indirectly activated by •NO (from eNOS), or whether neuronal processes controlled by Ras, such as synaptic plasticity, are regulated via eNOS or RasGEFs. A separate study recently provided evidence of H-ras S-nitrosation in mouse neurons, suggesting Ras regulation in these cells were influenced through •NO produced from active eNOS [92].
Ras GTPases also play a significant role in the survival and differentiation of neurons. In the PC12 neuronal cell lines, for example, nerve growth factor (NGF) binds to its receptor and mediates the survival of serum-starved cells. NGF-mediated differentiation of the cells is regulated by Ras activation via phosphorylation of Erk, PI3K and Shc [93]. NGF-mediated Ras activation of PI3K has also been implicated in neuronal survival [94]. An interesting study investigated whether •NO-mediated activation of Ras is linked to NGF-dependent phenotypes in neurons. PC12 cells were engineered to overexpress wild type or C118S H-ras. NGF-induced phosphorylation of Shc and Erk were unaffected in cells overexpressing the C118S mutant. These results suggest that Ras activation of the MAPK pathway, under these conditions, is not directly regulated by •NO or some other RNS or ROS. Likewise, NGF-induced differentiation occurred in both wild-type and C118S cells. However, only the cells overexpressing wild-type but not C118S H-ras were able to survive in low growth media. Furthermore, only wild-type but not C118S H-ras was able to bind PI3K in these cells. Both observations are consistent with a role for the direct activation of Ras by •NO leading to stimulation of PI3K activity and downstream pathways that promote cell survival [95]. This study importantly demonstrates differential effector signaling and phenotypes by RNS or ROS activated Ras (i.e. activation of the PI3K and survival, but not the MAPK pathway or differentiation). However, it was not determined which RNS or ROS was responsible. Interestingly, the data also suggests that NMDA receptor stimulation of PI3K activates Akt which then directly phosphorylates and activates nNOS. Once nNOS is activated (resulting in •NO production) in neurons [96], a possible positive feedback loop exists where PI3K and •NO signaling may reinforce each other (Figure 6). However, as there are different mechanisms besides Ras that can activate PI3K, it was unclear in this study if Ras-mediated activation of PI3K led to activation of NOS, as the use of the redox-insensitive C118S mutant was not used.
Figure 6.

Model of Ras activation in neurons by NGF. NGF promotes both differentiation and survival in neurons by activating Ras in different ways and through different effector pathways. Survival is facilitated by •NO-mediated Ras activation of the PI3K/Akt pathway, while differentiation is mediated through SOS1-dependent Ras activation of the Raf/Mek/Erk pathway.
While this may provide the first example of differential signaling of •NO versus RasGEF-mediated processes, many questions are left unanswered by this study. For example, is this a product of differential localization of •NO or RasGEFs, or is some other mechanism responsible for separate signaling responses? Additionally, since Ras is overexpressed ten-fold relative to the endogenous copy, could this affect the results? Overexpression of Ras can result in its mis-localization, which often renders Ras sensitive to activation by a different set of GEFs and/or RNS than it would be physiologically. Thus, in any system where Ras is overexpressed, caution must be made in interpreting activation mechanisms and downstream effector usage. Ideally, when using the C118S Ras mutant to investigate the role of RNS, the endogenous version should be knocked down with RNAi, and rescued with RNAi-resistant wild type or C118S at expression levels similar to the endogenous level.
Ras Activation and Cancer
Ras mutations occur in up to 30% of all human tumors. K-ras, in particular, is mutated at high rates in three of the four most deadly cancers (lung, colorectal and pancreatic) [97]. Besides mutation, Ras activity is often up-regulated in gliostoma and colorectal cancer through decreased expression of the RasGAPs, Neurofibromatosis1 and RASAL1 (Ras protein activator like 1), respectively [98, 99]. More recently an •NO-mediated pathway has been linked to tumor initiation and maintenance in Ras-driven pancreatic cancer [100]. In pancreatic cancer lines which have oncogenic K-ras mutations, it was demonstrated that endogenous H- and N-ras are highly activated. Knockdown of H-ras or N-ras by RNAi blocks anchorage-independent growth and xenograft tumor formation in mice. Furthermore, the defect in xenograft growth in mice is rescued by expression of RNAi-insensitive wild-type H-ras or N-ras, but not their redox-insensitive C118S variants. The authors found that endogenous H- and N-ras are both S-nitrosated in eNOS active, oncogenic K-ras driven pancreatic cancer. The authors also showed that knockdown of eNOS by RNAi not only blocks S-nitrosation of H-and N-ras, but also inhibits tumor xenograft formation in mice [100]. This study is, in our opinion, the strongest evidence for •NO-mediated activation of Ras in any biological process, since several key experiments are provided together. The authors successfully demonstrated endogenous Ras isoforms are S-nitrosated and active (i.e. high levels of GTP-bound Ras), and they were able to identify the endogenous •NO source responsible for Ras S-nitrosation. Furthermore, the biological consequence of Ras activation (in this case tumor growth) was determined to be dependent on an RNS process, as C118S mutants could not rescue the biological activity.
The study also showed that oncogenic K-ras causes activation of PI3K and Akt, as has been demonstrated in many cases. These pathways lead to direct phosphorylation and activation of eNOS. The authors demonstrate the most important effector pathway for pancreatic tumor maintenance by oncogenic K-ras is the PI3K/Akt/eNOS pathway. A proposed model from this study suggests oncogenic K-ras causes activation of eNOS (via PI3K and Akt), which then activates wild-type H- and N-ras isoforms directly through •NO production. Activation of eNOS, and subsequently H-ras and N-ras via RNS, may be a primary mechanism through which onocogenic K-ras maintains tumorigenesis (Figure 7).
Figure 7.

Model of Ras activation by •NO-mediated processes in pancreatic cancer. Pancreatic cancer growth and maintenance is dependent, in part, on activation of eNOS by oncogenic K-ras. NO subsequently activates wild-type H-ras and N-ras. H- and N-ras contribute to the effects of oncogenic K-ras, which include anchorage-independent growth and tumor formation in mice. N-ras and H-ras may also activate PI3K/Akt, which would constitute a positive feedback activation of the PI3K pathway.
While comprehensive in its scope, many intriguing questions arise from this study. Does •NO-mediated activation of H-ras or N-ras regulate specific Ras-dependent signaling pathways, while RasGEFs regulate others? Are eNOS and specific Ras isoforms co-localized in pancreatic cancer cells, allowing tight spatio-temporal control over Ras activation, as is the case in T-cells? The authors demonstrated that only two out of seven pancreatic cancer cell lines tested contained activated eNOS. Therefore, are H-ras and N-ras still necessary for the full transformed phenotype in the rest of the cell lines, and if so, does their activation depend on other NOS isoforms, or ROS? Other cancer types are also known to be driven by Ras signaling. Does full activation of Ras in these cancers require eNOS-mediated RNS or a different radical-based activation (i.e. ROS)? For example, K-ras mutations are common in lung cancer, and the redox state of the lung is very different than other organs [101]. Do RNS- or ROS-mediated regulation of Ras activity, contribute to Ras-driven lung cancers? Nevertheless, this study illustrated the significance of •NO-mediated Ras activation as it relates to tumor development and maintenance.
Future Directions
First, we wish to highlight some experiments that must be done to understand the physiological relevance of radical-based Ras activation in normal biology and in disease states. As highlighted in earlier sections, many studies, including those we have omitted due to space, demonstrate exogenous •NO-donors or endogenous •NO, via NOS isoforms, induce activation of Ras. In many cases, this activation may be indirect; acting through other targets (e.g. redox enzymes, phosphatases) or proteins that directly influence Ras activity (RasGEFs or RasGAPs). It has also become clear, that proper spatio-temporal regulation of Ras is critical to better understand the complexities of Ras signaling. Similarly, given the reactivity of many RNS, physiological regulation of biomolecules likely requires careful attention to the type of RNS and levels present at sites where Ras is localized, as well as the cellular reducing potential. Thus, to understand the physiological relevance of any RNS on the activation state of Ras, results obtained using endogenous RNS sources should be evaluated. While earlier studies have clearly established that exogenous •NO sources (i.e •NO gas and •NO-donors) can alter the activation state of Ras, they can react with a multitude of biological targets and thus alter Ras signaling by indirect means. The use of RNAi or specific inhibitors to block the action of endogenous •NO producers may prove helpful for establishing a link between a specific mode of RNS production and a specific Ras signaling pathway. Inhibitors fully specific to various NOS isoforms would also prove helpful. In many cases, however, only one of the isoforms are expressed at any given time, so general NOS inhibitors such as L-NAME are still useful. Knowing the sources of •NO in diseases involving hyperactive Ras may lead to novel pharmacological targeting efforts. This is exemplified by active eNOS in oncogenic Ras-driven pancreatic cancer [100].
The use of the C118S redox inactive Ras mutant is also crucial. RasGEF-mediated activation of Ras will be unaffected with this variant, while activation by a radical-based RNS or ROS mechanism will be blocked. Once it is established that •NO (or more accurately RNS) activates a given Ras isoform and biological phenotype is observed, RNAi should be used to test whether Ras is necessary for the phenotype. Subsequent rescue with RNAi-resistant wild-type or C118S mutant Ras would demonstrate whether an indirect or direct RNS-based mechanism is responsible, as demonstrated in the eNOS pancreatic cancer study [100]. This is the most direct way to demonstrate a functional role for RNS (or ROS) with regards to Ras activity.
While this review is focused on RNS/ROS modification of Cys118 in Ras, an interesting publication also determined that three C-terminal cysteines in addition to Cys118 in Ras were S-nitrosated and S-glutathiolated in vitro and in mouse fibroblasts when exogenous nitrocysteine and other RNS/ROS donors were added [102]. These cysteines are likely to be less accessible to modification by •NO compared to Cys118 in cells, as they undergo lipid modification and membrane association. While the physiological significance of these observation are unclear, modification of the C-terminal cysteines could potentially prevent proper membrane localization, and thus could be a RNS/ROS-mediated mechanism of downregulation of Ras, in contrast to the upregulation of Ras by RNS/ROS-mediated activation observed through Cys118. Future studies should address the relevance of RNS/ROS reactivity of these cysteines in a physiologically-relevant model system to determine their role in normal or pathological Ras signaling.
Immuno-spin trapping may also offer a viable avenue for future cell-based or in vivo studies aimed at detecting radical-based Ras activation events. Under physiological conditions, low molecular oxygen levels, coupled with the reducing environment of cells, would appear to hinder implementation of IST in a cellular context. However, cancer cells typically exhibit a well-established altered redox environment and possess altered levels of antioxidant enzymes and small molecule antioxidants [103, 104]. Therefore, implementing IST and detecting radical-mediated Ras activation in cancer cells may be feasible. While immuno-spin trapping offers direct identification of radical-mediated Ras activation, it could easily be coupled with simpler, less specific methods. Increased use of biotin switch methodologies [92] to detect Ras S-nitrosation, for example, could be utilized to determine the biological context in which Ras S-nitrosation is correlated to RNS activation. Perhaps most important, we stress the future use of the C118S mutant of Ras in cells and in vivo to determine whether there is a direct mechanism for activation of Ras by RNS/ROS.
While this review is dedicated to highlighting •NO-mediated activation of Ras and its contribution in disease processes, ROS have also been shown to activate Ras in vitro [58, 105] and in cells [106]. Endogenous sources of ROS, such as NADPH oxidases, play crucial roles in a variety of signaling processes [107]. ROS, like superoxide, also react with RNS (i.e. •NO2) to form other powerful oxidants, such as peroxynitrite. We hypothesize pathological activation of Ras through ROS may play a role in diseases such as cancer, where the redox environment of the cell is altered. Ras transformation has previously been demonstrated to be dependent on ROS, at least partially, through activation of NADPH oxidase by Rac (a Ras isoform) [108, 109]. Importantly, in pancreatic cancer cells which contain oncogenic K-ras mutations, Rac activation of NADPH oxidase increases ROS levels, which is crucial for tumor growth in vivo [110]. We speculate that oncogenic Ras activates ROS production by NADPH oxidase, which further activates other Ras isoforms in a positive feedback loop similar to that seen with eNOS-mediated activation of wild-type Ras isoforms.
Recently, addition of exogenous H2O2 was shown to alter Rho GTPase activity and stress fiber formation in rat fibroblasts [111]. Addition of exogenous H2O2 to cells, however, presents the same issues as bolus addition of exogenous •NO, in that other oxidizing species are generated. H2O2 can react with transition metals in cells, through Fenton chemistry, to produce other highly reactive free radical oxidants, such as the hydroxyl radical. To overcome these obstacles, Aghajanian et al. determined that endogenous as well as exogenous ROS sources activate RhoA and stimulate stress fiber formation in rat fibroblasts [111]. By knocking down endogenous RhoA and rescuing with wildtype or redox-inactive mutants, the authors demonstrated that stress fiber formation can occur by direct ROS-mediated activation of RhoA. This was the first example demonstrating that ROS can directly regulate RhoA activity and function. As ROS/RNS regulation of Rho GTPases is poorly understood, additional studies with RhoA and other Rho GTPases will be necessary. Moreover, additional oxidative mechanisms have been proposed to regulate Ras activity. For example, Adachi et al. suggest that Ras can be activated by H2O2 dependent S-glutathiolation of Ras [112]. Moreover, this group conducted additional studies, and proposed that stable S-gluthathione modification of Ras via two-electron oxidation mechanisms can regulate Ras activity in vitro and in cells [112–114]. Our studies, however, indicate only one-electron/radical-mediated oxidative mechanisms can drive Ras guanine nucleotide exchange. It is important to state that both radical and non-radical pathways can result in glutathiolation of Ras Cys118. Delineating whether or not radical pathways are contributing to S-gluthiolation, particularly in cultured cells, is a difficult task. Current efforts in our lab are aimed at understanding the consequences of one- versus two-electron Ras S-glutathiolation and its potential links to Ras activation.
Given the complex chemistry associated with RNS and ROS, many cell-based studies fail to properly mimic the oxidative stress of disease states. For example, the oxygen concentration in cell culture is very different than that in vivo, which can strongly affect redox processes. Furthermore, the circulatory system and various organs (such as lung) can have different oxygen concentrations than other organs and thus very distinct redox environments. Cell-based studies are convenient, however, and do offer an easy route for genetic manipulation. Focusing on animal models in future studies may be a better means for examining the effects of oxidative or nitrosative stress. As an example, if the endogenous H-ras gene is mutated to H-ras C118S, what functions of the protein will be changed? Also, K-ras is the only Ras isoform whose knockout in mice is embryonically lethal, so will a C118S mutant allow the mice to survive? Tissue-specific knock-in of a C118S variant of H-ras or N-ras C118S in a genetically engineered K-ras driven mouse model of pancreatic cancer could also advance our understanding of redox processes during pancreatic tumorigenesis. Along these same lines, immuno-spin trapping efforts have recently detected various protein radical intermediates in mouse models [69]. Future experiments could utilize this technique to directly detect radical-mediated activation of Ras in animal models.
Conclusion
An abundance of evidence clearly indicates RNS contribute to the activation of Ras, both in vitro and in cells. In this review, we have highlighted the mechanistic aspects and biological implications of RNS-mediated Ras activation. Understanding the full spectrum of Ras activation mechanisms, including activation through RNS, is paramount for a full appreciation of Ras signaling. Compared to protein-mediated and oncogenic activation, regulation of Ras by RNS remains relatively understudied. Advancement of the field ultimately rests in our ability to perform well controlled, physiological relevant studies that directly tie endogenous free radical oxidants to altered Ras activity. Thus far, the reactivity of redox active Cys118 in H-, N-, and K-Ras is the sole link between all studies which correlate RNS to Ras regulation. As studies continue to imply radical-mediated pathways in Ras activation, we anticipate new detection methods will be needed. Targeting RNS-mediated Ras activation through redox active Cys118 may spring new, novel therapeutic options. We hope these options ultimately open new avenues to combat hyperactive Ras in cancer and many other diseases states.
Abbreviations
- DMPO
5,5-dimethyl-1-pyrroline N-oxide
- Em7
midpoint oxidation-reduction potential at pH 7
- GAPs
GTPase activating proteins
- GDP
guanosine-5′-diphosphate
- GEFs
guanine exchange factors
- GTP
guanosine-5′-triphosphate
- MS
mass spectrometry
- NHE
normal hydrogen electrode
- N2O3
dinitrogen trioxide
- •NO
nitric oxide
- •NO2
nitrogen dioxide
- ONOO−
peroxynitrite anion
- NOS
nitric oxide synthase
Footnotes
This project was supported by NIH grant 5R01 GM075431-04.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 2.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 3.Heo J. Redox regulation of Ran GTPase. Biochem Biophys Res Commun. 2008;376:568–572. doi: 10.1016/j.bbrc.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 4.Heo J, Campbell SL. Mechanism of p21Ras S-nitrosylation and kinetics of nitric oxide-mediated guanine nucleotide exchange. Biochemistry. 2004;43:2314–2322. doi: 10.1021/bi035275g. [DOI] [PubMed] [Google Scholar]
- 5.Heo J, Campbell SL. Mechanism of redox-mediated guanine nucleotide exchange on redox-active Rho GTPases. J Biol Chem. 2005;280:31003–31010. doi: 10.1074/jbc.M504768200. [DOI] [PubMed] [Google Scholar]
- 6.Hill BG, Dranka BP, Bailey SM, Lancaster JR, Jr, Darley-Usmar VM. What part of NO don’t you understand? Some answers to the cardinal questions in nitric oxide biology. J Biol Chem. 2010;285:19699–19704. doi: 10.1074/jbc.R110.101618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartberger MD, Liu W, Ford E, Miranda KM, Switzer C, Fukuto JM, Farmer PJ, Wink DA, Houk KN. The reduction potential of nitric oxide (NO) and its importance to NO biochemistry. Proc Natl Acad Sci U S A. 2002;99:10958–10963. doi: 10.1073/pnas.162095599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Augusto O, Bonini MG, Amanso AM, Linares E, Santos CC, De Menezes SL. Nitrogen dioxide and carbonate radical anion: two emerging radicals in biology. Free Radic Biol Med. 2002;32:841–859. doi: 10.1016/s0891-5849(02)00786-4. [DOI] [PubMed] [Google Scholar]
- 9.Goldstein S, Czapski G. Kinetics of nitric oxide autoxidation in aqueous solution in the absence and presence of various reductants. The nature of the oxidizing intermediates. J Amer Chem Soc. 1995;117:12078–12084. [Google Scholar]
- 10.Keshive M, Singh S, Wishnok JS, Tannenbaum SR, Deen WM. Kinetics of S-nitrosation of thiols in nitric oxide solutions. Chem Res Toxicol. 1996;9:988–993. doi: 10.1021/tx960036y. [DOI] [PubMed] [Google Scholar]
- 11.Stubbe J, van Der Donk WA. Protein radicals in enzyme catalysis. Chem Rev. 1998;98:2661–2662. doi: 10.1021/cr980059c. [DOI] [PubMed] [Google Scholar]
- 12.Moller MN, Li Q, Vitturi DA, Robinson JM, Lancaster JR, Jr, Denicola A. Membrane “lens” effect: focusing the formation of reactive nitrogen oxides from the *NO/O2 reaction. Chem Res Toxicol. 2007;20:709–714. doi: 10.1021/tx700010h. [DOI] [PubMed] [Google Scholar]
- 13.Pryor WA, Lightsey JW. Mechanisms of nitrogen dioxide reactions: initiation of lipid peroxidation and the production of nitrous Acid. Science. 1981;214:435–437. doi: 10.1126/science.214.4519.435. [DOI] [PubMed] [Google Scholar]
- 14.Thomas HV, Mueller PK, Lyman RL. Lipoperoxidation of lung lipids in rats exposed to nitrogen dioxide. Science. 1968;159:532–534. doi: 10.1126/science.159.3814.532. [DOI] [PubMed] [Google Scholar]
- 15.Misiaszek R, Crean C, Geacintov NE, Shafirovich V. Combination of nitrogen dioxide radicals with 8-oxo-7,8-dihydroguanine and guanine radicals in DNA: oxidation and nitration end-products. J Am Chem Soc. 2005;127:2191–2200. doi: 10.1021/ja044390r. [DOI] [PubMed] [Google Scholar]
- 16.Niles JC, Wishnok JS, Tannenbaum SR. Peroxynitrite-induced oxidation and nitration products of guanine and 8-oxoguanine: structures and mechanisms of product formation. Nitric Oxide. 2006;14:109–121. doi: 10.1016/j.niox.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Bian K, Gao Z, Weisbrodt N, Murad F. The nature of heme/iron-induced protein tyrosine nitration. Proc Natl Acad Sci U S A. 2003;100:5712–5717. doi: 10.1073/pnas.0931291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas DD, Espey MG, Vitek MP, Miranda KM, Wink DA. Protein nitration is mediated by heme and free metals through Fenton-type chemistry: an alternative to the NO/O2- reaction. Proc Natl Acad Sci U S A. 2002;99:12691–12696. doi: 10.1073/pnas.202312699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonini MG, Fernandes DC, Augusto O. Albumin oxidation to diverse radicals by the peroxidase activity of Cu,Zn-superoxide dismutase in the presence of bicarbonate or nitrite: diffusible radicals produce cysteinyl and solvent-exposed and -unexposed tyrosyl radicals. Biochemistry. 2004;43:344–351. doi: 10.1021/bi035606p. [DOI] [PubMed] [Google Scholar]
- 20.Pryor WA, Lightsey JW, Church DW. Reaction of nitrogen dioxide with alkenes and polyunsaturated fatty acids: Addition and hydrogen abstraction mechansims. J Am Chem Soc. 1982;104:6685–6692. [Google Scholar]
- 21.Jiang H, Kruger N, Lahiri DR, Wang D, Vatele JM, Balazy M. Nitrogen dioxide induces cis-trans-isomerization of arachidonic acid within cellular phospholipids. Detection of trans-arachidonic acids in vivo. J Biol Chem. 1999;274:16235–16241. doi: 10.1074/jbc.274.23.16235. [DOI] [PubMed] [Google Scholar]
- 22.Ford E, Hughes MN, Wardman P. Kinetics of the reactions of nitrogen dioxide with glutathione, cysteine, and uric acid at physiological pH. Free Radic Biol Med. 2002;32:1314–1323. doi: 10.1016/s0891-5849(02)00850-x. [DOI] [PubMed] [Google Scholar]
- 23.Herold S, Rock G. Mechanistic studies of S-nitrosothiol formation by NO*/O2 and by NO*/methemoglobin. Arch Biochem Biophys. 2005;436:386–396. doi: 10.1016/j.abb.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 24.Keszler A, Zhang Y, Hogg N. Reaction between nitric oxide, glutathione, and oxygen in the presence and absence of protein: How are S-nitrosothiols formed? Free Radic Biol Med. 2010;48:55–64. doi: 10.1016/j.freeradbiomed.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldstein S, Czapski G. Mechanism of the nitrosation of thiols and amines by oxygenated center dot NO solutions: The nature of the nitrosating intermediates. J Amer Chem Soc. 1996;118:3419–3425. [Google Scholar]
- 26.Broszkiewicz RK. The pulse radiolysis study of NaNO2 and NaNO3 solutions. Bull Acad Pol Sci Sedr Sci Chim. 1976;24:221–229. [Google Scholar]
- 27.Grätzel M, Henglein A, Lilie J, Beck G. Pulsradiolytische untersuchung einigerelementarprozesse der oxidation und reduction des nitritions. Ber Bunsenges Phys Chem. 1969;73:646–653. [Google Scholar]
- 28.Wardman P. Nitrogen dioxide in biology: Correlating chemical kinetics with biological effects. UK: John Wiley & Sons; 1998. [Google Scholar]
- 29.Schwartz SE, White WH. Kinetics of reactive dissolution of nitrogen oxides into aqueous solution. Adv Environ Sci Technol. 1983;12:1–116. [Google Scholar]
- 30.Blough NV, Zafiriou OC. Reaction of superoxide with nitric-oxide to form peroxonitrite in alkaline aqueous solution. Inorg Chem. 1985;24:3502–3504. [Google Scholar]
- 31.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent Hydroxyl Radical Production by Peroxynitrite - Implications for Endothelial Injury from Nitric-Oxide and Superoxide. P Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and the ugly. Am J Physiol-Cell Ph. 1996;40:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 33.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Radi R, Peluffo G, Alvarez MN, Naviliat M, Cayota A. Unraveling peroxynitrite formation in biological systems. Free Radical Bio Med. 2001;30:463–488. doi: 10.1016/s0891-5849(00)00373-7. [DOI] [PubMed] [Google Scholar]
- 35.Squadrito GL, Pryor WA. Oxidative chemistry of nitric oxide: The roles of superoxide, peroxynitrite, and carbon dioxide. Free Radical Bio Med. 1998;25:392–403. doi: 10.1016/s0891-5849(98)00095-1. [DOI] [PubMed] [Google Scholar]
- 36.Goldstein S, Czapski G. The reaction of •NO and O2•− and HO2•−: A pulseradiolysis study. Free Rad Biol Med. 1995;25:392–403. doi: 10.1016/0891-5849(95)00034-u. [DOI] [PubMed] [Google Scholar]
- 37.Kissner R, Nauser T, Bugnon P, Lye PG, Koppenol WH. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem Res Toxicol. 1997;10:1285–1292. doi: 10.1021/tx970160x. [DOI] [PubMed] [Google Scholar]
- 38.Klug D, Rabani J, Fridovich I. A direct demonstration of the catalytic action of superoxide dismutase through the use of pulse radiolysis. J Biol Chem. 1972;247:4839–4842. [PubMed] [Google Scholar]
- 39.Denicola A, Souza JM, Radi R. Diffusion of peroxynitrite across erythrocyte membranes. Proc Natl Acad Sci U S A. 1998;95:3566–3571. doi: 10.1073/pnas.95.7.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koppenol WH, Moreno JJ, Pryor WA, Ischiropoulos H, Beckman JS. Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide. Chem Res Toxicol. 1992;5:834–842. doi: 10.1021/tx00030a017. [DOI] [PubMed] [Google Scholar]
- 41.Richeson CE, Mulder P, Bowry VW, Ingold KU. The complex chemistry of peroxynitrite decomposition: New insights. J Am Chem Soc. 1998;120:7211–7219. [Google Scholar]
- 42.Denicola A, Freeman BA, Trujillo M, Radi R. Peroxynitrite reaction with carbon dioxide/bicarbonate: kinetics and influence on peroxynitrite-mediated oxidations. Arch Biochem Biophys. 1996;333:49–58. doi: 10.1006/abbi.1996.0363. [DOI] [PubMed] [Google Scholar]
- 43.Lymar SV, Hurst JK. Rapid reaction between peroxonitrite ion and carbon dioxide: Implications for biological activity. J Am Chem Soc. 1995;117:8867–8868. [Google Scholar]
- 44.Bonini MG, Radi R, Ferrer-Sueta G, Ferreira AM, Augusto O. Direct EPR detection of the carbonate radical anion produced from peroxynitrite and carbon dioxide. J Biol Chem. 1999;274:10802–10806. doi: 10.1074/jbc.274.16.10802. [DOI] [PubMed] [Google Scholar]
- 45.Bonini MG, Augusto O. Carbon dioxide stimulates the production of thiyl, sulfinyl, and disulfide radical anion from thiol oxidation by peroxynitrite. J Biol Chem. 2001;276:9749–9754. doi: 10.1074/jbc.M008456200. [DOI] [PubMed] [Google Scholar]
- 46.Chen S, Hoffman MZ. Rate constant for the reaction of the carbonate radical with compounds of biochemical interest in neutral aqueous solution. Radiat Res. 1973;56:40–47. [PubMed] [Google Scholar]
- 47.Lander HM, Ogiste JS, Pearce SF, Levi R, Novogrodsky A. Nitric oxide-stimulated guanine nucleotide exchange on p21ras. J Biol Chem. 1995;270:7017–7020. doi: 10.1074/jbc.270.13.7017. [DOI] [PubMed] [Google Scholar]
- 48.Lander HM, Hajjar DP, Hempstead BL, Mirza UA, Chait BT, Campbell S, Quilliam LA. A molecular redox switch on p21(ras). Structural basis for the nitric oxide-p21(ras) interaction. J Biol Chem. 1997;272:4323–4326. doi: 10.1074/jbc.272.7.4323. [DOI] [PubMed] [Google Scholar]
- 49.Williams JG, Pappu K, Campbell SL. Structural and biochemical studies of p21Ras S-nitrosylation and nitric oxide-mediated guanine nucleotide exchange. Proc Natl Acad Sci U S A. 2003;100:6376–6381. doi: 10.1073/pnas.1037299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bosworth CA, Toledo JC, Jr, Zmijewski JW, Li Q, Lancaster JR., Jr Dinitrosyliron complexes and the mechanism(s) of cellular protein nitrosothiol formation from nitric oxide. Proc Natl Acad Sci U S A. 2009;106:4671–4676. doi: 10.1073/pnas.0710416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 52.Luchsinger BP, Rich EN, Gow AJ, Williams EM, Stamler JS, Singel DJ. Routes to S-nitroso-hemoglobin formation with heme redox and preferential reactivity in the beta subunits. Proc Natl Acad Sci U S A. 2003;100:461–466. doi: 10.1073/pnas.0233287100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stubauer G, Giuffre A, Sarti P. Mechanism of S-nitrosothiol formation and degradation mediated by copper ions. J Biol Chem. 1999;274:28128–28133. doi: 10.1074/jbc.274.40.28128. [DOI] [PubMed] [Google Scholar]
- 54.Kharitonov VG, Sundquist AR, Sharma VS. Kinetics of nitrosation of thiols by nitric oxide in the presence of oxygen. J Biol Chem. 1995;270:28158–28164. doi: 10.1074/jbc.270.47.28158. [DOI] [PubMed] [Google Scholar]
- 55.Raines KW, Bonini MG, Campbell SL. Nitric oxide cell signaling: S-nitrosation of Ras superfamily GTPases. Cardiovasc Res. 2007;75:229–239. doi: 10.1016/j.cardiores.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 56.Jourd’heuil D, Jourd’heuil FL, Feelisch M. Oxidation and nitrosation of thiols at low micromolar exposure to nitric oxide. Evidence for a free radical mechanism. J Biol Chem. 2003;278:15720–15726. doi: 10.1074/jbc.M300203200. [DOI] [PubMed] [Google Scholar]
- 57.Madej E, Folkes LK, Wardman P, Czapski G, Goldstein S. Thiyl radicals react with nitric oxide to form S-nitrosothiols with rate constants near the diffusion-controlled limit. Free Radic Biol Med. 2008;44:2013–2018. doi: 10.1016/j.freeradbiomed.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 58.Heo J, Campbell SL. Ras regulation by reactive oxygen and nitrogen species. Biochemistry. 2006;45:2200–2210. doi: 10.1021/bi051872m. [DOI] [PubMed] [Google Scholar]
- 59.Heo J, Prutzman KC, Mocanu V, Campbell SL. Mechanism of free radical nitric oxide-mediated Ras guanine nucleotide dissociation. J Mol Biol. 2005;346:1423–1440. doi: 10.1016/j.jmb.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 60.Kraulis PJ, Domaille PJ, Campbell-Burk SL, Van Aken T, Laue ED. Solution structure and dynamics of ras p21.GDP determined by heteronuclear three- and four-dimensional NMR spectroscopy. Biochemistry. 1994;33:3515–3531. doi: 10.1021/bi00178a008. [DOI] [PubMed] [Google Scholar]
- 61.Aubert C, Vos MH, Mathis P, Eker AP, Brettel K. Intraprotein radical transfer during photoactivation of DNA photolyase. Nature. 2000;405:586–590. doi: 10.1038/35014644. [DOI] [PubMed] [Google Scholar]
- 62.Detweiler CD, Deterding LJ, Tomer KB, Chignell CF, Germolec D, Mason RP. Immunological identification of the heart myoglobin radical formed by hydrogen peroxide. Free Radic Biol Med. 2002;33:364–369. doi: 10.1016/s0891-5849(02)00895-x. [DOI] [PubMed] [Google Scholar]
- 63.Mason RP. Using anti-5,5-dimethyl-1-pyrroline N-oxide (anti-DMPO) to detect protein radicals in time and space with immuno-spin trapping. Free Radic Biol Med. 2004;36:1214–1223. doi: 10.1016/j.freeradbiomed.2004.02.077. [DOI] [PubMed] [Google Scholar]
- 64.Bhattacharjee S, Deterding LJ, Jiang J, Bonini MG, Tomer KB, Ramirez DC, Mason RP. Electron transfer between a tyrosyl radical and a cysteine residue in hemoproteins: spin trapping analysis. J Am Chem Soc. 2007;129:13493–13501. doi: 10.1021/ja073349w. [DOI] [PubMed] [Google Scholar]
- 65.Deterding LJ, Ramirez DC, Dubin JR, Mason RP, Tomer KB. Identification of free radicals on hemoglobin from its self-peroxidation using mass spectrometry and immuno-spin trapping: observation of a histidinyl radical. J Biol Chem. 2004;279:11600–11607. doi: 10.1074/jbc.M310704200. [DOI] [PubMed] [Google Scholar]
- 66.Detweiler CD, Lardinois OM, Deterding LJ, de Montellano PR, Tomer KB, Mason RP. Identification of the myoglobin tyrosyl radical by immuno-spin trapping and its dimerization. Free Radic Biol Med. 2005;38:969–976. doi: 10.1016/j.freeradbiomed.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 67.Lardinois OM, Detweiler CD, Tomer KB, Mason RP, Deterding LJ. Identifying the site of spin trapping in proteins by a combination of liquid chromatography, ELISA, and off-line tandem mass spectrometry. Free Radic Biol Med. 2008;44:893–906. doi: 10.1016/j.freeradbiomed.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lardinois OM, Tomer KB, Mason RP, Deterding LJ. Identification of protein radicals formed in the human neuroglobin-H2O2 reaction using immuno-spin trapping and mass spectrometry. Biochemistry. 2008;47:10440–10448. doi: 10.1021/bi800771k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chatterjee S, Ehrenshaft M, Bhattacharjee S, Deterding LJ, Bonini MG, Corbett J, Kadiiska MB, Tomer KB, Mason RP. Immuno-spin trapping of a post-translational carboxypeptidase B1 radical formed by a dual role of xanthine oxidase and endothelial nitric oxide synthase in acute septic mice. Free Radic Biol Med. 2009;46:454–461. doi: 10.1016/j.freeradbiomed.2008.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 71.Groves JT, Kuriyan J. Molecular mechanisms in signal transduction at the membrane. Nat Struct Mol Biol. 2010;17:659–665. doi: 10.1038/nsmb.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raaijmakers JH, Bos JL. Specificity in Ras and Rap signaling. J Biol Chem. 2009;284:10995–10999. doi: 10.1074/jbc.R800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mitin N, Rossman KL, Der CJ. Signaling interplay in Ras superfamily function. Curr Biol. 2005;15:R563–574. doi: 10.1016/j.cub.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 74.Adari H, Lowy DR, Willumsen BM, Der CJ, McCormick F. Guanosine triphosphatase activating protein (GAP) interacts with the p21 ras effector binding domain. Science. 1988;240:518–521. doi: 10.1126/science.2833817. [DOI] [PubMed] [Google Scholar]
- 75.Zhang K, DeClue JE, Vass WC, Papageorge AG, McCormick F, Lowy DR. Suppression of c-ras transformation by GTPase-activating protein. Nature. 1990;346:754–756. doi: 10.1038/346754a0. [DOI] [PubMed] [Google Scholar]
- 76.Lancaster JR., Jr Protein cysteine thiol nitrosation: maker or marker of reactive nitrogen species-induced nonerythroid cellular signaling? Nitric Oxide. 2008;19:68–72. doi: 10.1016/j.niox.2008.04.028. [DOI] [PubMed] [Google Scholar]
- 77.Mor A, Philips MR, Pillinger MH. The role of Ras signaling in lupus T lymphocytes: biology and pathogenesis. Clin Immunol. 2007;125:215–223. doi: 10.1016/j.clim.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 78.Mor A, Philips MR. Compartmentalized Ras/MAPK signaling. Annu Rev Immunol. 2006;24:771–800. doi: 10.1146/annurev.immunol.24.021605.090723. [DOI] [PubMed] [Google Scholar]
- 79.Perez de Castro I, Bivona TG, Philips MR, Pellicer A. Ras activation in Jurkat T cells following low-grade stimulation of the T-cell receptor is specific to N-Ras and occurs only on the Golgi apparatus. Mol Cell Biol. 2004;24:3485–3496. doi: 10.1128/MCB.24.8.3485-3496.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Caloca MJ, Zugaza JL, Bustelo XR. Exchange factors of the RasGRP family mediate Ras activation in the Golgi. J Biol Chem. 2003;278:33465–33473. doi: 10.1074/jbc.M302807200. [DOI] [PubMed] [Google Scholar]
- 81.Malinski T, Bailey F, Zhang ZG, Chopp M. Nitric-Oxide Measured by a Porphyrinic Microsensor in Rat-Brain after Transient Middle Cerebral-Artery Occlusion. J Cerebr Blood F Met. 1993;13:355–358. doi: 10.1038/jcbfm.1993.48. [DOI] [PubMed] [Google Scholar]
- 82.Lander HM, Milbank AJ, Tauras JM, Hajjar DP, Hempstead BL, Schwartz GD, Kraemer RT, Mirza UA, Chait BT, Burk SC, Quilliam LA. Redox regulation of cell signalling. Nature. 1996;381:380–381. doi: 10.1038/381380a0. [DOI] [PubMed] [Google Scholar]
- 83.Mott HR, Carpenter JW, Campbell SL. Structural and functional analysis of a mutant Ras protein that is insensitive to nitric oxide activation. Biochemistry. 1997;36:3640–3644. doi: 10.1021/bi962790o. [DOI] [PubMed] [Google Scholar]
- 84.Ibiza S, Perez-Rodriguez A, Ortega A, Martinez-Ruiz A, Barreiro O, Garcia-Dominguez CA, Victor VM, Esplugues JV, Rojas JM, Sanchez-Madrid F, Serrador JM. Endothelial nitric oxide synthase regulates N-Ras activation on the Golgi complex of antigen-stimulated T cells. Proc Natl Acad Sci U S A. 2008;105:10507–10512. doi: 10.1073/pnas.0711062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ponimaskin E, Voyno-Yasenetskaya T, Richter DW, Schachner M, Dityatev A. Morphogenic signaling in neurons via neurotransmitter receptors and small GTPases. Mol Neurobiol. 2007;35:278–287. doi: 10.1007/s12035-007-0023-0. [DOI] [PubMed] [Google Scholar]
- 86.Buday L, Downward J. Many faces of Ras activation. Biochim Biophys Acta. 2008;1786:178–187. doi: 10.1016/j.bbcan.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 87.Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- 88.Cullen PJ, Lockyer PJ. Integration of calcium and Ras signalling. Nat Rev Mol Cell Biol. 2002;3:339–348. doi: 10.1038/nrm808. [DOI] [PubMed] [Google Scholar]
- 89.MacDonald JF, Jackson MF, Beazely MA. Hippocampal long-term synaptic plasticity and signal amplification of NMDA receptors. Crit Rev Neurobiol. 2006;18:71–84. doi: 10.1615/critrevneurobiol.v18.i1-2.80. [DOI] [PubMed] [Google Scholar]
- 90.Hardingham GE. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem Soc Trans. 2009;37:1147–1160. doi: 10.1042/BST0371147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yun HY, Gonzalez-Zulueta M, Dawson VL, Dawson TM. Nitric oxide mediates N-methyl-D-aspartate receptor-induced activation of p21ras. Proc Natl Acad Sci U S A. 1998;95:5773–5778. doi: 10.1073/pnas.95.10.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 93.Vaudry D, Stork PJ, Lazarovici P, Eiden LE. Signaling pathways for PC12 cell differentiation: making the right connections. Science. 2002;296:1648–1649. doi: 10.1126/science.1071552. [DOI] [PubMed] [Google Scholar]
- 94.Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- 95.Teng KK, Esposito DK, Schwartz GD, Lander HM, Hempstead BL. Activation of c-Ha-Ras by nitric oxide modulates survival responsiveness in neuronal PC12 cells. J Biol Chem. 1999;274:37315–37320. doi: 10.1074/jbc.274.52.37315. [DOI] [PubMed] [Google Scholar]
- 96.Rameau GA, Tukey DS, Garcin-Hosfield ED, Titcombe RF, Misra C, Khatri L, Getzoff ED, Ziff EB. Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J Neurosci. 2007;27:3445–3455. doi: 10.1523/JNEUROSCI.4799-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lau KS, Haigis KM. Non-redundancy within the RAS oncogene family: insights into mutational disparities in cancer. Mol Cells. 2009;28:315–320. doi: 10.1007/s10059-009-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McGillicuddy LT, Fromm JA, Hollstein PE, Kubek S, Beroukhim R, De Raedt T, Johnson BW, Williams SM, Nghiemphu P, Liau LM, Cloughesy TF, Mischel PS, Parret A, Seiler J, Moldenhauer G, Scheffzek K, Stemmer-Rachamimov AO, Sawyers CL, Brennan C, Messiaen L, Mellinghoff IK, Cichowski K. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell. 2009;16:44–54. doi: 10.1016/j.ccr.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ohta M, Seto M, Ijichi H, Miyabayashi K, Kudo Y, Mohri D, Asaoka Y, Tada M, Tanaka Y, Ikenoue T, Kanai F, Kawabe T, Omata M. Decreased expression of the RAS-GTPase activating protein RASAL1 is associated with colorectal tumor progression. Gastroenterology. 2009;136:206–216. doi: 10.1053/j.gastro.2008.09.063. [DOI] [PubMed] [Google Scholar]
- 100.Lim KH, Ancrile BB, Kashatus DF, Counter CM. Tumour maintenance is mediated by eNOS. Nature. 2008;452:646–649. doi: 10.1038/nature06778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Park HS, Kim SR, Lee YC. Impact of oxidative stress on lung diseases. Respirology. 2009;14:27–38. doi: 10.1111/j.1440-1843.2008.01447.x. [DOI] [PubMed] [Google Scholar]
- 102.Mallis RJ, Buss JE, Thomas JA. Oxidative modification of H-ras: S-thiolation and S-nitrosylation of reactive cysteines. Biochem J. 2001;355:145–153. doi: 10.1042/0264-6021:3550145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McEligot AJ, Yang S, Meyskens FL., Jr Redox regulation by intrinsic species and extrinsic nutrients in normal and cancer cells. Annu Rev Nutr. 2005;25:261–295. doi: 10.1146/annurev.nutr.25.050304.092633. [DOI] [PubMed] [Google Scholar]
- 104.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 105.Heo J, Campbell SL. Superoxide anion radical modulates the activity of Ras and Ras-related GTPases by a radical-based mechanism similar to that of nitric oxide. J Biol Chem. 2005;280:12438–12445. doi: 10.1074/jbc.M414282200. [DOI] [PubMed] [Google Scholar]
- 106.Lander HM, Ogiste JS, Teng KK, Novogrodsky A. p21ras as a common signaling target of reactive free radicals and cellular redox stress. J Biol Chem. 1995;270:21195–21198. doi: 10.1074/jbc.270.36.21195. [DOI] [PubMed] [Google Scholar]
- 107.Ushio-Fukai M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2009;11:1289–1299. doi: 10.1089/ars.2008.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 109.Yang JQ, Buettner GR, Domann FE, Li Q, Engelhardt JF, Weydert CD, Oberley LW. v-Ha-ras mitogenic signaling through superoxide and derived reactive oxygen species. Mol Carcinog. 2002;33:206–218. doi: 10.1002/mc.10037. [DOI] [PubMed] [Google Scholar]
- 110.Du J, Liu J, Smith BJ, Tsao MS, Cullen JJ. Role of Rac1-dependent NADPH oxidase in the growth of pancreatic cancer. Cancer Gene Ther. 2010 doi: 10.1038/cgt.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Aghajanian A, Wittchen ES, Campbell SL, Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One. 2009;4:e8045. doi: 10.1371/journal.pone.0008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Adachi T, Pimentel DR, Heibeck T, Hou X, Lee YJ, Jiang B, Ido Y, Cohen RA. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279:29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- 113.Pimentel DR, Adachi T, Ido Y, Heibeck T, Jiang B, Lee Y, Melendez JA, Cohen RA, Colucci WS. Strain-stimulated hypertrophy in cardiac myocytes is mediated by reactive oxygen species-dependent Ras S-glutathiolation. J Mol Cell Cardiol. 2006;41:613–622. doi: 10.1016/j.yjmcc.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 114.Clavreul N, Bachschmid MM, Hou X, Shi C, Idrizovic A, Ido Y, Pimentel D, Cohen RA. S-glutathiolation of p21ras by peroxynitrite mediates endothelial insulin resistance caused by oxidized low-density lipoprotein. Arterioscler Thromb Vasc Biol. 2006;26:2454–2461. doi: 10.1161/01.ATV.0000242791.28953.4c. [DOI] [PubMed] [Google Scholar]