

Abstract
Mandelalides A-D are variously glycosylated, unusual polyketide macrolides isolated from a new species of Lissoclinum ascidian collected from South Africa, Algoa Bay near Port Elizabeth and the surrounding Nelson Mandela Metropole. Their planar structures were elucidated on sub-milligram samples by comprehensive analysis of 1D and 2D NMR data, supported by mass spectrometry. The assignment of relative configuration was accomplished by consideration of homonuclear and heteronuclear coupling constants in tandem with ROESY data. The absolute configuration was assigned for mandelalide A after chiral GC-MS analysis of the hydrolyzed monosaccharide (2-O-methyl-α-L-rhamnose) and consideration of ROESY correlations between the monosaccharide and aglycone in the intact natural product. The resultant absolute configuration of the mandelalide A macrolide was extrapolated to propose the absolute configurations of mandelalides B-D. Remarkably, mandelalide B contained the C-4′ epimeric 2-O-methyl-6-dehydro-α-L-talose. Mandelalides A and B showed potent cytotoxicity to human NCI-H460 lung cancer cells (IC50, 12 and 44 nM, respectively) and mouse Neuro-2A neuroblastoma cells (IC50, 29 and 84 nM, respectively).
Introduction
Over the last 50 years, ascidians have been shown to be a prolific source of natural products with promising biomedical potential.1 Indeed, ascidian-derived natural products have yielded promising drug leads, among which ecteinascidin 743 (Yondelis®) and dehydrodidemnin B (Aplidin®) are in clinical use for the treatment of specific cancers.2 The diverse chemotypes reported from Lissoclinum species collected globally have important biological properties that range from cancer cell toxicity to antifungal and antibacterial activities,1 and include peptides, alkaloids, chlorinated diterpenes, polyether amides, lactones, and macrolides such as haterumalide B3 and patellazoles B and C.4,5 It has been noted that many secondary metabolites isolated from Lissoclinum, as well as other ascidians species, are present in bacteria, sponges and mollusks.6-8 This diversity of ascidian metabolites can be explained by the fact that tunicates are often hosts to cyanobacterial and heterotrophic bacterial symbionts, as well as being fed upon by predatory mollusks.7-10 Therefore, identification of the biogenetic origin of ascidian natural products is very often challenging.11 This is highlighted by the recent isolation of didemnin B (an analog of Aplidin®) from the α-proteobacterium Tistrella mobilis.8,12 This compound was originally identified from the ascidian Aplidium albicans, and is biosynthetically similar to peptides from the marine cyanobacterium Lyngbya majuscula8 (now Moorea producta).
In our search for new potential anticancer compounds, we encountered the highly cytotoxic organic extract (IC50 = 0.7 μg/mL in NCI-H460 lung cancer cells) of a new Lissoclinum species from Algoa Bay, South Africa. Bioassay-guided fractionation of this extract, yielded a series of new macrolides, named mandelalides A-D (1-4), two of which were tested and show good cytotoxicity to mouse Neuro-2A neuroblastoma and human NCI-H460 lung cancer cells. While the relative configuration of the compounds could be assigned from analysis of ROESY data in tandem with homonuclear (3JHH) and heteronuclear (2,3JCH) coupling constants, the assignment of absolute configuration relied on hydrolysis of the glycosylated mandelalide A and chiral GC-MS analysis of the released monosaccharide. Remarkably, mandelalide A contains 2-O-methyl-α-L-rhamnose, while mandelalide B contains the C-4′ epimer 2-O-methyl-6-dehydro-α-L-talose.
Results and Discussion
A new Lissoclinum ascidian species was collected from White Sands Reef in Algoa Bay, South Africa. The organic extract (1.45 g) was subjected to bioassay-guided fractionation through consecutive LH-20 and RP-HPLC chromatography to yield mandelalides A-D (1 - 4), of which mandelalide D degraded to deacylmandelalide D (4b).
The HR-ESI-MS data for mandelalide A (1) gave a pseudomolecular ion [M+Na]+ at m/z 647.3394, which is consistent with a molecular formula of C33H52O11, and implies 8 degrees of unsaturation. The 13C and multiplicity-edited HSQC NMR spectra for compound 1 (Table 1) indicated the presence of an ester carbonyl (δC 167.4), fourteen sp3 methines, twelve of which were oxygen-bearing (δC 94.2–68.1, 37.3 and 34.2), six olefinic methines (δC 147.1, 141.5, 131.3, 126.9, 123.9 and 123.1), eight methylenes (δC 66.1, 43.1, 39.7, 38.8, 37.6, 36.8, 34.1 and 31.1) and four methyl groups (δC 59.1, 18.3, 17.7 and 14.5). Interpretation of COSY and TOCSY correlations delineated two spin systems, one comprising 25 carbons from C-2 to C-26 (Figure 2a, fragment A), and the other of 6 carbons from C-1′ to C-6′ (Figure 1a, fragment B). The acquisition of a semi-phase sensitive HMBC13 optimized for a 4 Hz coupling constant (Figure S6), revealed a correlation between δH 5.23 (H-23) and δC 167.4 (C-1) that indicated cyclization of fragment A into a 23-carbon macrolactone. The C-1 carbonyl also showed HMBC correlations from the olefinic H-2 and H-3 multiplets, for which a 3JHH of 15.5 Hz indicated an E double bond geometry. An HMBC correlation between H-9 (δ 3.32) and C-5 (δ 73.9) delineated a tetrahydropyran ring, while a correlation between H-17 (δ 3.98) and C-20 (δ 83.2) was consistent with a tetrahydrofuran ring, both within fragment A (Table 1, Figure 1a). The four contiguous (TOCSY-coupled) olefinic methines CH-12 to CH-15 (δH 5.45, 6.28, 6.05 and 5.28 ppm) were consistent with a conjugated diene, situated β to the tetrahydrofuran and γ to the tetrahydropyran on the basis of HMBC correlations from H-15 to C-17 and H-12 to C-10, respectively. From homonuclear coupling constant values (3JHH = 14.8 and 10.8 Hz, respectively) it was evident that the geometry of H-12 and H-13 is trans, while H-14 and H-15 are cis. Knowing that 3JHH values are not sufficiently characteristic to assign the orientation between H-13 and H-14, ROESY data were examined.14 Signals confirming NOE enhancements between H-12 and H-10a, as well as between H-13 and H-16b, suggested an anti orientation for H-13 and H-14 (Figure 1b). In the case of fragment B, the downfield shift of C-1′ (δC 94.2), the presence of four midfield oxymethine 13C resonances, an upfield methyl doublet (CH3-6′, δC 17.7, δH 1.27) and an HMBC correlation between H-1′ (δ 5.02) and C-5′ (δ 68.1) suggested that fragment B is a 6-dehydro monosaccharide. An HMBC correlation between H-2′ (δ 3.40) and deshielded methyl C-7′ (δ 59.1) indicated methylation of the C-2′ hydroxyl group. An HMBC correlation from H-7 (δ 3.82) to C-1′ placed the monosaccharide (fragment B) at C-7 of fragment A.
Table 1.
NMR data for Mandelalide A (1) in CDCl3
| No. | δC (mult) | δH (J in Hz) | COSY | HMBC | TOCSY | ROESY |
|---|---|---|---|---|---|---|
| 1 | 167.4 (s) | |||||
| 2 | 123.1 (d) | 6.01 (dd, 15.5, 1.2) | 3, 4a | 1, 4 | 3, 4a, 4b, 5, 6ax, 6eq | 3b, 4a, 4b, 5 |
| 3 | 147.1 (d) | 6.97 (ddd, 15.2, 10.4, 4.6) | 2, 4a, 4b | 1, 2, 4, 5 | 2, 4a, 4b, 5, 6ax, 6eq | 3b, 4ab, 4bb, 5 |
| 4a | 38.8 (t) | 2.36 (m) | 2, 3 | 2, 3, 5, 6 | 2, 3, 5, 6ax, 6eq, 7 | 2, 3b, 5b, 6axb, 6eq |
| 4b | 2.39 (ddd, 14.1, 10.6, 10.6) | 3, 5 | 2, 3, 5, 6 | 2, 3, 5, 6ax, 6eq, 7 | 2, 3b, 5b, 6axb, 6eq | |
| 5 | 73.9 (d) | 3.36 (dddd, 11.4, 11.4, 2.3, 2.3) | 4a, 4b, 6ax, 6eq | 3, 4 | 2, 3, 4a, 4b, 6ax, 6eq, 7, 8ax, 8eq | 2, 3, 4a, 4b, 6eq, 7 |
| 6ax | 37.6 (t) | 1.20 (m) | 5, 6eq, 7 | 4, 5, 8 | 3, 4a, 4b, 5, 6eq, 7 | 4bb, 7 |
| 6eq | 2.02 (dddd, 12.6, 4.4, 2.3, 1.6)a | 5, 6ax, 7, 8eq | 7, 8 | 4a, 4b, 5, 6ax, 7, 8ax, | 6axb, 4a, 4b, 5, 7, 1′ | |
| 7 | 73.1 (d) | 3.82 (dddd, 11.1, 10.5, 4.4, 4.4)a | 6ax, 6eq, 8ax, 8eq | 8, 9, 1′ | 4a, 4b, 5, 6ax, 6eq, 8ax, 8eq, 9 | 5, 6axb, 6eq, 8axb, 8eq, 9, 1′ |
| 8ax | 39.7 (t) | 1.22 (m) | 7, 8eq, 9 | 6, 7, 9, 10 | 6eq, 7, 8eq, 9, 10b | 6eqb, 7, 8eqb |
| 8eq | 1.87 (m) | 6eq, 7, 8ax, 9 | 6, 7 | 6eq, 7, 8ax, 9, 10a, 10b | 7, 8axb, 9, 10b, 5′ | |
| 9 | 72.5 (d) | 3.32 (dddd, 11.2, 11.2. 2.2, 2.2)a | 8ax, 8eq, 10a, 10b | 5, 7 | 6eq, 7, 8ax, 8eq, 10a, 10b, 11, 12, 13, 25 | 7, 8eq, 10a, 10bb, 25 |
| 10a | 43.1 (t) | 1.21 (ddd, 15.2, 9.6, 2.2)a | 9, 10b, 11 | 11, 12, 25 | 8eq, 9, 10b, 12, 13, 14, 25 | 8eq, 9, 10bb, 11b, 12, 25 |
| 10b | 1.51 (ddd, 15.2, 11.2, 3.7)a | 9, 10a, 11 | 8, 9, 11, 12, 25 | 8ax, 8eq, 9, 10a, 11, 12, 13, 25 | 8eq, 9b, 10ab, 11, 25 | |
| 11 | 34.2 (d) | 2.37 (dqd, 9.6, 6.5, 3.7)a | 10a, 10b, 12, 25 | 10, 12, 25 | 8ax, 8eq, 9, 10a, 10b, 11, 12, 13, 14, 15 | 10ab, 10b, 12b, 13, 25b |
| 12 | 141.5 (d) | 5.45 (dd, 14.8, 9.7) | 11, 13 | 10, 11, 14, 25 | 9, 10a, 10b, 11, 13, 14, 15, 16a, 16b, 25 | 11b, 10a, 13b, 14b, 25 |
| 13 | 123.9 (d) | 6.28 (dd, 14.8, 11.0) | 12, 14 | 10, 11, 14, 15 | 10a, 10b, 11,12, 14, 15, 16a, 16b, 17, 25 | 11, 12b, 14b, 15b, 16b, 21 |
| 14 | 131.3 (d) | 6.05 (dd, 10.9, 10.9) | 13, 15 | 12, 13,16, 17 | 10a, 11, 12, 13, 15, 16a, 16b, 17, 25 | 12b, 13b, 15b, 16a |
| 15 | 126.9 (d) | 5.28 (ddd, 10.8, 10.8, 5.6) | 14, 16a, 16b | 13, 16, 17 | 12, 13, 14, 16a, 16b, 17, 18, 26 | 13b, 14b, 16a, 16bb, 17b |
| 16a | 31.1 (t) | 1.88 (m) | 14, 15, 16b, 17 | 14, 15, 17 | 12, 13, 14, 15, 16b, 17, 18, 19a, 26 | 14, 15, 16b, 17b, 26 |
| 16b | 2.28 (ddd, 13.1, 11.4, 11.4) | 15, 16a, 17 | 14, 15, 17, 18 | 12, 13, 14, 15, 16a, 17, 18, 19a, 19b, 26 | 13,15b,16a,17b,19a,21,26 | |
| 17 | 81.0 (d) | 3.98 (ddd, 11.1, 8.1, 1.8) | 16a, 16b, 18 | 15, 19, 20 | 14, 15,16a, 16b, 18, 19a, 19b, 20, 21, 26 | 15, 16ab, 16bb,18, 26 |
| 18 | 37.3 (d) | 2.52 (dddq, 12.0, 7.0, 7.0, 7.0)a | 17, 19a, 19b, 26 | 16, 17, 19, 26 | 15, 16a, 16b,17, 19a, 19b, 20,21, 22a, 26 | 17, 19ab, 19b, 20, 26b |
| 19a | 36.8 (t) | 1.17 (ddd, 11.9, 11.9, 10.3) | 18, 19b, 20 | 18, 20, 21, 26 | 17, 18, 19a, 20, 21, 22a, 22b, 26 | 16b, 18b, 21, 22b, 26 |
| 19b | 2.01 (ddd, 12.2, 7.0, 5.6)a | 18, 19a, 20 | 17, 18 | 16b, 17, 18, 19a, 20, 21, 22a, 26 | 18, 19a, 20, 22a, 22b, 26 | |
| 20 | 83.2 (d) | 3.63 (m) | 19a, 19b, 21 | 21, 22 | 17, 18, 19a, 19b, 21, 22a, 22b, 26 | 18, 19b, 21b, 22a, 22b |
| 21 | 73.0 (d) | 3.42 (ddd, 11.1, 8.8, 1.8) | 20, 22a, 22b | 20, 22, 23 | 17, 18, 19a, 19b, 20, 22a, 22b, 23, 24a, 24b, 26 | 13, 16b, 19a, 22ab, 22b, 23 |
| 22a | 34.1 (t) | 1.46 (ddd, 14.1, 11.1, 1.9) | 21, 22b, 23 | 20, 21 | 18, 19a, 19b, 20, 21, 22b, 23, 24a, 24b, 26 | 19b, 21b, 22bb, 23, 24a, 24b |
| 22b | 1.76 (ddd, 13.9, 11.7, 1.8) | 21, 22a, 23 | 23, 24 | 19a, 20, 21, 22a, 23,24a, 24b | 19a, 19b, 21, 22ab,23b, 24a, 24b | |
| 23 | 72.3 (d) | 5.23 (dddd, 11.7, 4.9, 2.9, 1.9) | 22a, 22b, 24a, 24b | 1 | 21, 22a, 22b, 24a, 24b | 21, 22a, 22bb, 24a, 24b |
| 24a | 66.1 (t) | 3.61 (m) | 23, 24b | 22, 23 | 23, 24b | 22a, 22b, 23, 24b |
| 24b | 3.81 (dd, 12.2, 2.9)a | 23, 24a | 22, 23 | 22b, 23, 24a | 22a, 22b, 23, 24a | |
| 25 | 18.3 (q) | 0.85 (d, 6.6) | 11 | 10, 11, 12 | 9, 10a, 10b, 11, 12, 13, 14 | 9, 10a, 10b, 11b, 12 |
| 26 | 14.5 (q) | 1.03 (d, 6.9) | 18 | 17, 18, 19 | 15, 16a, 16b, 17, 18, 19a, 19b, 20 21 | 16a, 16b, 18b, 19a, 19b |
| 1′ | 94.2 (d) | 5.02 (d, 1.1) | 2′ | 7, 2′, 3′, 5′ | 2′, 6′ | 6eq, 7, 8eq, 2′, 7′ |
| 2′ | 80.8 (d) | 3.40 (dd, 3.8, 1.4) | 1′, 3′ | 3′, 4′, 7′ | 1′, 3′, 4′, 5′, 6′, 8′, 9′ | 1′ |
| 3′ | 71.7 (d) | 3.68 (m) | 2′, 4′, 8′ | 2′, 4′, 5′, 6′, 8′, 9′ | 2′, 4′b | |
| 4′ | 74.3 (d) | 3.34 (dd, 9.4, 9.4) | 3′, 5′ | 3′, 5′, 6′ | 3′, 5′, 6′, 8′, 9′ | 3′b, 5′b, 6′ |
| 5′ | 68.1 (d) | 3.62 (m) | 4′, 6′ | 3′, 4′, 6′ | 4′, 6′, 8′, 9′ | 8eq, 4′b, 6′ |
| 6′ | 17.7 (q) | 1.27 (d, 6.3) | 4′, 5′ | 1′, 3′, 4′, 5′, 8′ | 4′, 5′b | |
| 7′ | 59.1 (q) | 3.45 (s) | 2′ | 1′ | ||
| OH-3′ | 2.24 (s) | 2, 4a, 4b | 2′, 3′, 4′, 5′, OH-4′ | |||
| OH-4′ | 1.54 (s) | 2, 3 | 3′, 4′, 5′, OH-3′ |
HMBC correlations are presented from proton to indicated carbon;
3JHH values obtained from DQFCOSY and/or NMRSim;
COSY artifacts observed in ROESY spectrum.
Figure 2.
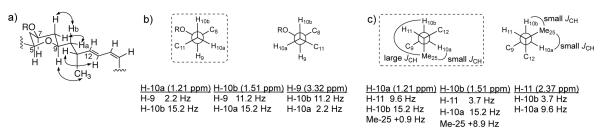
a) Key ROESY correlations indicated on the C-5/C-15 fragment of 1; b) Two possible rotamers around the C-9/C-10 bond; c) Most feasible rotamers around the C-10/C-11 bond of 1, with anticipated large/small heteronuclear coupling constant indicated on each. For b) and c) the JHH and JHC values (CDCl3) are listed below the Newman projections and the rotamer of best fit is indicated by a dashed outline.
Figure 1.
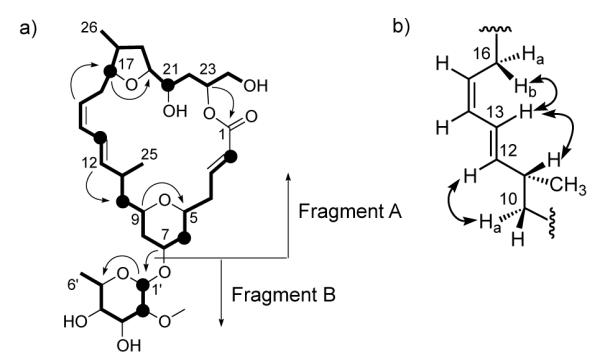
a) Planar structure of 1 showing TOCSY correlations from H-2, H-6, H-8, H-10b, H-13, H-17, H-21, H-1′, H-2′ (black circles) indicated as bolded lines and key HMBC correlations represented by single-headed arrows. b) The assignment of geometry around the C-13/C-14 bond in 1 using key ROESY correlations indicated by double-headed arrows.
The second compound characterized, mandelalide B (2), was assigned a molecular formula of C37H58O13, based on HR-ESI-MS data for [M+Na]+ m/z 733.3730, which is consistent with 9 degrees of unsaturation. Inspection of the 1H and 13C NMR spectra for mandelalide B (2) suggested that it was structurally related to mandelalide A (1). However, the 1H NMR spectrum for 2 lacked the coupled olefinic signals at δ 6.01 (H-2) and 6.97 (H-3) and the relatively deshielded diastereotopic H2-24 signals at δ 3.81 and 3.61. Instead an additional oxymethine double doublet at δ 5.48 was evident, as well as a methyl triplet (δH 0.95) indicative of an aliphatic chain. Correspondingly, comparison of the 13C NMR spectra for 1 and 2 revealed the absence of olefinic 13C resonances at δ 123.1 (C-2) and 147.1 (C-3) from the spectrum for 2, and the presence of two additional midfield 13C resonances (δ 79.3 and 69.5), as well as a second carbonyl resonance (δC 173.6). These data accounted for 8 of the 9 degrees of unsaturation, implying the presence of an additional cycle in 2. The analysis of HSQC and HMBC data for 2 permitted assignment of an oxygenated quaternary C-2 (δ 79.3) and an oxymethine CH-3 (δ 69.5, δ 5.48). While the same C-1/C-23 macrolactone linkage was apparent in both compounds, two- and three-bond HMBC correlations from H2-24 (δ 1.93, 2.39) to C-2 and C-3, respectively, indicated a C-2/C-24 bond in 2. These data described a γ-butyrolactone ring around the ester linkage of the mandelalide B macrocycle. Placement of the second carbonyl carbon (δC 173.6) in a butyrate substituent at C-3 was facilitated by HMBC correlations to δC 173.6 from δH 5.48 (H-3), as well as 2.35 (H2-2″) and 1.67 (H2-3″). Further analysis of NMR data confirmed the presence of a 2-O-methyl-6-dehydro sugar in 2. However, differences in both 1H and 13C shifts for CH-3′ to CH-6′ compared to compound 1 suggested a different relative configuration for the monosaccharide in 2 (Table 2).
Table 2.
1H and 13C NMR data for mandelalides B-D (2-4) in CDCl3
| Mandelalide B (2) | Mandelalide C (3) | Mandelalide D (4) | ||||
|---|---|---|---|---|---|---|
| No. | δC (mult) | δH (J in Hz) | δC (mult) | δH (J in Hz) | δC (mult) | δH (J in Hz) |
| 1 | 175.6 (s) | - | 174.7 (s) | - | 172.9 (s) | - |
| 2 | 79.3 (s) | - | 82.0 (s) | - | 81.3 (s) | - |
| 3 | 69.5 (d) | 5.48 (dd, 6.0, 1.7) | 68.3 (d) | 5.51 (dd, 6.6, 0.9) | 68.6 (d) | 5.56 (dd, 6.6, 0.7) |
| 4a | 36.3 (t) | 1.59 (m) | 36.4 (t) | 1.65 (m) | 36.2 (t) | 1.68 (m) |
| 4b | 2.14 (ddd, 15.0, 11.7, 1.5) | 2.14 (ddd, 15.6, 11.7, 1.0) | 2.18 (ddd, 15.4, 11.5, 0.9) | |||
| 5 | 72.7 (d) | 3.30 (dddd, 11.4, 11.4, 1.5, 1.5) |
72.3 (d) | 3.25 (dddd, 11.4, 11.4, 1.8, 1.8) | 72.2 (d) | 3.21 (dddd, 11.2, 11.2, 1.3, 1.3) |
| 6ax | 37.4 (t) | 1.10 (ddd, 11.7, 11.7, 11.7) | 41.2 (t) | 1.11 (ddd, 12.5, 11.4, 11.4) | 41.2 (t) | 1.12 (ddd, 11.3, 11.3, 11.3) |
| 6eq | 1.89 (m) | 1.86 (m) | 1.85 (dddd, 12.2, 4.6, 1.5, 1.5) | |||
| 7 | 73.3 (d) | 3.75 (m) | 68.3 (d) | 3.76 (dddd, 10.9, 10.9, 4.6, 4.1) | 68.4 (d) | 3.75 (m) |
| 8ax | 39.6 (t) | 1.23 (m) | 41.8 (t) | 1.13 (ddd, 12.5, 11.2, 11.2) | 41.8 (t) | 1.12 (ddd, 11.3, 11.3, 11.3) |
| 8eq | 1.81 (dddd, 12.4, 4.8, 1.8, 1.8) |
1.83 (m) | 1.83 (dddd, 12.2, 4.6, 1.5, 1.5) | |||
| 9 | 72.4 (d) | 3.39 (dddd, 11.0, 11.0, 2.2, 2.2) |
72.3 (d) | 3.37 (dddd, 10.9, 10.9, 2.1, 2.1) | 72.2 (d) | 3.36 (dddd, 11.0, 11.0, 2.2, 2.2) |
| 10a | 42.2 (t) | 1.18 (ddd, 14.1, 12.3, 2.8) | 42.1 (t) | 1.19 (ddd, 14.2, 12.1, 2.8) | 42.1 (t) | 1.19 (ddd, 13.9, 11.5, 2.8) |
| 10b | 1.57 (m) | 1.57 (m) | 1.57 (ddd, 14.0, 11.5, 4.0) | |||
| 11 | 33.9 (d) | 2.50 (m) | 34.0 (t) | 2.49 (m) | 34.2 (d) | 2.48 (m) |
| 12 | 142.1 (d) | 5.50 (dd, 15.1, 9.8) | 142.2 (d) | 5.50 (dd, 14.6, 9.6) | 142.3 (d) | 5.50 (dd, 14.8, 9.7) |
| 13 | 123.3 (d) | 6.40 (dd, 14.7, 11.3) | 123.2 (d) | 6.39 (dd, 14.6, 11.4) | 123.1 (d) | 6.38 (dd, 15.0, 11.3) |
| 14 | 131.0 (d) | 6.10 (dd, 11.1, 11.1) | 131.1 (d) | 6.10 (dd, 11.0, 11.0) | 131.1 (d) | 6.10 (dd, 11.0, 11.0) |
| 15 | 127.1 (d) | 5.28 (ddd, 11.1, 11.1, 5.2) | 127.1 (d) | 5.28 (ddd, 11.0, 11.0, 5.4) | 127.1 (d) | 5.28 (ddd, 11.3, 11.3, 5.4) |
| 16a | 30.7 (t) | 1.90 (m) | 30.7 (t) | 1.90 (ddd, 13.6, 5.2, 1.1) | 30.6 (t) | 1.90 (ddd, 13.6, 5.3, 1.0) |
| 16b | 2.31 (ddd, 13.6, 11.6, 11.5,) | 2.30 (ddd, 13.8, 11.9, 11.9) | 2.29 (ddd, 13.1, 11.3, 11.3) | |||
| 17 | 81.5 (d) | 3.95 (ddd, 11.6, 7.46, 1.6) | 81.6 (d) | 3.95 (ddd, 11.5, 7.5, 1.1) | 81.6 (d) | 3.94 (ddd, 12.0, 7.5, 1.1) |
| 18 | 38.3 (d) | 2.52 (m) | 38.3 (d) | 2.53 (m) | 38.3 (d) | 2.54 (dddq, 12.3, 7.5, 7.02, 6.8) |
| 19a | 35.6 (t) | 1.33 (ddd, 12.2, 12.2, 8.2) | 35.7 (t) | 1.33 (ddd, 12.2, 12.2, 9.1) | 35.7 (t) | 1.34 (ddd, 12.3, 12.3, 9.2) |
| 19b | 2.05 (ddd, 12.8, 6.9, 6.9) | 2.10 (ddd, 12.5, 7.0, 6.8) | 2.08 (ddd, 12.2, 7.02, 7.02) | |||
| 20 | 82.1 (t) | 3.77 (m) | 82.4 (d) | 3.82 (ddd, 9.2, 9.2, 7.0) | 82.2 (d) | 3.79 (ddd, 9.3, 9.3, 7.0) |
| 21 | 74.5 (d) | 3.76 (m) | 74.4 (d) | 3.73 (ddd, 10.8, 9.5, 1.2) | 74.3 (d) | 3.73 (ddd, 10.0, 10.0, 1.6) |
| 22a | 37.9 (t) | 1.51 (m) | 32.1 (t) | 1.59 (m) | 32.5 (t) | 1.47 (ddd, 14.2, 10.5, 1.0) |
| 22b | 1.64 (m) | 1.82 (ddd, 14.4, 11.0, 1.0) | 1.65 (m) | |||
| 23 | 74.9 (d) | 4.84 (dddd, 10.4, 10.4, 4.9, 1.0) |
78.9 (d) | 5.01 (ddd, 11.2, 2.0, 2.0) | 76.7 (d) | 5.13 (ddd, 10.8, 3.3, 0.8) |
| 24a | 39.7 (t) | 1.93 (dd, 12.7, 10.7) | 72.2 (d) | 3.98 (d, 2.8) | 74.0 (d) | 5.17 (d, 3.3) |
| 24b | 2.39 (dd, 12.8, 4.9) | - | - | - | - | |
| 25 | 18.5 (q) | 1.07 (d, 6.6) | 18.4 (q) | 1.06 (d, 6.2) | 18.3 (q) | 1.07 (d, 6.6) |
| 26 | 14.2 (q) | 1.03 (d, 6.9) | 14.2 (q) | 1.03 (d, 6.9) | 14.3 (q) | 1.04 (d, 6.8) |
| OH-21 | 2.70 (br s) | OH | 2.80 (br s) | OH | 2.71 (br s) | |
| OH-2 | 2.87 (s) | - | - | OH | 2.78 (br s) | |
| 1′ | 94.8 (d) | 5.04 (s) | 173.4 (s) | - | 173.6 (s) | - |
| 2′a | 80.9 (d) | 3.33 (ddd, 3.0, 1.5. 1.3) | 36.3 (t) | 2.34 (td, 7.5, 1.2) | 36.3 (t) | 2.40 (dt, 15.5, 7.3) |
| 2′b | - | - | - | - | 2.43 (dt, 15.5, 7.3) | |
| 3′ | 66.6 (d) | 3.74 (m) | 18.7 (t) | 1.65 (m) | 18.6 (t) | 1.70 (m) |
| 4′ | 73.3 (d) | 3.49 (ddt, 11.8, 3.3, 1.2, 1.2) | 13.9 (q) | 0.94 (t, 7.5) | 13.9 (q) | 0.96 (t, 7.4) |
| 5′ | 67.4 (d) | 3.85(br q, 6.6) | - | - | - | - |
| 6′ | 16.6 (q) | 1.23 (d, 6.5) | - | - | - | - |
| 7′ | 59.6 (q) | 3.45 (s) | - | - | - | - |
| OH-3′ | 2.86 (d, 11.6) | - | - | - | - | |
| OH-4′ | 2.74 (d, 11.6) | - | - | - | - | |
| 1″ | 173.6 (s) | - | - | - | 173.1 (s) | - |
| 2″a | 36.3 (t) | 2.35 (t, 7.5) | - | - | 36.0 (t) | 2.31 (dt, 12.8, 7.5) |
| 2″b | - | - | - | - | 2.33 (dt, 12.8, 7.5) | |
| 3″ | 18.8 (t) | 1.67 (tq, 7.4, 7.4) | - | - | 18.6 (t) | 1.63 (m) |
| 4″ | 14.0 (q) | 0.95 (t, 7.4) | - | - | 13.8 (q) | 0.93 (t, 7.3) |
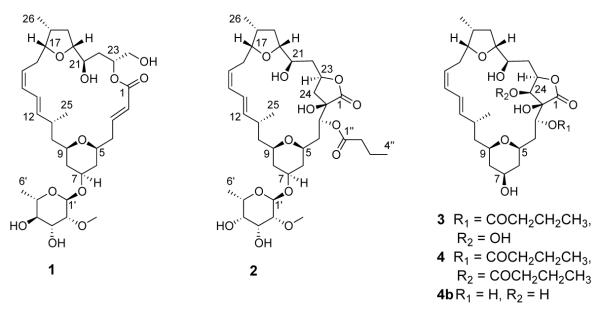
The HR-ESI-MS data ([M+Na]+ m/z 589.2970) for mandelalide C (3) supported a molecular formula of C30H46O10, for 8 degrees of unsaturation. The 1H NMR spectrum for 3 was reminiscent of that for 2. However, a careful comparison of the two spectra revealed the absence of midfield glycosidic signals in the 1H spectrum for 3. Similarly, inspection of the 13C NMR spectrum for 3 showed a lack of midfield resonances for a methoxy methyl (δ 59.6, C-7′ in 2), anomeric carbon (δ 94.8, C-1′ in 2), and other glycosidic carbons. Together, the MS and 1D NMR data for compound 3 indicated that the structure of 3 is related to the aglycone of mandelalide B (2, Table 2). Indeed, the only difference between the 13C NMR signals for the aglycones of compounds 2 and 3 was a relatively downfield methine resonance at δ 72.2 in the spectrum for 3, compared to the methylene signal at δ 37.9 (C-24) in the spectrum for 2. This methine was HSQC-correlated to a 1H doublet at δH 3.98, and could be assigned to an oxymethine CH-24 on the basis of HMBC correlations from the latter 1H doublet to C-1, C-2, C-3 and C-23. Thus, considering its MS and NMR data, mandelalide C (3) could be assigned as the C-24 hydroxylated aglycone of mandelalide B (2). Unfortunately, an attempt to crystallize mandelalide C (0.5 mg) was unsuccessful and degradation of the compound over several months prevented the acquisition of additional spectroscopic or biological data.
The molecular formula of mandelalide D (4) was assigned by HR-ESI-MS ([M+Na]+ m/z 659.3326) as C34H52O11, implying 9 degrees of unsaturation. Comparison of the similar 1H NMR spectra (Table 2) for mandelalides C (3) and D (4) indicated a second aglycone structure. A key difference between the 1H spectra for the two compounds was the further deshielded H-24 doublet (δH 5.17) for 4 relative to that for 3 (δH 3.98). In the HMBC spectrum for 4, the H-24 doublet was correlated to a carbonyl 13C resonance (δC 173.1, C-1″), which also showed correlations from H-2a”, H-2b” and H-3″, consistent with an additional butyrate substituent at C-24. Thus, mandelalide D (4) is 24-butyro mandelalide C (3). Over a period of 12 months, we observed changes in the 1H NMR spectrum of 4 that were consistent with the loss of both butyrate moieties, resulting in the new compound deacylmandelalide D (4b, m/z 519.3 [M+Na]+, Table S6).
Consideration of the differences in the planar macrocyclic structures, and available amounts, of mandelalides A-D (1-4) lead to the conclusion that the assignment of relative configuration should be performed on mandelalide A and one of mandelalides B-D. Similarities in coupling constant (3JHH) values for mandelalides B-D suggested retention of the relative configuration of the macrocycle between these structures (Table 2). Therefore besides mandelalide A, mandelalide B (2) was selected for further analysis because it also possesses a monosaccharide moiety, although it lacks the C-24 chiral center present in 3 and 4. For both mandelalides A (1) and B (2), the relative configuration of the macrocycle could be assigned fully using a combination of ROESY and J-based configuration analysis of data acquired in CDCl3 and pyridine-d5, which were consistent between the two solvents.
Considering the tetrahydropyran ring of 1, ROESY correlations between H-5, H-7 and H-9 supported their axial orientation, consistent with a chair conformation and equatorial C-7 glycosidic bond (Figure 2a). The relative configuration for fragment C-9 to C-11 (-CHCH2CH-) was assigned on the basis of coupling constants obtained from the 1H spectrum and a DQFCOSY. In CDCl3, 3JHH values of 2.2 and 11.2 Hz for H-9/H-10a and H-9/H-10b, respectively, localized H-10a gauche and H-10b anti to H-9 in two possible rotamers (Figure 2b), the correct one of which should be distinguishable from ROESY data. 1H signal overlap in the CDCl3 ROESY spectrum for mandelalide A (1) obscured ROE interactions in the region of interest. However, acquisition of NMR data for 1 in py-d5 provided sufficient resolution (0.16 ppm, Figure S20) between H-8ax and H-10a to reveal ROESY correlations between H-8ax and H-10b, and H-8eq and H-10a. This lead to the conclusion that rotamer A is correct, orienting H-10a to the outside of the macrocycle, (Figure 2). The assignment of relative configuration around the C-10/C-11 bond required direct measurement of 3JHH values from DQFCOSY, again due to significant overlap. In CDCl3, 3JHH values of 9.6 Hz for H-10a/H-11 and 3.7 Hz for H-10b/H-11 indicated anti and gauche orientations, respectively (Figure 2c). Finally, ROESY correlations between H-9 and H3-25, and H-11 and H-13 were consistent with the relative configuration shown in Figure 2a. This assignment was supported by HETLOC data in CDCl3 (Figures S11), which provided very weak, but measurable 3JHC between H-10a and C-25 (0.9 Hz), and between H-10b and C-25 (8.9 Hz).
For the tetrahydrofuran ring of 1, H-17 and H-20 could be in a cis or trans configuration. There are two close conformational minima (envelope and half-chair) for THF rings.15 However, in the envelope conformation CH3-26, H-18, H-19a and H-19b in 1 would be eclipsed, while these same moieties would be gauche in the half-chair conformation and there would be no eclipsed interaction along the C-18/19 bond.15 Therefore we decided to analyze ROESY correlations in the context of a half-chair THF ring to assign the relationship of H-17 and H-20. The relatively large 3JHH couplings between vicinal proton pairs H-16b/H-17 (11.4 Hz) and H-20/H-21 (8.8 Hz) supported their anti orientation in each case (Figure 3a). Moreover, ROESY correlations between H-16b and H-19a, H-16b and H-21, and H-19a and H-21 were consistent with a cis localization of H-17 and H-20 (Figure 3b), despite that no direct ROESY correlation was observed between H-17 and H-20. Instead, the proposed conformation, consistent with ROESY correlations between H3-26/H2-16 and H-16b/H-21, brings ROE-correlated H-18 and H-20 into close proximity (Figure 3b). For the C-21 chiral center, the large values of 3JHH and 2JHC (8.8 and -4.0 Hz, respectively), in parallel with the lack of NOE contact between H-20 and H-21 (resolved in py-d5), are consistent with their anti orientation (Figure 3a), as stated above. Analysis of 3JHH between H-21/H-22a (11.1 Hz), and H-21/H-22b (1.8 Hz), placed H-22a anti and H-22b gauche with respect to H-21 (Figure 3c). ROESY correlations between H-19a and H-22b, H-19b and H-22a, and H-19b and H-22b suggested that H2-22 are directed away from the center of macrocycle, with the C-21 hydroxyl pointing towards the center. Analogous reasoning was used to resolve the relative configuration at C-23. The respective large and small 3JHH values between H-22b/H-23 (11.7 Hz) and H-22a/H-23 (1.9 Hz) suggested anti and gauche orientations, respectively (Figure 3d).
Figure 3.
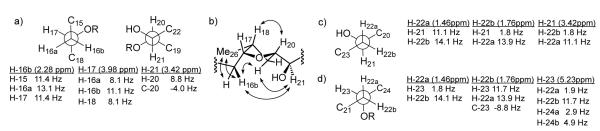
a) Newman projections along the C-16/C-17 and C-20/C-21 bonds in 1; b) The most feasible cis orientation of the THF ring with double-headed arrows indicating ROESY correlations; c) Newman projection along the C-21/C-22 bond with applicable JHH and JHC values (CDCl3) indicated on the right. d) Newman projection along the C-22/C-23 bond and relevant coupling constants.
Configurational assignment of the mandelalide A monosaccharide (C-1′ to C-6′) relied on 3JHH values, obtained from the 1H NMR and assigned by DQFCOSY, given the overlap for H-3′ to H-5′ signals in both CDCl3 and py-d5 ROESY spectra. Although the small value of 3JH-1′/H-2′ = 1.1 Hz was inconclusive, H-2′ was assigned as equatorial based on 3JH-2′/H-3′ = 3.8 Hz. The H-4′ multiplet indicated couplings of 9.4 Hz with both H-3′ and H-5′, indicative of axial hydrogens, although the direct analysis of H-3′ and H-5′ multiplets was impeded by their overlap with other 1H shifts. Overall, these data for CH-2′ to CH-5′ were consistent with the relative configuration of 2-O-methylrhamnose. To confirm this and to assign the monosaccharide as α- or β-2-O- methylrhamnose, we decided to analyze the magnetization transfer pattern of TOCSY signals originating from the anomeric center (C-1′), following the method proposed by Gheysen et al.16 This “TOCSY matching” approach takes advantage of the fact that the size of the 3JHH scalar couplings affect the rate of magnetization transfer through a 1H spin system during the TOCSY spin-lock period. The acquisition of a TOCSY with a spin-lock of 100 ms (Figure S9) affords a differential presence or absence of the CH3-6′ signal in α- and β-rhamnose, respectively. In the TOCSY acquired for mandelalide A (1), the intensity of peaks fit to previously reported data for α-rhamnose (Figure S9).16 Additional confirmation that the monosaccharide attached to 1 is 2-O- methyl-α-rhamnose was provided by the measurement of 1JHC = 167.2 Hz for the anomeric CH- 1′ from a HETLOC experiment (Figure S12). This value is in agreement with that reported previously for 2-O-methyl-α-rhamnose, although is smaller than in the case of nonmethylated common sugars (~170 Hz).17,18
As in the case of 1, the coupling constants values and ROESY correlations measured from both CDCl3 and py-d5 data for mandelalide B (2) were comparable, indicating similar conformations in both solvents. The chair conformation of the THP ring in 2 (C-5 to C-9) was confirmed by the presence of diaxial ROESY correlations between H-5, H-7 and H-9 (Figure 4a). For the C-9/C-11 segment, coupling constants of 2.2 and 11 Hz, between H-9/H-10a, and H-9/H- 10b confirmed gauche and anti relationships, respectively. Similarly for H-10a/H-11 and H- 10b/H-11, large (12.3 Hz) and small (4.1 Hz in CDCl3, 3.8 Hz in py-d5) coupling constants assigned from DQFCOSY supported anti and gauche relationships, respectively, overall leading to the same relative configuration in this region as for 1. For the C-3/C-5 fragment of 2, 3JHH values of 1.5 and 11.7 Hz between H-4a/H-5 and H-4b/H-5 supported gauche and anti orientations, respectively. A small coupling of 1.6 Hz between H-3 and H-4b indicated their gauche relationship (Figure 4b), and was in agreement with large H-4a/C-3 (-5.6 Hz) and small H-4b/C-3 (0 Hz) heteronuclear couplings. However, medium couplings for H-3/H-4a (6.0 Hz) and H-5/C-3 (4.3 Hz) suggested that the staggered orientation along C-3/4 may be distorted or that more than one conformation is present along this bond. Clear ROESY correlations between H-3 and each of H-4b, H-5, H-9, H-11, H-23 and H-24b, but not H-4a (apparent COSY artifact only), support a distorted rotamer in which 138° and -102° angles are present between H-3/H-4a and H-3/H-4b, as calculated using MestReJ for 3JHH 6.0 and 1.6 Hz respectively (Figure 4b).
Figure 4.
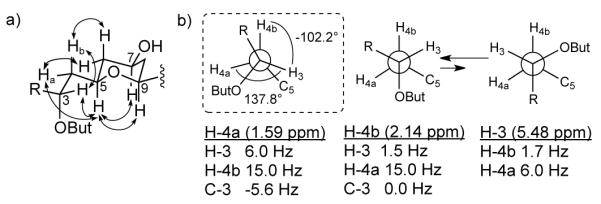
a) The most probable rotamer along the C-3/C-4 bond highlighted by the dashed outline and alternative equilibrium of two rotamers explaining medium size coupling constants. b) Key ROESY correlations for the C-3/C-5 fragment are indicated by double-headed arrows on the partial structure. But = butyrate.
The presence of a ROESY correlation between H-18 and H-20 on the THF ring in mandelalide B (2) placed CH3-26 quasi-equatorial, consistent with ROESY correlations from the latter methyl to H2-16, (as for 1, Figure 3). ROESY correlations (Table S2) and comparative analysis of coupling constant values (Table 2) around the C-16/C-21 fragment confirmed that a cis conformation of the THF ring is conserved in mandelalides A (1) and B (2).
Given extensively overlapped CDCl3 1H chemical shifts for the C-20/C-23 fragment in 2, analysis of 1H NMR and DQFCOSY in py-d5 was used to assign the relative configuration of this region. The 3JHH values for 2 were similar to those for mandelalide A (1), despite the presence of the γ-lactone ring in 2. However, in contrast to 1, in which the more shielded H-22a (δ 1.46) is anti to H-21 and gauche to H-23 (Figure 3), for 2 the more shielded H-22a (δ 1.65) is gauche to H-21 (3JHH <1 Hz) and H-22b (δ 1.77) is anti to H-21 (3JHH = 9.4 Hz), while H-20 and H-21 remain antiperiplanar (3JHH = 9.4 Hz) (Figure 5a). Coupling constant values obtained directly from the oxymethine H-23 multiplet, and assigned by DQFCOSY, indicate anti relationships with H-22a (JHH = 11.2 Hz), and a gauche relationship with H-22b (JHH = 1.0 Hz). The orientation of the ester bond in the butyrolactone ring could be assigned knowing the localization of H-23 anti to H-22a. The ester oxygen must then be positioned gauche or anti to H-22b. The ROEs observed between H-24a, H-24b and H-22b in py-d5, in parallel with the absence of ROEs between H2-24 and H-21 suggested that the ester bond is anti to H-22b (Figure 5a). Finally, knowing the relative configuration of the C-3/C-11 and C-20/C-23 fragments, the chirality of the quaternary C-2 in the butyrolactone ring was considered. ROESY correlations between H-24b and both H-3 and H-23 were consistent with an α-oriented H-24b, while H-24a, showing ROESY correlations to H2-22, is oriented above the butyrolactone ring in a β configuration. Similarly, the C-2 hydroxyl could be oriented to the outside of the macrocycle due to the presence of weak ROESY signals between this OH-2 and H-4b and H-24a, suggesting a 2R configuration (Figure 5b).
Figure 5.
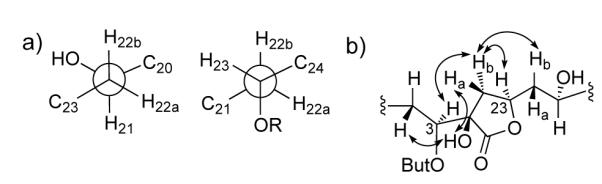
a) Assigned rotamers along the C-21/C-22 and C-22/C-23 bonds in 2. b) ROESY correlations around the γ-butyrolactone of 2. But = butyrate.
The relative configuration of the 2′-O-methyl, 6′-deoxy monosaccharide moiety (C1′-C7′) in 2 was established analogously to that in 1. A very weak ROESY correlation between H-1′ and H-5′ together with 1JHC = 167 Hz for anomeric CH-1′ suggested the presence of an α-sugar. A ROESY correlation between H-3′ and H-5′ localized these protons axial, while the small 3JHH between H-2′ and H-3′ (2.8 Hz) localized H-2′ equatorial (Figure 6). The presence of a W coupling (4JHH = 0.98 Hz) between H-2′ and H-4′, as well as 3JHH’ = 3.2 Hz between H-3′ and H- 4′ localized H-4′ equatorial, overall leading to the conclusion that mandelalide B (2) contains 2- O-methyl-6-dehydro-α-L-talose, the C-4′ epimer of the monosaccharide in mandelalide A.
Figure 6.
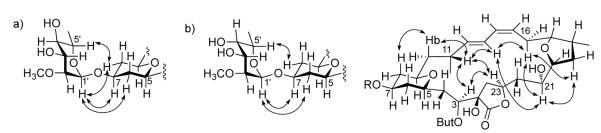
a) Key ROE correlations (CDCl3) between the monosaccharide and the macrolide THP moiety in mandelalide A; b) Key ROE correlations between the monosaccharide and the macrolide, as well as through ring correlations for mandelalide B.
Given the assignment of relative configuration of the macrocycle and monosaccharide units of mandelalides A (1) and B (2), it remained to establish the absolute configuration of these glycosidic macrolides. Despite our desire to conserve the limited sample quantities available for further biological investigations, a chemical degradation/derivatization approach to the absolute configuration would be most rigorous and was facilitated by the contiguous nature of the stereogenic fragments throughout the molecular framework. Importantly, inspection of the CDCl3 and py-d5 ROESY spectra for 1 and 2 revealed clear correlations between protons of the monosaccharide and THP of the macrocycle (Figure 6a, 6b). Given the well-defined solution structures of monosaccharides and glycosidic bond conformations,19 assignment of the rhamnose absolute configuration would permit subsequent relay of configurational assignments around the macrocycle, as shown for mandelalide B (Figure 6b). Therefore, a portion (100 μg) of mandelalide A (1) was sacrificed for hydrolysis of the glycosidic bond and chiral GC-MS comparison of the liberated derivatized monosaccharide with permethylated and silylated D- and L-rhamnose standards. During co-injection of each standard with the derivatized natural product hydrolysate, the natural product sugar coeluted with the L (97.4 min) and not the D (96.4 min) synthetic standard, confirming that the sugar substituent of 1 is 2-O-methyl-α-L-rhamnose. The absolute configuration of this sugar could then be extrapolated to the aglycone of 1 based on the ROESY correlations between H-1′ and H-7, H-1′ and H-6eq, and H-5′ and H-8eq to provide an assignment of the mandelalide A (1) aglycone as 2E, 5S, 7S, 9R, 11R, 12E, 14Z, 17R, 18R, 20R, 21R, 23R. Given retention of the macrocycle configuration (C-5 to C-21) between 1 and 2, the absolute configuration of 2 could be assigned similarly by considering ROESY correlations between the monosaccharide and THP moiety, and key correlations across this more rigid macrocycle (Figure 6b). Thus the aglycone of mandelalide B (2) could be assigned as 2R, 3R, 5R, 7S, 9R, 11R, 12E, 14Z, 17R, 18R, 20R, 21R, 23R.
Computational modeling of mandelalide B was used to examine possible mandelalide B conformations (Figure 7). An attempt to investigate the fit of calculated conformations to experimental data was based on measurements of ROE distances across the macrocycle in py- d5.20 However, as could be expected for a flexible macrolide, for the ten lowest energy conformations of mandelalide B (2), the differences between the calculated and experimental average distances between all analyzed protons except H-21/H-11 and H-21/H-13 were significantly different (0.2 Å or more). Nevertheless, the lowest energy computational model assists in visualizing the through ring ROE contacts that facilitated assignment of the absolute configuration (Figure 7).
Figure 7.
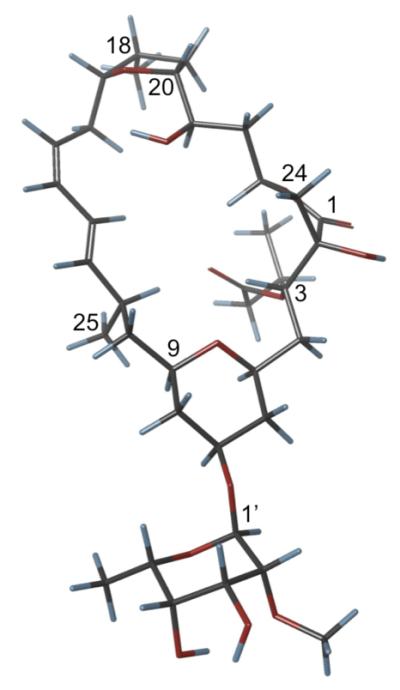
Computational model of the lowest energy conformer for mandelalide B (2).
The 3JHH values acquired in CDCl3 for mandelalides B-D (2-4) were all similar, suggesting that the relative configuration of the macrocycle incorporating a γ-butyrolactone is retained. The configuration at the remaining C-24 stereocenter present in mandelalides C (3) and D (4) was assigned from ROESY data. In each case, the H-24 doublet (δH 3.98 and 5.17 in 3 and 4, respectively) is correlated to the H-3 double doublet, localizing the C-24 hydroxyl/butyrate moiety antiperiplanar to H-3 and H-23, in an S configuration.
Cytotoxicity of the organic extract from Lissoclinum sp. was examined against mouse Neuro-2A neuroblastoma, and human MDA-MB-231 breast and NCI-H460 lung cancer cell lines following 48 h exposure. In all cases low μg/mL IC50 values were obtained (Table 3). The Neuro-2A cell line was chosen to perform activity-guided fractionation leading to the purification of mandelalides A–D (1-4). The pure compounds mandelalides A and B yielded nanomolar IC50 values against Neuro-2A (44.0 and 83.8, respectively) and NCI-H460 (12.0 and 29.4 respectively) cell lines (Table 3, Figure S57). The potent cytotoxicities of these mandelalides are somewhat surprising given the reported minimal cytotoxicity of the related glycosylated polyketides madeirolides A and B.21 The latter metabolites caused less than 50% inhibition of AsPC-1 and PANC-1 pancreatic cancer cells at 10 μg/mL, but showed potent fungicidal activity against Candida albicans. Madeirolides A and B share essentially the same western hemisphere of the mandelalides (C-5 to C-22), including a THP (alternatively substituted), diene, THF and neighboring hydroxymethine. The two series of compounds vary only in the closure of the macrocycle: the position of the lactone and additional cycle. Given that the madeirolides were isolated from a deep-water lithistid Leiodermatium sponge, their structural relatedness is consistent with a microbial biogenetic origin. Side-by-side evaluation of the biological properties of all four mandelalides and the two madeirolides would provide insight into the structure-activity relationships that result in potent cytotoxicity and fungicidal activity, respectively. However, the inaccessible supply of both source organisms means that further investigation of these metabolites will likely await their total syntheses.
Table 3.
Cytotoxicity of Lissoclinum sp. organic extract and mandelalides A (1) and B (2) to mouse Neuro-2A neuroblastoma and human NCI-H460 lung and MDA-MB-231 breast cancer cells.
| aIC50 (at 48 h) in Cancer Cell Lines: | |||
|---|---|---|---|
| NCI-H460 | Neuro-2A | MDA-MB-231 | |
| organic extract | 0.7 μg/mL | 5.6 μg/mL | 22.1μg/mL |
| mandelalide A (1) | 12 nM | 44 nM | - |
| mandelalide B (2) | 29 nM | 84 nM | - |
Cell viability was assessed by MTT assay and IC50 values were derived using nonlinear regression analysis.
Experimental Section
General Experimental Methods
were as described previously.22 Additionally NMR data in CDCl3 were acquired at 700 (1H) and 175 (13C) MHz on a 5 mm inverse cryogenic probe. The NMR data in py-d5, TOCSY (mixing time 100 ms), HETLOCs and gated-decoupled 13C NMR experiments were obtained at 700 (1H) and 175 (13C) MHz on a 5 mm 13C cryogenic probe. The spectra were referenced to internal residual solvent signals in ppm (1H NMR: CDCl3, 7.24, py-d5, 8.74, CD3OD, 3.31; 13C NMR: CDCl3, 77.23, py-d5, 150.35, CD3OD, 49.15). High-resolution MS data were acquired using an orthogonal acceleration time of flight (oa-TOF) mass analyzer and electrospray ionization (ESI).
Extraction and Isolation
The ascidian, Lissoclinum sp. (Ascidiacea, Aplousobranchia, Didemnidae) was collected by hand using SCUBA at a depth of 18 m (July 20, 2004) from White Sands Reef in Algoa Bay, Eastern Cape Province, South Africa (33:59.916S, 25:42.573W). The type specimen for this new ascidian species is housed at the South African Institute for Aquatic Biodiversity (SAIAB), Grahamstown, South Africa.
South Africa
The freeze-dried organism (15.1 g) was extracted with 2:1 CH2Cl2-MeOH yielding 1.45 g of organic extract. This organic extract was fractionated on Sephadex LH-20 (CH2Cl2- MeOH, 1:3) to give eight fractions, of which fractions six and seven were subjected to reversed phase C18 solid phase extraction (RP-SPE) using a stepped gradient of 50-100% MeOH in H2O. The 75% MeOH-H2O and 100% MeOH fractions were further separated by RP-HPLC (C18 column, 250 mm x 10 mm, 7:3 MeOH-0.1% FA in H2O) to yield mandelalides A (1, 0.8 mg), B (2, 0.5 mg), C (3, 0.8 mg) and D (4, 0.6 mg).
Mandelalide A (1)
Amorphous solid; [α]23D –9 (c = 0.25, MeOH); UV (MeOH) λmax (log ε) 279 (2.7), 217 (4.1); LR-ESI-MS m/z [M+Na]+ 647.4; HR-ESI-MS m/z [M+Na]+ 647.3394, (calcd for C33H52O11Na, 647.3407); 1H and 13C NMR, COSY, HMBC, TOCSY, ROESY (Tables 1, S1).
Mandelalide B (2)
Amorphous solid; [α]24D –13 (c = 0.5, MeOH); UV (MeOH) λmax (log ε) 279 (3.0), 229 (4.1); LR-ESI-MS m/z [M+Na]+ 733.5; HR-ESI-MS m/z [M+Na]+ 733.3773, (calcd for C37H58O13Na, 733.3775); 1H and 13C NMR, COSY, HMBC, ROESY (Tables 2, S2, S3).
Mandelalide C (3)
Amorphous solid; UV (MeOH) λmax (log ε) 280 (2.7), 229 (4.0); LR-ESI- MS m/z [M+Na]+ 589.5; HR-ESI-MS m/z [M+Na]+ 589.2970, (calcd for C30H46O10Na, 589.2989); 1H and 13C NMR, COSY, HMBC, TOCSY, ROESY (Tables 2, S4).
Mandelalide D (4)
Amorphous solid; [α]25D –50 (c = 0.2, MeOH); UV (MeOH) λmax (log ε) 280 (2.7), 229 (4.0); LR-ESI-MS m/z [M+Na]+ 659.5; HR-ESI-MS m/z [M+Na]+ 659.3424, (calcd for C34H52O11Na, 659.3407); 1H and 13C NMR, COSY, HMBC, TOCSY, ROESY (Tables 2, S5).
Deacylmandelalide D (4b)
Amorphous solid; [α]27D –10 (c = 0.2, MeOH); UV (MeOH) λmax (log ε) 280 (2.8), 227 (3.8); LR-ESI-MS m/z [M+Na]+ 519.3; HR-ESI-MS m/z [M+H]+ 497.2752, (calcd for C26H41O9, 497.2751); 1H and 13C NMR, COSY, HMBC (Table S6).
Measurement of 1,2JHC coupling constants
The sensitivity and gradient-enhanced HETLOC (ω1-hetero half-filtered TOCSY) experiment was employed to measure JHC coupling constants, with DIPSI-2 spin-lock set to 60 ms.23 Spectral widths of 6229 Hz and 5597 Hz, with a data matrix of 4K (F2) x 128 (F1), and 146 or 96 scans were employed in py-d5 and CDCl3, respectively for mandelalide A. In the case of mandelalide B, a spectral width of 5597 Hz with a data matrix of 4K (F2) x 128 (F1) and 136 scans were implemented in CDCl3. The 1D spectra that were obtained after extraction of F2/ F1 slices were subjected to inverse Fourier transform. The resulting FIDs were multiplied by the exponential window function prior to linear prediction processing.24
TOCSY data acquisition
To determine the magnetization transfer pattern in 2-O-methyl-L-rhamnose a 2D TOCSY was acquired with a DIPSI-2 spin-lock sequence in CDCl3. The spectra were recorded with a spin-lock mixing time of 100 ms, 5597 Hz spectral width at 2K (F2) x 512 (F1) data matrix for 40 scans. Data were processed with a sine-bell squared function with 1.5 Hz (F2) and 0.3 Hz (F1) line broadening before measurement of the absolute volumes of peaks. The following designation was applied to depict intensity of peaks; grey ovals indicate intensity or signal greater than 1.5% of intensity measured for the signal of the anomeric proton, white ovals indicate intensity of at least 0.5% but no more than 1.5% intensity measured for the anomeric 1H signal.16
Calculation of Dihedral Bond Angles in MestReJ
25 The calculation of angles between H-3/H- 4a and H-3/H-4b was performed using the Altona equation with substituents at C-3 defined as CH2OR and OCOR, and substituents at C-4 as H and CH2OR. Resulting 3JHH values for H-3/H- 4a (138.7°) and H-3/H-4b (-102.2°) were 6.0 and 1.6 Hz, respectively.
Computational Modeling of Mandelalide B (2)
Computational modeling was performed using the 2009 version of a contemporary software package. Minimization using the Amber* force field with PRCG algorithm, in pyridine (ε = 12.9, 10000 steps, maximum derivative less than 0.05 kcal/mol) and constrained torsion angles H-3/H-4a (138°), H-3/H-4b (-102.2°), H-4b/H-5, H-9/H-10b, H-20/H-21, H-21/H-22b, H-22a/H-23 and distance H-4b/H-24b = 5 Å was first performed. All torsions were restrained based on the 3JHH coupling constants values. The minimized structure was subjected to conformational search using Amber* force field via the low mode sampling method with an energy cut off of 21 kJ/mol and 1000 steps (100 steps per rotatable bond).26,27 Torsion restraints H-3/H-4a (138°), H-3/H-4b (-102.2°), H-4b/H-5, H-9/H- 10b, H-20/H-21, H-21/H-22b, H-22a/H-23 were applied to obtain ground state conformations. Optimization of the ten lowest energy conformations using DFT with the B3LYP functional and 6-31G** basis set in the gas phase resulted in the structure presented in Figure 7.
Methanolysis of Mandelalide A (1)
Compound 1 (0.1 mg) was treated with 1 N methanolic HCl (1.0 mL) and heated for 24 h at 70 °C with stirring. The mixture was concentrated in vacuo, redissolved in 0.75 mL of 1-(TMS)-imidazole/py (1:4) and the reaction was continued for 40 min at 70 °C with stirring.28 The solution was concentrated, the final residue partitioned between CH2Cl2 and H2O (1:1), and the organic fraction used for GC-MS analysis.
Preparation of 1,2-di-O-methyl-3,4-di-O-TMS-α-L-rhamnose and 1,2-di-O-methyl-3,4-di-O- TMS-α-D-rhamnose standards
L-rhamnose (50 mg) was dissolved in 1N methanolic HCl, heated for 24 h at 70 °C with stirring, and concentrated in vacuo. The residue was redissolved in dioxane (0.6 mL) and 2 M TMS-diazomethane in diethyl ether (1.5 mL) was added, with H3BO3 as catalyst. The solution was maintained at room temperature with stirring for 5 h and then evaporated to dryness in vacuo.29 Column chromatography on Si gel GF (15 μm) with 5% MeOH in CH2Cl2 as mobile phase afforded a 1:3 mixture of 1,2-di-O-methyl-α-L-rhamnose and 1,3-di-O-methyl-α-L-rhamnose iñ65% yield from this rate limiting reaction: equivalent to ~16% yield of the desired 1,2-di-O-methyl-α-L-rhamnose to be carried forward for silylation. This 1:3 mixture of methylation products was treated with 1-(TMS)-imidazole/py (1:4) for 40 min at 60 °C and concentrated in vacuo.28 Finally, the residue was dissolved in CH2Cl2, loaded on a Si gel column, washed with 100% hexanes, and eluted with 3% EtOAc in hexanes to obtain pure 1,2-di-O-methyl-3,4-di-O-TMS-α-L-rhamnose in a final yield of 5.4%. The same procedure was applied to produce 1,2-di-O-methyl-3,4-di-O-TMS -α-D-rhamnose in 6.4% yield. 1H NMR (700 MHz, CDCl3): δ 0.12 (s, 9H), 0.15 (s, 9 H), 3.31 (s, 3H), 3.32 (dd, J = 1.7, 3.2 Hz, 1H), 3.48 (s, 3H), 3.52 (m, 1H), 3.53 (m, 1H), 3.78 (dd, J = 3.6, 8.4 Hz, 1H), 4.61 (d, J = 1.3, 1H); 13C NMR (175 MHz, CDCl3): δ 0.7, 1.2, 18.5, 54.8, 60.0, 69.1, 73.1, 74.3, 81.6, 98.8, ppm; CI-LR- MS: m/z 305 calculated for oxonium ion C13H29O4Si2.
Absolute configuration of 2-O-methyl-α-rhamnose from mandelalide A (1)
Analyses of the synthetic standards and permethylated and silylated mandelalide A hydrolysate were performed by GC-MS using a 30 m × 0.25 mm i.d. Cyclosil-B column, and electron impact (EI) ionization. The injector and detector were operated at 250 °C, while the temperature gradient was set to 75 – 175 °C at 0.5 °/min. The retention times for 1,2-di-O-methyl-3,4-di-O-TMS-α-D-rhamnose and 1,2-di-O-methyl-3,4-di-O-TMS-α-L-rhamnose injected separately were 96.5 and 98.3 min, respectively. The retention time for the monosaccharide in the derivatized mandelalide A hydrolysate injected separately was 97.5 min. (Figure S56). Therefore co-injection of the natural product hydrolysate with each standard was performed, yielding two peaks with retention times of 96.4 and 97.4 min, which is indicative of the L glycoside in mandelalide A (Figure S56).
Cell viability assays
Cytotoxicity of the organic extract and crude fractions was evaluated in mouse Neuro-2a neuroblastoma, and human MDA-MB-231 breast and NCI-H460 lung cancer cells (ATCC, Manassas, VA) using a previously described protocol subjected to slight modifications.22 Cells were seeded into 96-well plates (20,000 cells per well for MDA-MB-231 and NCI-H460, 25,000 cells per well for Neuro-2A) in 50 μL of medium twelve hours before treatment. Each test sample was added in a 25 μL aliquot generated by serial dilution in serum-free medium on the day of the experiment, after prior removal of 25 μL of media from the treated well. Aliquots were generated from stock solutions of 6 mg/mL compound in 100% DMSO. Pure compounds 1 and 2 were evaluated in Neuro-2A and NCI-H460 cells after 48 h treatment, as described above. Each compound was tested at final concentrations ranging from 0.001 μg/mL to 10 μg/mL. In all cases, cell viability was determined after 48 h treatment using a standard 3-(4,5-dimethylthiazol-2-yl)-2,5,diphenyl tetrazolium bromide (MTT) assay.22 The cytotoxicity of each pure compound was assessed in at least three independent cultures with the viability of vehicle-treated control cells defined as 100% in all experiments. Dose response curves (Figure S57) were plotted using contemporary biostatistics and curve fitting software.
Supplementary Material
Acknowledgments
We thank Professor Bill Fenical of Scripps Institution of Oceanography, San Diego, USA for the generous donation of funding which enabled the first large scale SCUBA collection of marine ascidians in Algoa Bay, South Africa. We are also grateful to Rodger Kohnert for NMR technical assistance, and Brian Arbogast and Jeff Morre of the Environmental Health Sciences Center at OSU for mass spectrometric data acquisition (NIEHS P30 ES00210). The National Science Foundation (CHE-0722319) and the Murdock Charitable Trust (2005265) are acknowledged for their support of the OSU Natural Products and Small Molecule Nuclear Magnetic Resonance. Additional funding was provided by the OSU College of Pharmacy.
Footnotes
Supporting Information Available. Tables of 1D and 2D NMR data and NMR spectra for compounds 1-3 in CDCl3 and py-d5, for compound 4 in CDCl3, for compound 4b in CD3OD. Computational data for mandelalide B (2). GC-MS chromatograms for derivatized α-L- and α-D- rhamnose standards and mandelalide A hydrolysate. Dose response curves for mandelalides A and B (1 and 2) in Neuro-2A and NCI-H460 cell lines. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Blunt JW, Copp BR, Keyzers RA, Munro MHG, Prinsep MR. Nat. Prod. Rep. 2012;29:144. doi: 10.1039/c2np00090c. [DOI] [PubMed] [Google Scholar]
- (2).Molinski TF, Dalisay DS, Lievens SL, Saludes JP. Nat. Rev. Drug Discov. 2009;8:69. doi: 10.1038/nrd2487. [DOI] [PubMed] [Google Scholar]
- (3).Ueda K, Hu Y. Tetrahedron Lett. 1999;40:6305. [Google Scholar]
- (4).Corley DG, Moore RE, Paul VJ. J. Amer. Chem. Soc. 1988;110:7920. [Google Scholar]
- (5).Zabriskie TM, Mayne CL, Ireland CM. J. Amer. Chem. Soc. 1988;110:7919. [Google Scholar]
- (6).Schupp P, Poehner T, Edrada R, Ebel R, Berg A, Wray V, Proksch P. J. Nat. Prod. 2002;66:272. doi: 10.1021/np020315n. [DOI] [PubMed] [Google Scholar]
- (7).Fu X, Palomar AJ, Hong EP, Schmitz FJ, Valeriote FA. J. Nat. Prod. 2004;67:1415. doi: 10.1021/np0499620. [DOI] [PubMed] [Google Scholar]
- (8).Tsukimoto M, Nagaoka M, Shishido Y, Fujimoto J, Nishisaka F, Matsumoto S, Harunari E, Imada C, Matsuzaki T. J. Nat. Prod. 2011;74:2329. doi: 10.1021/np200543z. [DOI] [PubMed] [Google Scholar]
- (9).Schmidt EW, Sudek S, Haygood MG. J. Nat. Prod. 2004;67:1341. doi: 10.1021/np049948n. [DOI] [PubMed] [Google Scholar]
- (10).Ogi T, Margiastuti P, Teruya T, Taira J, Suenaga K, Ueda K. Mar. Drugs. 2009;7:816. doi: 10.3390/md7040816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Schmidt EW, Donia MS, McIntosh JA, Fricke WF, Ravel J. J. Nat. Prod. 2012;75:295. doi: 10.1021/np200665k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Xu Y, Kersten RD, Nam S-J, Lu L, Al-Suwailem AM, Zheng H, Fenical W, Dorrestein PC, Moore BS, Qian P-Y. J. Am. Chem. Soc. 2012 doi: 10.1021/ja301735a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cicero DO, Barbato G, Bazzo R. J. Magn. Reson. 2001;148:209. doi: 10.1006/jmre.2000.2234. [DOI] [PubMed] [Google Scholar]
- (14).Tanaka J.-i., Higa T. Tetrahedron Lett. 1996;37:5535. [Google Scholar]
- (15).Grindley TB. In: Glycoscience: Chemistry and Chemical Biology. 2 ed Fraser-Reid BO, Tatsuta K, Thiem J, Coté GL, Flitsch S, Ito Y, Kondo H, Nishimura S.-i., Yu B, editors. Vol. 1. Springer-Verlag; Germany: 2008. p. 11. [Google Scholar]
- (16).Gheysen K, Mihai C, Conrath K, Martins JC. Chem. – Eur. J. 2008;14:8869. doi: 10.1002/chem.200801081. [DOI] [PubMed] [Google Scholar]
- (17).Bock K, Pedersen C. J. Chem. Soc., Perkin Trans. 2. 1974:293. [Google Scholar]
- (18).Motohashi K, Takagi M, Shinya K. J. Nat. Prod. 2010;73:755. doi: 10.1021/np9007409. [DOI] [PubMed] [Google Scholar]
- (19).Seo S, Tomita Y, Tori K, Yoshimura Y. J. Am. Chem. Soc. 1978;100:3331. [Google Scholar]
- (20).Chini MG, Jones CR, Zampella A, D’Auria MV, Renga B, Fiorucci S, Butts CP, Bifulco G. J. Org. Chem. 2012;77:1489. doi: 10.1021/jo2023763. [DOI] [PubMed] [Google Scholar]
- (21).Winder PL. Ph.D. Thesis. Florida Atlantic University; 2009. [Google Scholar]
- (22).Thornburg CC, Thimmaiah M, Shaala LA, Hau AM, Malmo JM, Ishmael JE, Youssef DTA, McPhail KL. J. Nat. Prod. 2011;74:1677. doi: 10.1021/np200270d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Uhrín D, Batta G, Hruby VJ, Barlow PN, Kövér KE. J. Magn. Reson. 1998;130:155. doi: 10.1006/jmre.1997.1308. [DOI] [PubMed] [Google Scholar]
- (24).Sugahara K, Kitamura Y, Murata M, Satake M, Tachibana K. J. Org. Chem. 2011;76:3131. doi: 10.1021/jo102585k. [DOI] [PubMed] [Google Scholar]
- (25).Navarro-Vázquez A, Cobas JC, Sardina FJ, Casanueva J, Díez E. J. Chem. Inf. Comput. Sci. 2004;44:1680. doi: 10.1021/ci049913t. [DOI] [PubMed] [Google Scholar]
- (26).Parish C, Lombardi R, Sinclair K, Smith E, Goldberg A, Rappleye M, Dure M. J. Mol. Graphics Modell. 2002;21:129. doi: 10.1016/s1093-3263(02)00144-4. [DOI] [PubMed] [Google Scholar]
- (27).Foloppe N, Chen I-J. Curr. Med. Chem. 2009;16:3381. doi: 10.2174/092986709789057680. [DOI] [PubMed] [Google Scholar]
- (28).Lu Z, Van Wagoner RM, Harper MK, Baker HL, Hooper JNA, Bewley CA, Ireland CM. J. Nat. Prod. 2011;74:185. doi: 10.1021/np100613p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Evtushenko EV. Carbohydr. Res. 1999;316:187. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.