

Abstract
Water-soluble gold nanoparticles (AuNPs) have gained considerable attention because they offer a myriad of potential applications, especially in the fields of biology and medicine. One method to prepare such gold nanoparticles is through the well-known Murray place-exchange reaction. In this method, precursor gold nanoparticles, bearing labile ligands and with very good size distribution, are synthesized first, and then reacted with a large excess of the desired ligand. We report a comparison of the reactivity of several known precursor gold nanoparticles (citrate-stabilized, pentanethiol-stabilized, tetraoctylammonium bromide-stabilized, and 4-dimethylaminopyridine-stabilized) to several biologically relevant ligands, including amino acids, peptides, and carbohydrates. We found that citrate-stabilized and 4-dimethylaminopyridine-stabilized gold nanoparticles have broader reactivities than the other precursors studied. Citrate-stabilized gold nanoparticles are more versatile precursors because they can be prepared in a wide range of sizes and are very stable. The hydrophobic pentane-stabilized gold nanoparticles made them “inert” toward highly water-soluble ligands. Tetraoctylammonium bromide-stabilized gold nanoparticles exhibited selective reactivity, especially for small, unhindered and amphiphilic ligands. Depending on the desired ligand and size of AuNPs, a judicious selection of the available precursors can be made for use in place-exchange reactions. In preparing water-soluble AuNPs with biologically relevant ligands, the nature of the incoming ligand and the size of the AuNP should be taken into account in order to choose the most suitable place-exchange procedure.
Keywords: Gold nanoparticles, place-exchange reaction, water-soluble ligands, peptides, carbohydrates, amino acids
1. Introduction
Water-soluble gold nanoparticles (AuNPs) have been of great interest for a number of years for their wide range of potential applications, such as sensing,[1] catalysis,[2] drug delivery,[3-5] and imaging.[6, 7] After the discovery of the synthesis of size-controllable citrate-stabilized AuNPs by Turkhevich[8] and Frens,[9] there has been a constant effort to improve and complement the preparation of AuNPs. For example, Brust and co-workers[10, 11] have paved the way in synthesizing stable, size-controllable (1.5 to 5.2 nm size), and functionalizable AuNPs. In one of these reactions,[11] AuNPs were synthesized by mixing an organothiol and a gold salt (AuCl4−) in the same solvent under reducing conditions. Another method involves the formation of precursor AuNPs bearing weakly bound ligands, followed by the reaction with an excess of the desired organothiol to form the AuNPs.[10] The latter method, which is also known as the Murray place-exchange reaction,[12] is very attractive because it offers both more control over particle size and more flexibility in the nature of the coating ligands employed. Murray and co-workers have extensively studied place exchange reactions, especially those involving the formation of alkylthiolate-stabilized AuNPs.[12-15] The precursor AuNPs for place-exchange reactions can vary in size and/or passivating ligand. Several known AuNPs, such as the citrate-stabilized,[8, 9] alkanethiolate-stabilized,[10] dimethylaminopyridine (DMAP)-stabilized,[16, 17] and tetraoctylammonium bromide (TOAB)-stabilized AuNPs,[10] are potential precursors or starting materials for ligand-exchange reactions. Although numerous studies have examined this process in detail,[12-15] the mechanism is still not fully understood. Nevertheless, this reaction provides access to water-soluble AuNPs with very uniform size distributions and ligand control.
In the advent of a multitude of applications of water-soluble AuNPs, the need for robust ligand-exchange methods has never been more crucial. A better understanding of the reaction may prove useful in choosing the appropriate method of AuNP preparation. In this study, we evaluated four precursors (citrate-stabilized, pentanethiolate-stabilized, TOAB-stabilized, DMAP-stabilized AuNPs) for place-exchange reaction using a wide variety of biologically relevant water-soluble ligands (Figure 1). Representative ligands included the amino acids cysteine (Cys) and Methionine (Met), the tripeptide biological antioxidant, glutathione (GSH), 4-mercaptobenzoic acid (MBA), thioglucose (Glc-SH), the Thomsen-Friedenreich tumor antigen disaccharide attached to a polyethylene glycol (PEG) linker[18] (TF-PEG-SH) and 16-mer peptide derived from the tandem repeat sequence of the membrane associated mucin MUC4[19] attached to a similar PEG linker (MUC4-PEG-SH).[20] These compounds were chosen to represent different sizes, classes, and polarity of water-soluble ligands that are of biological importance. We were able to classify the degree of reactivity of the precursors based on the number of ligands that bound to the AuNP. In addition, we were also able to deduce some structural and physical requirements of the ligands for a particular precursor (i.e. size, polarity, amphiphilicity, charge).
Figure 1.
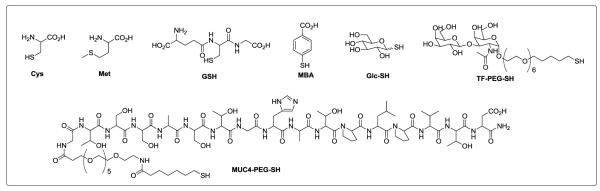
Model water-soluble ligands used for place-exchange reactions.
2. Experimental
2.1 General Remarks
HAuCl4·XH2O was purchased from Strem Chemicals and used as received. Sodium borohydride, pentanethiol, tetraoctylammonium bromide, cysteine, methionine, thioglucose sodium salt, 4-mercaptobenzoic acid, and dimethylaminopyridine were purchased from Sigma-Aldrich Corp. and were used as received. Compounds TF-PEG-SH and MUC4-PEG-SH were synthesized following the literature procedure.[18, 20] Water was obtained from a Millipore Milli-Q plus purification system (resistivity of 18.2 MΩ/cm). UV-visible spectra were recorded on an Agilent 8453 spectrophotometer. Citrate-stabilized AuNPs (5 nm) were purchased from Ted Pella, Inc. and used as received. 1H NMR spectra were recorded on a Varian Inova-400 spectrometer at 25°C. TEM samples were prepared by placing an aqueous solution (~2μl) of the AuNPs on the grid with a carbon-coated support film that was previously treated with glow discharge. The excess liquid was blotted with a filter paper, allowed to dry, and rinsed with distilled water twice. TEM images were taken using a Hitachi H7650 TEM (Tokyo, Japan) operating at 80 kV with a 2k×2k CCD camera (AMT; Danvers, MA). The sizes of Au NPs were analyzed using the camera’s measurement software (AMT).
2.2 Synthesis of Pentanethiol-Stabilized Au NPs
Pentanethiol-stabilized AuNPs were prepared following a literature procedure[21] adapted from the original Brust protocol.[10]. Briefly, a solution of aqueous HAuCl4 (30 mM, 30 mL) was mixed with a toluene solution of tetraoctylammonium bromide (50 mM, 80 mL). Pentanethiol (170 mg, 0.84 mmol) was then added, followed by the slow addition of NaBH4 solution (0.4 M, 25 mL). The mixture was stirred for 10 min at room temperature, the organic layer was collected and concentrated by rotary evaporation to ~10 mL. Ethanol (500 mL) was added, and the mixture was kept at 4°C overnight. The precipitate was collected by centrifugation, redissolved in toluene, and re-precipitated by the addition of ethanol. After cooling the mixture at 4°C, the dark brown precipitate was collected and dried under vacuum overnight.
2.3 Synthesis of 4-(Dimethylamino)pyridine-Stabilized Au NPs
DMAP-stabilized Au NPs were prepared a following literature procedure.[22] A toluene solution of TOAB (612 mg in 20 mL) was mixed with an aqueous solution of HAuCl4 (100 mg in 8 mL). The heterogeneous mixture was vigorously stirred until all of the gold salt had transferred to the organic phase. A freshly prepared sodium borohydride solution (105 g in 6 mL) was added slowly and the mixture was stirred for 12 h at room temperature. The organic layer was collected; washed with water 3×; and dried with anhydrous Na2SO4. The volume of solution was adjusted to 50 mL by addition of toluene. A solution of 4-(dimethylamino)pyridine in water (610 g, 50 mL) was added and the mixture was stirred until phase transfer was complete (i.e., all of the color transferred to the aqueous phase). The dark red mixture was isolated and stored at 4°C.
2.4 Synthesis of Tetraoctylammonium bromide-stabilized Au NPs
The preparation of tetraoctylammonium bromide-stabilized Au NPs was done according to the method used of Brust et al.[10] as described above in the preparation of pentanethiol-stabilized Au NPs.
2.5 General Procedure for the Reaction of Precursor Nanoparticles with Various Ligands
2.5.1 Using Citrate-Stabilized Au NPs
Commercially-available citrate-stabilized gold nanoparticles (5 nm, ~1 nM) was mixed with a 200-fold excess of the ligands (relative to the concentration of the AuNPs), and the resulting mixture was stirred for 1-72h. The progress of reaction was monitored by UV-Vis spectroscopy.
2.5.2 Using Pentanethiol-Stabilized Au NPs
The pentanethiol-stabilized Au NPs (1 mL of 10 mg/mL solution in toluene) were reacted with the model ligands in water for 1 h at 25°C. The reaction was monitored by visually inspecting for the phase transfer of the AuNPs
2.5.3 Using DMAP-Stabilized AuNPs
DMAP-AuNPs (15mL) were washed with water (3×) by ultrafiltration (Amicon MWCO 30kDa), resuspended in water (~15 ml), and used immediately in the next step. The thiol ligand was dissolved in water and the pH of the solution was adjusted to ~10. DMAP-Au NPs (167 μl) were mixed with the model ligand (0.45 mM, 250 μl) and agitated overnight at 25°C. The mixture was ultrafiltered (Amicon, MWCO 30kDa) for 10 min at 10k×g. The amount of DMAP released in the filtrate was determined by analyzing an aliquot of the filtrate using analytical HPLC with UV detection at 300 nm (column: Phenomenex Gemini 5μ C18, Gradient 100% A – 100% B over 20 min; Solvent A: 0.1% TFA in water, Solvent B: acetonitrile with 0.1% TFA) and comparing it against a standard DMAP calibration curve.
2.5.4 Using Tetraoctylammonium bromide-stabilized AuNPs
The freshly prepared TOAB-Au NPs (2.4 mL) were reacted with the model ligand (0.1 M solution, 2.4 mL) for 30 min. The reaction was monitored by the phase transfer of the dark brown-colored Au NPs from the organic layer to the aqueous layer.
3. Results
3.1 Citrate-Stabilized AuNPs as Precursor
Since the early discovery of methods to prepare size-controllable AuNPs by Turkevich and Frens, the applications of AuNPs have increased dramatically. Several procedures are now available in preparing citrate AuNPs with sizes ranging from 3 to 64 nm.[23] In addition, several commercial companies (i.e., Sigma-Aldrich, TedPella) are now supplying these AuNPs with very narrow size-distribution. This facile access to a broad range of sizes have made citrate-AuNPs very attractive precursor in preparing water-soluble AuNPs for many applications.
Representative water-soluble ligands (Figure 1) were reacted with citrate-stabilized AuNPs (5 nm) for 72 h, and the reaction was monitored by UV-vis spectroscopy. Figure 2 shows the progress of reaction of selected model ligands with the citrate-stabilized AuNPs. AuNPs exhibit a characteristic surface plasmon band at 520 nm, which is due to the collective oscillation of electrons in the conduction band for AuNPs.[24] A slight shift in the plasmon band (~520 nm), usually between 2-5 nm,[25, 26] indicates place-exchange reaction occurring. This is due to the change in dielectric permittivity of the AuNPs brought about by the formation of the thiolate monolayer.[26] AuNPs treated with the ligands: Met, GSH, MBA, TF-PEG-SH, and MUC4-PEG-SH showed a slight shift in the plasmon band over a 72-h period indicating successful place exchange to AuNPs (Figures 2 and S3-S6). On the other hand, AuNPs treated with the ligands Cys and Glc-SH exhibited broadening of and a larger shift (≥ 20 nm) in the plasmon absorption band (Figure 2C and 2D). These changes were much more rapid for the Cys-treated AuNPs (~3 h) as compared to the very slow aggregation of the Glc-SH-treated AuNPs(~24 hours). The large shift in the plasmon band for aggregating particles can be attributed to the decrease in the interparticle distance during the process of self-assembly.[27] In the case of Cys as ligand, where the shift was large (> 50 nm), its zwitterionic nature may be responsible for the fast aggregation of the resulting AuNPs, whereby interparticle charge neutralization may facilitate aggregation.[28] For the Glc-SH-treated AuNPs, for which the magnitude of the shift was not as great (~20 nm, illustrated by dotted lines at the absorption maxima, Figure 2D), attractive interparticle H-bonding of the hydroxyl groups could explain its aggregation aptitude. The reactivity of TF-PEG-SH and MUC4-PEG-SH could be attributed to their propensity to form monolayers due to the presence of a nonpolar spacer before the thiol group in the form of a fatty chain. The fatty chain or alkyl spacer presumably behaves the same way as the alkyl chains in alkanethiols on surfaces or nanoparticles, where the chains are stretched out and the alkanethiols exist in a monolayer assembly.[29] As an example of a successful place exchange reaction, the AuNPs formed from the reaction of MUC4-PEG-SH and citrate-stabilized AuNPs were isolated by ultrafiltration and purified by size exclusion chromatography. The 1H NMR spectrum of the purified AuNPs (see Figure S1; Supporting Information) resembled that of the spectrum of free MUC4-PEG-SH ligand, indicating the successful passivation of the Au core. MBA ligand is known to stabilize AuNPs, not only by imparting a negative charge but also by orienting itself in such a way that there are ring-ring and sulfur-ring interactions as observed in the crystal structure of a 1.1 nm Au-MBA.[30] The reactivity of GSH is similar to Cys except that the GSH-stabilized AuNPs have better stability, especially at very high or low pH, because there is either a net negative or positive charge on the ligand. In the case of Met, the interaction between the surface of the AuNP and the sulfur of Met thioether gives a net positive charge, which contributes to the interparticle repulsion. In general, citrate-AuNPs seem to be reactive with a broad range of ligands. They can be a powerful precursor for making water-soluble AuNPs because their size can be varied, their size distribution is excellent, and their stability is very good.
Figure 2.

Ligand-exchange reaction of citrate-AuNPs and the different water-soluble ligands as monitored by UV-vis spectroscopy; A: Control (citrate-AuNPs), B: GSH-treated AuNPs, C: Cys-treated AuNPs, D: Glc-SH-treated AuNPs. The dotted lines in D indicate the maxima from 0 hours (black) to 72 hours (pink)
3.2 Pentanethiol-Stabilized Au NPs as Precursor
Alkanethiolate-stabilized AuNPs are also an attractive precursor for the preparation of water-soluble AuNPs because of their ease of preparation and stability. These ligands have been used to prepare important AuNPs that have applications, for example, in sensing[31, 32] and drug delivery.[33, 34] Water-soluble AuNPs for these applications were typically prepared using ligands that were amphiphilic, and so the place-exchange reaction occurred in a suitable organic solvent (i.e., methylene chloride/methanol mixture).[31-34] In the present work, most of our ligands were insoluble in organic solvent and so we performed the reaction of our model ligands with pentanethiol-stabilized AuNPs in a biphasic mixture of toluene and water. Expectedly, only MBA reacted, as observed in the transfer of color from organic phase to aqueous phase. The relatively large nonpolar phenyl ring makes MBA soluble in toluene and reactive with the organic-soluble AuNPs. Although the other ligands contain alkyl moieties, their large polar headgroups (sugar, peptide and polyethyleneglycol moieties) render them insoluble in nonpolar organic solvents such as toluene. Even with the use of DMF as solvent or crown-8-ether as a phase-transfer catalyst the reaction still did not proceed. Pentanethiolate-AuNPs may not be the best precursor for making water-soluble AuNPs through place-exchange reaction because of their inherent hydrophobicity and their relative instability over time.
3.3 DMAP-stabilized AuNPs as Precursor
The DMAP-stabilized AuNPs have found utility, for example, in the fabrication of metallopolymer-gold nanoparticles composites with enhanced electrochemiluminescence[35] and the construction of capillaries modified with gold nanoparticles coated on polyelectrolyte multilayer for open-tubular capillary electrochromatography. [36] Lennox and coworkers have extensively studied the reactivity of the DMAP-AuNPs in place-exchange reactions at various conditions to produce water-soluble AuNPs.[22] However, the structural contribution of the incoming ligand was not emphasized. Here we used our diverse family of ligands to address how various ligand structures will affect exchange propensity for these precursors. We followed the place-exchange protocol developed by Lennox,[16] and in this case, quantitatively determined the amount of displaced DMAP in the filtrate by through its UV signature from HPLC chromatograms, to follow the course of the exchange protocol (Figure 3). Small amino acids, such as Cys and Met, were generally reactive; albeit, their resulting AuNPs flocculated over time.
Figure 3.

Concentration of the displaced DMAP in the filtrate after the filtration of the reaction between DMAP-AuNPs and the representative ligands.
Cys seemed to be the more reactive than Met, presumably because of the formation of the relatively stable S-Au bond. MBA was the least reactive among the smaller ligands, perhaps due to its borderline solubility in water. The ligands containing sugar moieties, such as Glc-SH and TF-PEG-SH, were very reactive, but the smaller Glc-SH gave unstable AuNPs after concentration by ultrafiltration. TF-PEG-SH gave stable AuNPs possibly because of the presence of the PEG-fatty chain thiol linker. The peptide ligands, GSH and MUC4-PEG-SH both gave stable AuNPs. Aside from the stable Au-S bond that GSH forms with the core, the presence of flexible carboxylic acid functional groups and an amino group possibly allow for additional stabilizing interactions. MUC4-PEG-SH has the advantage of having the PEG-fatty chain thiol linker that may stabilize the AuNP core. DMAP-AuNPs, like citrate-stabilized AuNPs can also be a reasonable precursor for place-exchange reaction, due to their reactivity with a wide range of water-soluble ligands. One of the disadvantages of this precursor is that the size of the AuNPs is limited to the 4-6 nm range.
3.4 TOAB-Stabilized AuNPs as precursor
Although not as common as the DMAP- or citrate-stabilized AuNPs, TOAB-stabilized AuNPs have been used previously to prepare water-soluble AuNPs.[37] The long-term stability of TOAB-stabilized AuNPs[38] and the difficulty to purify their place-exchange products from TOAB[39] may be some of the reasons why they are not very commonly used as precursors. However, they have been primarily used in applications like modification of the surface of Au electrodes for sensing small molecules[40, 41] and also forming multi-layered thin film assembly.[42, 43] In this study, we investigated the reactivity of the model ligands with TOAB-AuNPs using a two-phase system (water-toluene). It was found that complete phase transfer occurred with Cys, Glc-SH, and TF-PEG-SH; and partial phase transfer occurred with MUC4-PEG-SH. Since TOAB is known to transfer Au3+ ions from the aqueous to organic phase, the relatively small zwitterionic Cys may be transferred in a similar fashion to the organic phase by the excess TOAB present in the AuNP toluene solution, followed by a standard exchange reaction of the Cys thiol group to the AuNP surface. Similar to the AuNPs formed from using citrate-stabilized AuNP precursor, the Cys-AuNPs formed from the reaction between TOAB-AuNPs and Cys were only stable for a few minutes and showed extensive and irreversible precipitation. The instability of Cys-coated AuNPs was also observed by Mocanu and coworkers.[28] Analogous to Cys, the zwitterionic Met could also be transferred from the aqueous phase to the organic phase by TOAB. However, the sulfur atom on Met does not form a multi-coordinate bond with Au, and so this interaction may be weaker than the interaction between TOAB and Au. Thus, no reaction was observed. Another relatively small ligand that reacted with TOAB-AuNPs is Glc-SH. Glc-SH formed AuNPs that were stable enough to be isolated by size-exclusion chromatography, and characterized by NMR and TEM (see Figs. S7 and S8; Supporting Information). Slightly larger and polar ligands, such as GSH, probably undergo TOAB-assisted phase transfer, but their relatively large size reduces their effective access to the surface of the AuNP which is covered with bulky TOAB ligands. Amphiphilic ligands, such as the TF-PEG-SH, can presumably partition between organic and aqueous phases without the need for a surfactant. In addition, the thiol group on TF-PEG-SH is unhindered, and so the interaction with the AuNP surface would be more effective. MUC4-PEG-SH can also be considered amphiphilic since it contains the PEG-fatty chain thiol linker. However, its 16-residue peptide moiety is probably too big and polar to dissolve completely in the toluene layer. This is why it partitioned partially between the aqueous and organic layers. Lastly, MBA ligand was found to be inert towards TOAB-AuNPs. Although this ligand is relatively small, the steric hindrance around the thiol functional group probably limits its reactivity towards the also sterically hindered AuNP surface.
As an illustration of the successful ligand-exchange reaction, Figure 4A (upper spectrum) shows the NMR spectrum of the purified TF-PEG-SH-coated AuNPs obtained from the reaction of TOAB-AuNP precursor with TF-PEG-SH. Even though extensive broadening was observed due to reduction in relaxation times of these high molecular weight entities, the NMR spectrum of the purified TF-PEG-SH-coated AuNP retained the major features of that of the free ligand (Figure 4A lower spectrum), which indicates successful exchange of ligands from TOAB to TF-PEG-SH. The resulting AuNPs had good size distribution centered about 3.5 nm diameter (Figure 4B). While the histogram indicates a symmetric bell-shaped curve for size, uniformity was not ideal, but this level of homogeneity was similar to several other studies with particles made with similar methods.[37, 44] This methodology may be an alternative to the place-exchange reaction using citrate-stabilized AuNPs as precursor, however, with the shortcomings stated above.
Figure 4.

A. 1H-NMR spectra of the free TF-PEG-SH (upper) and its AuNP form (lower); B. TEM image of the TF-PEG-SH coated AuNPs with histogram of size distribution (inset).
The standard experimental procedure for place exchange reactions may be adjusted to account for differences in the physical or chemical properties of the ligands. The reactions can be 1) performed at various pH values (usually basic) or 2) with the assistance of reverse micelle formation to assist in the transfer of ligands to the self-assembled monolayer surface. We discounted adjustment 2 (reverse micelle assisted reactions) since the standard reagents used for these experiments are sodium bis(2-ethylhexyl) sulfosuccinate (NaAOT) or cetyltrimethylammonium bromide (CTAB); the former possibly reacting with the formed gold nanosphere and the latter used primarily for production of nanorods of different aspect ratios. Thus, we performed the reaction of the TOAB precursor with Cys, GSH and Met and pentanethiol precursors with GSH and Met all at pH 10. No reaction was observed for any of these conditions, with the Cys reaction with TOAB causing aggregation after 72 h.
4. Discussion
In the last decade, gold nanoparticles, in all their various forms, have been the subject of an unprecedented amount of research, due primarily to the ease in which these versatile multivalent platforms can be prepared and coated with ligands from virtually every molecular family.[45, 46] Several methods have been explored for their synthesis, and the use of the Murray place exchange reaction[12] has a special place in this repertoire. High quality precursor particles for ligand exchange may be prepared with a variety of passivating agents in a wide range of sizes. Thus the synthesis of particles with the ligand of choice can start from materials of very uniform and reproducible sizes and stabilities. Here we have explored the use of several precursors for the synthesis of water soluble gold nanoparticles with potential biological utility. Ligands were taken from a wide range of molecular subtypes, from amino acids to sugars to glycopeptides, with and without PEG linkers, from both commercial sources and our own research interests.
All precursors work to some degree with different ligands. The decades-old “tried-and true” citrate-stabilized Turkevich precursor was the overall winner, with the ability to exchange ligands of most molecular families successfully. Free amino acid ligands like cysteine were troublesome, undoubtedly due to their zwitterionic character and the pH-dependent state of ionization. The amino and acid groups could form “head-to-tail” ionic salt bridges between particles, causing enhanced aggregation, akin to that of gold nanoshell coated magnetic particles passivated by homocysteine.[47]
The unsuccessful reactions were primarily due to the incompatibility of the precursors and the ligands to be exchanged. For example, pentanethiol coated AuNPs have been used with remarkable success by Rotello and co-workers.[21] Exchange to selective ligands occurs primarily from an organic/water-miscible interface with amphiphilic molecules that can partition well in a variety of milieus. Our attempts to transfer water soluble ligands essentially failed for most molecules, and most likely this is the simple physical difficulty in phase transfer for two entities with solubilities on extreme opposite ends of the spectrum. Thus, although pentanethiol precursors are quite versatile in their own right, they have limited use in the cases studied here.
DMAP-AuNPs have been employed in a limited set of place exchange reactions, primarily from the Lennox group.[16, 22, 48] Although not as widely used as citrate-stabilized AUNPs, these precursors are extremely versatile and should find more widespread use. DMAP precursors worked almost as well as the AuNPs from the Turkevich precursor, and the fact that the DMAP ligand is quite labile and somewhat amphiphilic in nature makes exchange reactions proceed faster with lower concentrations of thiol ligands and in a kinetically controlled manner. Recent work by the Lennox groups showed that liquid crystal ligand-capped AuNPs can be prepared from DMAP-AuNP precursors, with control of miscibility and ligand composition in binary and ternary systems, where 2 or 3 different ligands are attached to the gold surface.[49] This expands the utility of these particles and they may be poised for use in other potentially clinically useful therapeutic venues. There is a limitation on the size range that one can prepare with this precursor, but they are promising for the preparation of AuNPs with ligand of varying solubilities
The TOAB precursors were of mixed utility in our hands, with our specific set of water soluble ligands although also useful. One issue with these precursors is purification away from excess TOAB, which could be highly detrimental in subsequent biological experiments. In addition, some of the reactions we performed obviously were superior in single phase systems as opposed to two-phase transfer protocols. One possibility is the nature of our ligands: Partitioning of large, multifunctional peptide and sugars containing PEG units may hinder transfer as the duplicitous solubility adds to the complexity of these systems. Further adjustments to the general procedure may more efficiently facilitate reaction with complex ligands. With all the known methods that have been evaluated for producing AuNPs, most water soluble systems still need to be evaluated on a case-by-case basis. In 2005, Kornberg and co-workers surveyed the synthesis of water soluble gold clusters via thiolate chemistry with 36 different ligands.[50] They used the Brust or Hutchison[51] procedures for particle synthesis and also used ligands employed in the present study (Cysteine, methionine, Glc-SH, GSH and a Cys-containing peptide). Success of the synthesis was highly dependent on the size and chemical makeup of the ligand, with stabilities of resultant particles ranging from days to months. These results along with those presented here suggest that although citrate- and DMAP-stabilized precursors generally work well, there may not be a “one-size-fits-all” protocol for water soluble AuNP synthesis: The factors described above all need to be accounted for before the optimum conditions are revealed.
5. Summary and conclusion
In summary, both citrate-stabilized and DMAP-stabilized AuNPs showed the broadest reactivity toward the model water-soluble ligands in place-exchange reaction (Table 1). The citrate-AuNPs are very good precursors because they are water-soluble and they can be prepared in a wide range of sizes. DMAP-AuNPs have similar reactivity to that of the citrate-stabilized AuNPs, but they are limited to making AuNPs in the 4-6 nm size range. Pentanethiolate-AuNPs are generally not reactive, except with the relatively hydrophobic MBA ligand, to the model water-soluble ligands in a two-phase reaction. Even in the presence of phase transfer catalyst, most of the model ligands did not react, even at elevated pH. The reactivity of TOAB-AuNPs depends on the size and the hydrophobicity/hydrophilicity of the incoming ligands. Smaller and unhindered ligands tend to displace TOAB easier than sterically hindered ligands. Among the four methods, the one-phase place-exchange reaction involving citrate-AuNPs and DMAP-AuNPs appear to be the reactive precursors for making water-soluble AuNPs, especially involving ligands such as amino acids, peptides, and carbohydrates. Since water-soluble AuNPs have a multitude of potential applications in biology and medicine, this study serves as a guideline in choosing the appropriate precursor and ligand for making biologically relevant water-soluble AuNPs.
Table 1.
Summary of the place-exchange reactivity of the various nanoparticle precursors with representative water-soluble ligands
| Ligand | Precursor AuNPs (reactive = +; unreactive = −) | |||
|---|---|---|---|---|
| Citrate | DMAP | TOAB | Pentanethiol | |
| Cys | − | +a | +b | − c |
| Met | + | +a | − c | − c |
| GSH | + | + | − c | − c |
| MBA | + | − | − | + |
| Glc-SH | − | +a | + | − |
| TF-PEG-SH | + | + | + | − |
| MUC4-PEG-SH | + | + | +/−d | − |
Precipitation observed after concentration of the mixture.
Precipitation occurred after a few minutes of mixing.
Also performed at pH 10.0, with similar results (no exchange).
Partial phase transfer observed.
Supplementary Material
Research Highlights.
Four AuNP precursors were studied for their reactivity to place-exchange reaction
A panel of varied water-soluble ligands was used to react with the AuNP precursors
Specific precursor AuNPs have broad reactivities compared with the others
Some precursor reactivity is selective to small unhindered and amphiphilic ligands
Acknowledgement
We are grateful to Dr. Kunio Nagashima for performing the TEM analyses. We also appreciate the assistance of Dr. Sergei Tarasov and Ms. Marzena Dyba with the UV-Vis spectrophotometer and Souvik Biswas for help with the high pH exchange reactions. This work was supported by funds from the Intramural program of the National Cancer Institute at the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Kim N, Kim DK, Cho YJ. Current Applied Physics. 2010;10:1227–1230. [Google Scholar]
- [2].Zhang ZM, Fu QA, Li XQ, Huang X, Xu JY, Shen JC, Liu JQ. J. Biol. Inorg. Chem. 2009;14:653–662. doi: 10.1007/s00775-009-0478-8. [DOI] [PubMed] [Google Scholar]
- [3].Hwu JR, Lin YS, Josephrajan T, Hsu MH, Cheng FY, Yeh CS, Su WC, Shieh DB. J. Am. Chem. Soc. 2009;131:66. doi: 10.1021/ja804947u. + [DOI] [PubMed] [Google Scholar]
- [4].Podsiadlo P, Sinani VA, Bahng JH, Kam NWS, Lee J, Kotov NA. Langmuir. 2008;24:568–574. doi: 10.1021/la702782k. [DOI] [PubMed] [Google Scholar]
- [5].Aryal S, Grailer JJ, Pilla S, Steeber DA, Gong SQ. J. Mater. Chem. 2009;19:7879–7884. [Google Scholar]
- [6].Hainfeld JF, Slatkin DN, Focella TM, Smilowitz HM. Br. J. Radiol. 2006;79:248–253. doi: 10.1259/bjr/13169882. [DOI] [PubMed] [Google Scholar]
- [7].Li J, Chaudhary A, Chmura SJ, Pelizzari C, Rajh T, Wietholt C, Kurtoglu M, Aydogan B. Phys. Med. Biol. 2010;55:4389–4397. doi: 10.1088/0031-9155/55/15/013. [DOI] [PubMed] [Google Scholar]
- [8].Turkevich J, Stevenson PC, Hillier J. Discuss. Faraday Soc. 1951:55. &. [Google Scholar]
- [9].Frens G. Nature-Physical Science. 1973;241:20–22. [Google Scholar]
- [10].Brust M, Walker M, Bethell D, Schiffrin DJ, Whyman R. Journal of the Chemical Society-Chemical Communications. 1994:801–802. [Google Scholar]
- [11].Brust M, Fink J, Bethell D, Schiffrin DJ, Kiely C. Journal of the Chemical Society-Chemical Communications. 1995:1655–1656. [Google Scholar]
- [12].Hostetler MJ, Wingate JE, Zhong CJ, Harris JE, Vachet RW, Clark MR, Londono JD, Green SJ, Stokes JJ, Wignall GD, Glish GL, Porter MD, Evans ND, Murray RW. Langmuir. 1998;14:17–30. [Google Scholar]
- [13].Templeton AC, Wuelfing MP, Murray RW. Acc. Chem. Res. 2000;33:27–36. doi: 10.1021/ar9602664. [DOI] [PubMed] [Google Scholar]
- [14].Song Y, Murray RW. J. Am. Chem. Soc. 2002;124:7096–7102. doi: 10.1021/ja0174985. [DOI] [PubMed] [Google Scholar]
- [15].Donkers RL, Song Y, Murray RW. Langmuir. 2004;20:4703–4707. doi: 10.1021/la0497494. [DOI] [PubMed] [Google Scholar]
- [16].Rucareanu S, Gandubert VJ, Lennox RB. Chem. Mater. 2006;18:4674–4680. [Google Scholar]
- [17].Gittins DI, Caruso F. Angewandte Chemie-International Edition. 2001;40:3001–3004. doi: 10.1002/1521-3773(20010817)40:16<3001::AID-ANIE3001>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- [18].Svarovsky SA, Szekely Z, Barchi JJ. Tetrahedron-Asymmetry. 2005;16:587–598. [Google Scholar]
- [19].Brocke C, Kunz H. Synthesis-Stuttgart. 2004:525–542. [Google Scholar]
- [20].Sundgren A, Barchi JJ. Carbohydr. Res. 2008;343:1594–1604. doi: 10.1016/j.carres.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].You CC, De M, Rotello VM. Org. Lett. 2005;7:5685–5688. doi: 10.1021/ol052367k. [DOI] [PubMed] [Google Scholar]
- [22].Gandubert VJ, Lennox RB. Langmuir. 2005;21:6532–6539. doi: 10.1021/la050195u. [DOI] [PubMed] [Google Scholar]
- [23].Handley DA. Methods for Synthesis of Colloidal Gold. In: Hayat M, editor. Colloidal Gold: Principles, Methods, and Applications. Academic Press; New York: 1989. pp. 13–32. [Google Scholar]
- [24].Eustis S, El-Sayed MA. Chem. Soc. Rev. 2006;35:209–217. doi: 10.1039/b514191e. [DOI] [PubMed] [Google Scholar]
- [25].Li DX, He Q, Cui Y, Duan L, Li JB. Biochem. Biophys. Res. Commun. 2007;355:488–493. doi: 10.1016/j.bbrc.2007.01.183. [DOI] [PubMed] [Google Scholar]
- [26].Levy R, Thanh NTK, Doty RC, Hussain I, Nichols RJ, Schiffrin DJ, Brust M, Fernig DG. J. Am. Chem. Soc. 2004;126:10076–10084. doi: 10.1021/ja0487269. [DOI] [PubMed] [Google Scholar]
- [27].Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ. Nature. 1996;382:607–609. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- [28].Mocanu A, Cernica I, Tomoaia G, Bobos LD, Horovitz O, Tomoaia-Cotisel M. Colloids and Surfaces a-Physicochemical and Engineering Aspects. 2009;338:93–101. [Google Scholar]
- [29].Love JC, Estroff LA, Kriebel JK, Nuzzo RG, Whitesides GM. Chem. Rev. 2005;105:1103–1169. doi: 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- [30].Jadzinsky PD, Calero G, Ackerson CJ, Bushnell DA, Kornberg RD. Science. 2007;318:430–433. doi: 10.1126/science.1148624. [DOI] [PubMed] [Google Scholar]
- [31].Phillips RL, Miranda OR, You CC, Rotello VM, Bunz UHF. Angewandte Chemie-International Edition. 2008;47:2590–2594. doi: 10.1002/anie.200703369. [DOI] [PubMed] [Google Scholar]
- [32].You CC, Miranda OR, Gider B, Ghosh PS, Kim IB, Erdogan B, Krovi SA, Bunz UHF, Rotello VM. Nature Nanotechnology. 2007;2:318–323. doi: 10.1038/nnano.2007.99. [DOI] [PubMed] [Google Scholar]
- [33].Kim B, Han G, Toley BJ, Kim CK, Rotello VM, Forbes NS. Nature Nanotechnology. 2010;5:465–472. doi: 10.1038/nnano.2010.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kim C, Agasti SS, Zhu ZJ, Isaacs L, Rotello VM. Nature Chemistry. 2010;2:962–966. doi: 10.1038/nchem.858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Devadoss A, Spehar-Deleze AM, Tanner DA, Bertoncello P, Marthi R, Keyes TE, Forster RJ. Langmuir. 2010;26:2130–2135. doi: 10.1021/la902676p. [DOI] [PubMed] [Google Scholar]
- [36].Qu QS, Liu DP, Mangelings D, Yang C, Hu XY. J. Chromatogr. A. 2010;1217:6588–6594. doi: 10.1016/j.chroma.2010.08.057. [DOI] [PubMed] [Google Scholar]
- [37].Azzam T, Bronstein L, Eisenberg A. Langmuir. 2008;24:6521–6529. doi: 10.1021/la703719f. [DOI] [PubMed] [Google Scholar]
- [38].Isaacs SR, Cutler EC, Park JS, Lee TR, Shon YS. Langmuir. 2005;21:5689–5692. doi: 10.1021/la050656b. [DOI] [PubMed] [Google Scholar]
- [39].Waters CA, Mills AJ, Johnson KA, Schiffrin DJ. Chem. Commun. 2003:540–541. doi: 10.1039/b211874b. [DOI] [PubMed] [Google Scholar]
- [40].John SA, Sagara T. J. Electroanal. Chem. 2009;633:175–181. [Google Scholar]
- [41].Nair SS, John SA, Sagara T. Electrochim. Acta. 2009;54:6837–6843. [Google Scholar]
- [42].Baum T, Bethell D, Brust M, Schiffrin DJ. Langmuir. 1999;15:866–871. [Google Scholar]
- [43].Brust M, Bethell D, Kiely CJ, Schiffrin DJ. Langmuir. 1998;14:5425–5429. [Google Scholar]
- [44].Thomas KG, Zajicek J, Kamat PV. Langmuir. 2002;18:3722–3727. [Google Scholar]
- [45].Mout R, Moyano DF, Rana S, Rotello VM. Chem. Soc. Rev. 2012;41:2539–2544. doi: 10.1039/c2cs15294k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yeh YC, Creran B, Rotello VM. Nanoscale. 2012;4:1871–1880. doi: 10.1039/c1nr11188d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lo CK, Xiao D, Choi MMF. J. Mater. Chem. 2007;17:2418–2427. [Google Scholar]
- [48].Rucareanu S, Maccarini M, Shepherd JL, Lennox RB. J. Mater. Chem. 2008;18:5830–5834. [Google Scholar]
- [49].Milette J, Toader V, Reven L, Lennox RB. J. Mater. Chem. 2011;21:9043–9050. [Google Scholar]
- [50].Ackerson CJ, Jadzinsky PD, Kornberg RD. J. Am. Chem. Soc. 2005;127:6550–6551. doi: 10.1021/ja046114i. [DOI] [PubMed] [Google Scholar]
- [51].Brown LO, Hutchison JE. J. Am. Chem. Soc. 1997;119:12384–12385. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.