

Abstract
Background. Clostridium difficile is a major cause of nosocomial diarrhea, with 30-day mortality reaching 30%. The cell surface comprises a paracrystalline proteinaceous S-layer encoded by the slpA gene within the cell wall protein (cwp) gene cluster. Our purpose was to understand the diversity and evolution of slpA and nearby genes also encoding immunodominant cell surface antigens.
Methods. Whole-genome sequences were determined for 57 C. difficile isolates representative of the population structure and different clinical phenotypes. Phylogenetic analyses were performed on their genomic region (>63 kb) spanning the cwp cluster.
Results. Genetic diversity across the cwp cluster peaked within slpA, cwp66 (adhesin), and secA2 (secretory translocase). These genes formed a 10-kb cassette, of which 12 divergent variants were found. Homologous recombination involving this cassette caused it to associate randomly with genotype. One cassette contained a novel insertion (length, approximately 24 kb) that resembled S-layer glycosylation gene clusters.
Conclusions. Genetic exchange of S-layer cassettes parallels polysaccharide capsular switching in other species. Both cause major antigenic shifts, while the remainder of the genome is unchanged. C. difficile genotype is therefore not predictive of antigenic type. S-layer switching and immune escape could help explain temporal and geographic variation in C. difficile epidemiology and may inform genotyping and vaccination strategies.
Keywords: Clostridium difficile, S-layer, S-layer glycosylation, immunodominant antigen, recombination, switching, multilocus sequence type, genotype, evolution
The severity of an infectious disease is determined by 2 key opposing factors: the virulence of the invading pathogen and the success of the host defense. Both factors are influenced by the outer surface of the bacterial cell, which is under strong immune selection for high antigenic diversity and rapid evolutionary change. As this process can quickly transform the epidemiology of an infection [1, 2], it is important to understand bacterial antigenic repertoires and the mechanisms by which they evolve.
The high diversity of genes encoding cell surface antigens can preclude their large-scale characterization by conventional Sanger DNA sequencing, since to be performed at high throughput this technique requires prior knowledge of gene content and nucleotide sequence. Recent advances in DNA sequencing technology [3] offer many new opportunities to clinical microbiology [4], including the chance to understand the diversity and evolution of previously intransigent species. An example is Clostridium difficile, a Gram-positive sporulating anaerobe that has become an important nosocomial and community-acquired human pathogen [5]. The loci encoding 2 of its major cell surface antigens exhibit high genetic diversity between strains [6, 7], but they have only been investigated on a small scale [7, 8].
C. difficile infections (CDIs) range in severity from mild diarrhea to pseudomembranous colitis and toxic megacolon, with a crude 30-day mortality that can exceed 30% [9]. C. difficile virulence is multifactorial and includes variable toxigenicity and transmissibility [10]. Two hypervirulent strains (polymerase chain reaction [PCR] ribotypes 027 and 078) associated with poor clinical outcome have been identified in recent years [11, 12]. However, the contribution of the outer cell surface to C. difficile virulence is poorly understood.
C. difficile is one of many bacterial species surrounded by a paracrystalline S-layer, which self-assembles from multiple copies of a single protein [13]. The C. difficile S-layer is encoded by the slpA gene, located within a 36.6-kb cell wall protein (cwp) gene cluster [14]. Bacterial S-layers are in many cases glycosylated, a post-translational modification that is encoded by a characteristic gene cluster located near the S-layer gene [15]. However, neither genetic nor phenotypic evidence of S-layer glycosylation has been found in C. difficile to date [14, 16].
The C. difficile S-layer protein is an immunodominant antigen, activating both innate and adaptive immunity, and providing the basis for C. difficile serological typing [17–19]. The S-layer precursor protein is cleaved after translation into high-molecular-weight and low-molecular-weight (LMW) fragments [8]. The LMW fragment exhibits extremely high genetic diversity between serogroups, which consequently lack immunological cross-reactivity [6, 8, 13]. Within a serogroup, the slpA nucleotide sequences show little variability [19]. These observations are consistent with mathematical models predicting that intense immune selection will polarize strains into distinct, non–cross-reactive antigenic types [20]. A second major C. difficile cell surface antigen is Cwp66, an adhesin that is also a member of the cwp cluster, exhibits high genetic diversity, and elicits a strong immune response [7, 21]. Both Cwp66 and S-layer proteins are exported from the cell by a secretory system, the specificity of which is redirected by the protein SecA2 [22]. SecA2 is encoded within the cwp cluster between cwp66 and slpA [14] (Figure 1A).
Figure 1.

Density of polymorphism in the cwp cluster and phylogeny of whole genomes and S-layer. A, Genetic organization in and around the cwp cluster. B, Density of polymorphism across the cwp cluster. C, Phylogeny constructed from the whole genomes. Solid black shapes indicate the clades. D, Phylogeny constructed from the slpA gene. Shapes indicate the clade, and colors indicate the S-layer variant with the exception of 2 hybrids, designated H and indicated by a star. Numbers indicate bootstrap support. Abbreviation: ORF, open reading frame.
Meaningful investigation of the diversity and evolution of bacterial antigenic determinants requires the rational choice of representative isolates. This is facilitated by combining molecular epidemiology and clinical phenotype data with population structure information [23]. We used this approach to select isolates for detailed genomic analysis [3] to discover the extent of cwp cluster genetic diversity and investigate how the cwp cluster evolves within the species.
METHODS
Isolates and Genotyping
Isolates were cultured from C. difficile positive stool samples identified by enzyme immunoassay (EIA; Premier Toxins A&B Enzyme Immunoassay; Meridian Bioscience Europe, Milan, Italy) at the Clinical Microbiology Laboratory, Oxford University Hospitals NHS Trust, Oxford, and by cytotoxin testing at the Leeds Teaching Hospitals NHS Trust, Leeds. A total of 1001 Oxford isolates were cultured between 16 September 2006 and 6 August 2010, and 84 Leeds isolates cultured between 12 October 2006 and 12 March 2008, together with five additional PCR-ribotype reference isolates were included (Table 1). Primary culture and genotyping by multilocus sequence typing (MLST) and PCR-ribotyping were performed as described elsewhere [24]. The notation ST1[4] was adopted to indicate ST1[S-layer cassette variant], and ST1(027) was adopted to indicate ST1 (PCR ribotype).
Table 1.
Isolates, Genotypes, and EMBL Short Read Archive (SRA) Sample Numbers
| Genotype |
SRA Sample No. | |||||||
|---|---|---|---|---|---|---|---|---|
| Genome ID | ST | S-Layer | Clade | PCR Rib | Tox | Isolate ID | Isolation Date | |
| 0090 | 1 | 4 | 2 | 027 | + | Ox160 | 29 Jan 2007 | ERS139371 |
| 0098 | 2 | 10 | 1 | 020 | + | Ox709 | 26 Sep 2007 | ERS139373 |
| 6417 | 2 | 12 | 1 | 014 | + | Ox1475 | 6 Sep 2008 | ERS139420 |
| 6019 | 3 | 1 | 1 | 305 | + | Ox428 | 14 Jun 2007 | ERS139415 |
| 0193 | 3 | 4 | 1 | 001/072 | + | Ox1867 | 13 Apr 2009 | ERS139380 |
| 0551 | 3 | 8 | 1 | 262 | − | Ox1003 | 23 Jan 2008 | ERS139388 |
| 0039 | 4 | 7 | 1 | 137 | + | Ox1145 | 20 Mar 2008 | ERS139368 |
| 5010 | 5 | 10 | 3 | 069 | + | Ox2183 | 18 Oct 2009 | ERS139411 |
| 0568 | 5 | 11 | 3 | 023 | + | Ox1523 | 26 Sep 2008 | ERS139391 |
| 0034 | 6 | 11 | 1 | 005 | + | Ox247 | 12 Mar 2007 | ERS139367 |
| 0561 | 7 | 5 | 1 | 026 | + | Ox1437a | 14 Aug 2008 | ERS139389 |
| 0117 | 8 | 9 | 1 | 002 | + | Ox1192c | 14 Apr 2008 | ERS139374 |
| 0096 | 10 | 4 | 1 | 015 | + | Ox660 | 11 Sep 2007 | ERS139372 |
| 2448 | 11 | 3 | 5 | 033 | + | L033a | 7 Jun 2008b | ERS139399 |
| 2471 | 11 | 8 | 5 | 066 | + | 7721734 | 24 Nov 2009 | ERS139402 |
| 2490 | 11 | 8 | 5 | 045 | + | L045a | 7 Jun 2008b | ERS139405 |
| 1729 | 11 | H2/6 | 5 | 078 | + | Ox575 | 14 Aug 2007 | ERS139395 |
| 2387 | 11 | H2/6 | 5 | 193 | + | L16/26 | 19 Oct 2006 | ERS139396 |
| 0562 | 12 | 1 | 1 | 003 | + | Ox1424 | 1 Aug 2008 | ERS139390 |
| 0149 | 13 | 10 | 1 | 129 | + | Ox1533 | 1 Oct 2008 | ERS139376 |
| 0160 | 14 | 6 | 1 | 014 | + | Ox1463 | 27 Aug 2008 | ERS139377 |
| 5172 | 15 | 12 | 1 | 010 | − | Ox1342 | 10 Jun 2008 | ERS139412 |
| 4139 | 16 | 2 | 1 | 029 | + | Ox858 | 30 Nov 2007 | ERS139409 |
| 6483 | 16 | 11 | 1 | 050 | + | Ox590 | 30 Oct 2008 | ERS139421 |
| 0143 | 17 | 6 | 1 | 018 | + | Ox1192e | 14 Apr 2008 | ERS139375 |
| 1506 | 22 | 11 | 3 | 023 | + | Ox561 | 7 Aug 2007 | ERS139393 |
| 6282 | 23 | 10 | 4 | 138 | − | Ox904 | 18 Dec 2007 | ERS139416 |
| 2431 | 32 | 9 | 2 | 153 | + | L28/45 | 31 May 2007 | ERS139397 |
| 0182 | 35 | 3 | 1 | 284 | + | Ox1121 | 9 Mar 2008 | ERS139379 |
| 2958 | 36 | 4 | 1 | 011 | + | Ox1916 | 22 May 2009 | ERS139407 |
| 4350 | 36 | 8 | 1 | 011 | + | Ox2553 | 7 May 2010 | ERS139410 |
| 0089 | 37 | 7 | 4 | 017 | + | Ox920 | 26 Dec 2007 | ERS139370 |
| 6346 | 39 | 2 | 4 | 085 | − | L085a | 7 Jun 2008b | ERS139418 |
| 0224 | 41 | 4 | 2 | 106 | + | Ox1307 | 30 May 2008 | ERS139382 |
| 1728 | 41 | 5 | 2 | 194 | + | Ox2287 | 10 Dec 2009 | ERS139394 |
| 2439 | 41 | 10 | 2 | 321 | + | Ox1802b | 28 Feb 2009 | ERS139398 |
| 0175 | 42 | 10 | 1 | 106 | + | Ox687 | 19 Sep 2007 | ERS139378 |
| 0516 | 43 | 11 | 1 | 054 | + | Ox1844 | 28 Mar 2009 | ERS139385 |
| 0477 | 44 | 5 | 1 | 015 | + | Ox990 | 21 Jan 2008 | ERS139383 |
| 0024 | 44 | 9 | 1 | 015 | + | Ox466 | 6 Jul 2007 | ERS139366 |
| 5740 | 45 | 1 | 1 | 454 | + | Ox2294 | 15 Dec 2009 | ERS139413 |
| 0061 | 45 | 10 | 1 | 013 | + | Ox887 | 12 Dec 2007 | ERS139369 |
| 5984 | 47 | 11 | 2 | 375 | + | Ox2621b | 17 Jun 2010 | ERS139414 |
| 6353 | 48 | 7 | 1 | 038 | − | Ox2167 | 12 Oct 2009 | ERS139419 |
| 6325 | 49 | 10 | 1 | 014 | + | Ox593 | 14 Aug 2007 | ERS139417 |
| 0526 | 54 | 1 | 1 | 012 | + | Ox1567 | 24 Oct 2008 | ERS139386 |
| 2479 | 54 | 7 | 1 | 012 | + | Ox2478 | 26 Mar 2010 | ERS139403 |
| CD630 | 54 | 7 | 1 | 012 | + | CD630 | 1982 | NC009089 |
| 0529 | 54 | 8 | 1 | 012 | + | Ox1396 | 16 Jul 2008 | ERS139387 |
| 0509 | 58 | 8 | 1 | 056 | + | Ox707 | 26 Sep 2007 | ERS139384 |
| 1463 | 59 | 3 | 1 | 316 | + | Ox1893 | 4 May 2009 | ERS139392 |
| 2482 | 63 | 1 | 1 | 053 | + | Ox1918 | 25 May 2009 | ERS139404 |
| 2975 | 66 | 11 | 1 | 022 | + | Ox2124 | 11 Sep 2009 | ERS139408 |
| 2493 | 67 | 7 | 2 | 019 | + | Ox2163 | 7 Oct 2009 | ERS139406 |
| 0197 | 77 | 11 | 1 | 011 | + | Ox2017 | 22 Jul 2009 | ERS139381 |
| 2460 | 95 | 11 | 2 | 075 | + | L075a | 7 Jun 2008b | ERS139401 |
| 2452 | 96 | 11 | 3 | 058 | + | L058a | 7 Jun 2008b | ERS139400 |
| 6877 | 160 | 11 | 1 | 015 | + | L86/78 | 27 Feb 2009 | ERS139422 |
The project Web site is available at: http://www.ebi.ac.uk/ena/data/view/ERP001417.
Abbreviations: ID, identification; L, Leeds; Ox, Oxford; PCR Rib, polymerase chain reaction ribotype; ST, sequence type; Tox, toxigenic; –, negative; +, positive.
a PCR-ribotyping reference library isolate.
b Date received at Leeds laboratory.
DNA Sequencing
DNA was extracted from bacterial cells that had been cultured anaerobically on Columbia blood agar for 48 hours at 37°C, using commercial kits (FastDNA, MP Biomedicals, CA; QuickGene, Fujifilm Europe, Dusseldorf, Germany; or QIAamp, Qiagen, Hilden, Germany). DNA was sequenced using Illumina (San Diego, CA) sequencing-by-synthesis technology [3], frequently referred to as “next-generation DNA sequencing.” A combination of standard Illumina and in-house protocols were used to produce multiplexed paired-end libraries with an average insert size of approximately 200 bp. Twelve-plex pooled libraries were sequenced on the Genome Analyzer II (GAII) or GAIIx platforms to produce 51 bp or 100–108 bp reads, respectively, and 96-plex pooled libraries were sequenced on the HiSeq2000 platform to produce 99 bp or 100 bp paired reads.
De Novo Assembly of Sequence Data
All genomes were assembled de novo using Velvet [25]. Velvet Optimiser was used to determine optimal parameter values. Assemblies had an average of 703 contigs. We assembled the genomes de novo, without using a reference genome template, so that insertions and deletions would not be missed and regions of high genetic diversity would be assembled correctly.
Analysis of Whole-Genome and Partial-Genome Sequences
A total of 57 isolates were chosen for detailed phylogenetic analysis; they represented the 5 clades of the C. difficile population structure and a range of recognized clinical phenotypes [10–12, 23]. A whole-genome Neighbor-joining tree was constructed as follows, on the basis of de novo sequence assemblies. BLAST was used to search the 57 genomes for 3742 genes previously annotated in CD630 [14]. A total of 1639 of these genes were found to be present (with ≥90% sequence coverage) in all 57 genomes, and these sequences were used to build the tree (Figure 1C).
The entire de novo genome assemblies were uploaded into a Bacterial Isolate Genome Sequence Database (BIGSdb) [26]. BIGSdb allows simultaneous BLAST searching of user-defined loci in the genome assemblies of multiple isolates. In this way, open reading frames (ORFs) of interest, such as slpA, were tagged, extracted, and then aligned using ClustalW [27]. Neighbor-joining trees were then constructed from this alignment, using MEGA version 4 [28].
The 24-kb insertion of S-layer cassette 11 was translated using BioEdit Sequence Alignment Editor [29] and Artemis genome browser and annotation tool [30]. A BLAST search of the predicted translation products against GenBank was used to identify related proteins, thus predicting functions of the ORFs and the species in which they occurred (Table 2).
Table 2.
Predicted Functions of Proteins Encoded by 19 Open Reading Frames (ORFs) of the Putative S-Layer Glycosylation Gene Cluster
| ORFa (nt) | Gene | Putative Function | Closest Species Match | GenBank Accession No. | Amino Acid (%) |
GC Content (%) | |
|---|---|---|---|---|---|---|---|
| Identity | Similarity | ||||||
| 1 (408) | … | Hypothetical protein | Clostridium difficile | ZP_05400366.1 | 57 | 84 | 29.9 |
| 2 (1395) | … | Undecaprenyl-phosphate glucose phosphotransferase | Clostridium lentocellum | YP_004310697.1 | 57 | 74 | 23.9 |
| 3 (969) | … | Glycosyl transferase | Lachnospiraceae bacteria | ZP_08602110.1 | 56 | 75 | 24.8 |
| 4 (891) | … | Phosphoribose diphosphate: decaprenyl-phosphate phosphoribosyltransferase | C. difficile | YP_001087463.1 | 65 | 81 | 24.5 |
| 5 (1797) | … | Hypothetical protein | Tolumonas auensis | YP_002893223.1 | 26 | 45 | 26.2 |
| 6 (1332) | … | Glycosyl transferase | Xanthomonas perforans | ZP_08189725.1 | 49 | 65 | 26.6 |
| 7 (3486) | |||||||
| N-terminus | … | Multidomain glycosyl transferase | Bacteroides species | ZP_08584442.1 | 32 | 54 | 26.3b |
| C-terminus | … | Multidomain glycosyl transferase | Polynucleobacter necessarius | YP_001155105.1 | 51 | 68 | 26.3b |
| 8 (1161) | … | Glycosyl transferase | Aromatoleum aromaticum | YP_157889.1 | 50 | 65 | 28.7 |
| 9 (1383) | … | Glycosyl transferase | Clostridium beijerinckii | YP_001311811.1 | 34 | 58 | 22.4 |
| 10 (909) | … | Glycosyl transferase | C. beijerinckii | YP_001311812.1 | 67 | 81 | 22.4 |
| 11 (1283) | wzt | ATP binding cassette (ABC) transporter: ATPase | C. beijerinckii | YP_001311813.1 | 65 | 81 | 26.6 |
| 12 (789) | wzm | ABC transporter: permease | Eubacterium yurii | ZP_07454366.1 | 68 | 84 | 25.6 |
| 13 (1023) | rmlBc | dTDP-glucose 4,6-dehydratase | Turicibacter species | ZP_08166203.1 | 73 | 88 | 27.7 |
| 14 (573) | rmlCc | dTDP-4-dehydrorhamnose 3,5-epimerase | Clostridium perfringens | ZP_02863393.1 | 72 | 86 | 28.0 |
| 15 (924) | … | Phosphogluconolactonase; provisional | Lachnospiraceae bacteria | ZP_08605193.1 | 34 | 59 | 25.4 |
| 16 (879) | rmlAc | Glucose-1-phosphate thymidylyltransferase | Turicibacter species | ZP_08166199.1 | 83 | 93 | 30.3 |
| 17 (681) | rmlDc | dTDP-4-dehydrorhamnose reductase | C. difficile | ZP_05328363.1 | 69 | 86 | 27.2 |
| 18 (816) | rfbNc | Rhamnosyltransferase | Thermoanaerobacter pseudethanolicus | YP_001665609.1 | 50 | 70 | 25.5 |
| 19 (1608) | wzyC | O-Antigen polymerase/ligase | Geobacillus thermoglucosidasius | YP_004586423.1 | 33 | 54 | 23.2 |
C. difficile was found as the closest species match to 3 ORFS. However, although the low level of amino acid identity indicates a similar function for the ORF (eg, an alternative rhamnose pathway in the case of rmlD), the 3 ORFs were widely dispersed in the CD630 reference genome [14]. Thus, these low-level matches do not indicate the presence of another putative S-layer glycosylation cluster. Putative functions of the predicted ORFs were inferred using the entire results of the BLAST query, rather than just the closest match shown in Table 2.
Abbreviations: nt, nucleotide; ORF, open reading frame.
a Numbered as in Figure 3.
b Includes both the N-terminus and C-terminus species matches.
c Rhamnose pathway.
The Artemis Comparison Tool (ACT) [31] was used to represent the relative position of the genes in the reference genome CD630 [14] and in a genome containing the glycosylation cluster (Figure 3).
Figure 3.
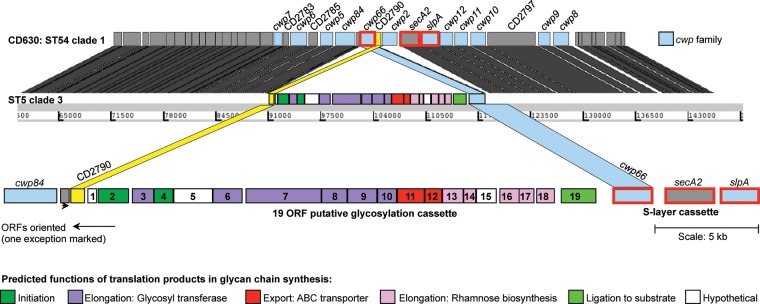
Annotation of the putative S-layer glycosylation cluster within ST5 relative to the reference genome CD630 [14]. The annotation of CD630 is shown at the top, and the location of each CD630 gene in the ST5 genome is shown using grey blocks. Color has been used to highlight the glycosylation cluster insertion, the rearrangement of the 2 flanking open reading frames (ORFs), and the deletion of cwp2. Annotation of the genes in the glycosylation cluster is given at the bottom. Numbers 1–19 indicate the ORFs of the glycosylation cluster as in Table 2.
Analyses Using a Reference-Anchored Multiple Alignment
Each de novo assembled genome was aligned separately against the reference genome CD630 [14] by using progressive Mauve [32], and reference-anchored alignments were extracted and combined to produce a multiple-way alignment of all genomes, in which each alignment column corresponded to a genomic position of CD630. The density of polymorphic sites in the part of this alignment corresponding to the cwp cluster of CD630 was computed. Distributions of polymorphisms between pairs of isolates were plotted along the whole CD630 genome, using DNAplotter [33] (Figure 4A), and around the cwp cluster (Figure 4B). Ratios of nonsynonymous-to-synonymous mutations (dN/dS) were calculated for selected genes of the cwp cluster on the basis of the reference-anchored alignment, using the method of Nei and Gojobori [34].
Figure 4.

S-layer switching at the chromosomal scale and at the scale of the cwp cluster. A, Whole-genome distributions of polymorphism between pairs of isolates very closely related in terms of whole-genome phylogeny (Figure 1C) but with distinct S-layer cassettes (Figure 1D and Figure 2A and 2B). The 2 outer rings composed of small red lines indicate the open reading frames (ORFs) annotated on the forward and reverse strands of reference genome CD630 [14]. The left-hand plot shows, from inside out, pairs ST10[4]/44[5], ST54[8]/54[1], and ST14[6]/49[10]; the middle plot shows, from inside out, ST41[5]/41[10], ST11(078)[H]/ST11(033)[3], and ST11(078)[H]/ST11(066)[8]; and the right-hand plot shows, from inside out, ST2[10]/ST2[12],ST36[4]/ST36[8], and ST160 [11]/ST44[5]. Blue highlighting indicates the location of the cwp cluster within the chromosome. B, Distribution of polymorphism between the pairs of genomes shown in (A) within the region of the genome containing the cwp cluster. By enlarging and comparing only the region of the cwp cluster involved in S-layer switching (shown in blue in panel A), the distribution of polymorphisms can be used to estimate the size of recombination events. Each row represents a pairwise comparison of 2 isolates, and polymorphisms are shown in red. Abbreviation: SNV, single nucleotide variant.
Nucleotide Sequence Accession Numbers
The 57 genomes that underwent detailed phylogenetic analysis (Table 1) were submitted to the EBI short read archive under the project accession number ERP001417, available at http://www.ebi.ac.uk/ena/data/view/ERP001417. Accession numbers for individual genomes are listed in Table 1. The annotated DNA sequence containing the putative S-layer glycosylation gene cluster (Figure 3) was submitted to EMBL (accession number HE980331).
Ethics Statement
This study included bacterial isolates for which no corresponding patient data were used. However, the isolates were collected without written informed consent as part of a larger study of C. difficile transmission across the health economy, for which ethical permission was obtained from the Berkshire Ethics Committee (10/H0505/83) and the UK National Information Governance Board (8–05(e)/2010).
RESULTS
Choice of Isolates and Genomes
Whole-genome sequences (WGS) were available for 1085 recent clinical isolates (1001 from Oxford and 84 from Leeds) and 5 PCR-ribotype reference isolates (Table 1). Genotyping was performed by MLST; the sequence types (STs) were determined by conventional MLST [23, 24] or were extracted from WGS using BIGSdb [26]. STs were consistent by both methods. The slpA gene of each isolate was also extracted from WGS by using BIGSdb, or it was identified from its genetic context within the cwp cluster if it was genetically divergent from known sequences (Figure 1A) [14].
The genotype and slpA sequence data were used to choose 57 isolates for detailed phylogenetic study (Table 1). Isolate choice was based on 2 considerations. First, isolates were chosen to represent known characteristics of different genotypes, including the spectrum of virulence and clinical prevalence [11, 12, 35, 36] and their evolutionary context within the C. difficile population structure [23]. Second, if a ST occurred with >1 highly genetically divergent slpA variant, a representative of each combination was included. Hence, 6 STs with 2 highly divergent slpA variants and 4 STs with 3 slpA variants were included (Table 1).
cwp Cluster Genetic Diversity
The diversity of the cwp cluster and flanking region (total, 63.5 kb; Figure 1A) was examined using a nucleotide sequence alignment generated from the 57 isolates and reference strain CD630 [14]. Diversity peaked across the central approximately 10 kb, which included cwp66, secA2, and slpA (Figure 1B). The ratio of nonsynonymous to synonymous point mutations (dN/dS) was equal to 1.03 in the outer LMW fragment of the S-layer protein, indicating a strong relaxation of the purifying selection at work elsewhere in the genome.
Phylogenetic Relationships
The phylogeny of the 58 isolates based on the core genome (genes common to all) showed the same 5 clades of the population structure as determined using MLST data (Figure 1C) [23]. This structure was also preserved in genes flanking the cwp cluster. However, with increasing proximity to the cwp66, secA2, and slpA genes, an entirely different interstrain relationship emerged. A neighbor-joining tree for the slpA gene of the 57 isolates and CD630 identified 12 distinct clusters (Figure 1D), rather than the usual 5 clades. Furthermore, neighbor-joining trees for the cwp66 adhesin and secA2 translocase clustered these 58 isolates identically to slpA (Figure 2A and 2B). These data suggest that slpA, cwp66, and secA2 coevolve as an approximately 10-kb gene cassette, which has at least 12 genetically divergent variants. Most variants of this S-layer cassette occurred in multiple clades, indicating independent dispersal of the cassette throughout the C. difficile population by frequent horizontal genetic exchange. Only 2 isolates (ST11(078) and ST11(193)) contained a hybrid cassette comprising the cwp66 gene of one cassette and the secA2 and slpA genes of another (Figure 1D and Figure 2A and 2B). A comparison of cwp cluster sequences from 4 genomes (ST1(027), ST3(001), ST37(017), ST54(012)) with older isolates of the same ST and S-layer cassette [14, 37] indicated a very low rate of point mutation accumulation over time.
Figure 2.

Phylogeny of cwp66 and secA2. A and B, Phylogeny constructed from cwp66 (A) and secA2 (B). Shapes indicate the clade of each genome (Figure 1C). S-layer cassette variants are shown using colors, with the exception of 2 hybrids, indicated by a star. Numbers indicate bootstrap support.
Glycosylation Gene Cluster
A novel insertion of 23.8 kb containing 19 predicted ORFs was identified within one of the S-layer cassettes (variant 11, associated with 11 STs; Figure 3). The insertion represented a putative S-layer glycosylation gene cluster, encoding enzymes required to initiate, extend membrane translocate and ligate a glycan chain to a substrate molecule (Table 2). These components are typical of the 4 S-layer glycosylation gene clusters described to date, which are found in nonpathogenic Gram-positive bacilli [15]. No such clusters have been described in Clostridium species. The putative C. difficile S-layer glycosylation cluster exhibited detectable amino acid sequence homologies with 2 of the Bacillus species S-layer glycosylation gene clusters (Supplementary Figure 1) [15].
The order of 2 cwp cluster genes flanking the 23.8-kb insertion (cwp66 and CD2790) was reversed, and the gene cwp2 was deleted (Figure 3), bringing together the 3 genes (cwp66, secA2, and slpA) of the S-layer cassette. The slpA gene of cassette 11 was also uniquely short, at 611 amino acids, compared with 720 amino acids for CD630. Cassette 11 occurred in 3 different clades (1, 2, and 3), indicating that horizontal genetic exchange had occurred involving a DNA fragment at least 34 kb containing cwp66, secA2, slpA, and the associated glycosylation cluster.
Scale of Recombination Events
The uniqueness and scale of the recombination events causing S-layer cassettes to be exchanged were visualized at the chromosome level for selected pairs of isolates (Figure 4A). By comparing the polymorphisms between the cwp clusters of these otherwise closely related pairs, the size of the recombination events was inferred as 12.9–35.2 kb (Figure 4B). A region of approximately 12 kb containing the S-layer cassette was identified, which remained unbroken (Figure 4B). This is consistent with the congruence of neighbor-joining trees for cwp66, secA2, and slpA (Figure 1D), further supporting the genetic coevolution of these loci as 12 independently reassorting S-layer cassettes.
DISCUSSION
Although evolutionary changes in immunodominant cell surface antigens are likely critical to host-pathogen interactions, the extremely high level of genetic polymorphisms in the C. difficile slpA and cwp66 genes has precluded large-scale population-wide comparisons by conventional Sanger sequencing. We have overcome this limitation by using newly available DNA sequencing methods [3] to determine whole pathogen genome sequences. These data have confirmed the existence of 5 clades in C. difficile and identified 12 stable variants of the approximately 10-kb S-layer cassette. The cassettes contain the highly diverse (Figure 1B) and functionally interrelated slpA, cwp66, and secA2 genes [6, 7, 22]. Unexpectedly, the clades and S-layer cassettes associated randomly, effectively behaving as independent components of the genome in linkage equilibrium (Figure 1). The underlying mechanism is the genetic replacement of one S-layer cassette with another by homologous recombination involving DNA fragments of 12–35 kb (Figure 4). We refer to this process as S-layer switching because it parallels polysaccharide capsular switching in other bacterial species [1, 2]. Both S-layer and capsular switching result in major antigenic shifts, while the remainder of the genome is unchanged.
The extreme genetic diversity between S-layer cassettes contrasts with the low rate of point mutation accumulation within each cassette, with isolates of the same variant showing very little diversity. These observations are consistent with well-established mathematical models of strong immune selection [20], which predict that the collective immune responses of the host population shape pathogen antigenic diversity into nonoverlapping, independently transmitted antigenic types. Such models predict that intermediate antigenic types that cross-react serologically will be lost from the population through competitive exclusion. The high diversity of the slpA and cwp66 genes and higher dN/dS ratio for the LMW S-layer domain are consistent with this region being under intense diversifying selective pressure. In addition to host immunity, the high selective pressure may also reflect the need for successful strains to avoid bacteriophage attack [38].
An interesting question is whether there is any evidence of association between S-layer cassette and hypervirulent phenotype. We found that the S-layer cassettes of ST1(027) (a major hypervirulent strain identified in 2005 [11]) and ST3(001) (a previous United Kingdom epidemic strain [39]) were very closely related (Figure 1D), consistent with previous reports [36]. However, this S-layer cassette also occurred in a third clinically abundant genotype, ST10(015), and in 2 clinically rare genotypes (Figure 1D, and Figure 2; unpublished data) [35]. The fact that 3 clinically important genotypes share closely related S-layer cassettes suggests that the hypervirulence of ST1(027) may be influenced but not solely conferred by this feature. The S-layer cassette of the more recently identified hypervirulent ST11(078) [12] is noteworthy since it represents the only example of a hybrid cassette; its cwp66 gene is homologous to cassette 2 cwp66, and its slpA and secA2 genes are similar to those of cassette 6 (Figure 1D and Figure 2). On the basis of nucleotide sequence data, this hybrid would be predicted to cross-react immunologically with its 2 parent cassettes [7, 19]. This contradicts model predictions [20] that cross-reactive variants should be removed from the population by competitive exclusion. The hybrid was also found in clinically rare ST11(193) [35], which may have evolved from ST11(078) (Figure 1C). Its separate cwp66 and secA2/slpA elements were closely related to variants found in nonhypervirulent STs (Figure 1D and Figure 2A and 2B), arguing against a role for the hybrid cassette in the hypervirulent phenotype of PCR-ribotype 078. However, the recombination affecting the ST11(078) S-layer cassette seems to have occurred relatively recently because the chromosomes of the ST11 genomes showed few differences outside this region (Figure 4). This recombination may be coincident with the emergence of ST11(078) as a causative agent of severe CDI. Overall, our data do not exclude the possibility that S-layer variants can affect clinical phenotype.
This study contradicts previous smaller studies suggesting that traditional genotyping predicts antigenic type [36, 40]. By including a large number of genomes (1090, of which 1085 were from recent clinical isolates) in our initial screen, we were able to demonstrate that isolates sharing the same ST and PCR-ribotype can carry multiple distinct S-layer cassettes. For example, PCR-ribotype 015 was found in association with 4 different S-layer cassettes, and 4 STs (ST3, ST11, ST41, and ST54) occurred with 3 different S-layer cassettes (Figure 1 and Table 1). This demonstrates that the rate of S-layer switching exceeds the rate at which new genotypes (as defined by PCR ribotype or ST) are generated. It also establishes conclusively that genotype is an unreliable predictor of antigenic type. Our observations have important implications for the way in which isolates and subsequently antigens are identified for potential inclusion in C. difficile vaccines and for the development of C. difficile phage therapy [41].
The use of de novo assembled sequences in this study (rather than reference-based assemblies) revealed an unexpected insertion of approximately 24 kb in one S-layer cassette (variant 11). Its 19 ORFs were predicted to encode all of the components that characterize an S-layer glycosylation cluster (Figure 3) [15], including a rhamnose biosynthesis pathway [42]. This sugar is unique to bacteria and therefore represents an attractive drug target. The genes flanking cassette 11 were swapped, bringing together the 3 genes cwp66, secA2, and slpA (Figure 3). The 19 ORFs of the insertion were present and intact in all 11 STs in which it was found, which represented 3 of the 5 clades. This suggests that its approximately 34-kb sequence participates in S-layer switching as a single genetic element (Figure 4). S-layer cassette 11 is so unusual that it may have originated in a distinct and as yet unidentified species and then incorporated into the C. difficile chromosome by interspecies recombination. This is the first description of a putative S-layer glycosylation gene cluster in a Gram-positive human pathogen or in any Clostridium species. It may significantly alter the characteristics of the outer cell surface, with potentially important effects on antigenicity, antibiotic permeability, and virulence.
Our findings demonstrate the novel insights to be gained by exploiting WGS for the cross-population analysis of important bacterial pathogens. Knowledge of the evolutionary process of S-layer switching could help explain temporal changes and geographic differences in the epidemiology of CDI [35, 43] and will potentially influence future genotyping schemes, as well as therapeutic and vaccination strategies. Further insights into the impact of C. difficile evolution on the epidemiology of this clinically important human pathogen will require longitudinal isolate collections from different geographical areas, concomitant with changes in incidence and the clinical severity of CDI.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org/). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank the staff of the Clinical Microbiology Laboratory and Infection Control, John Radcliffe Hospital, Oxford, and the staff of the Infection Control Laboratory staff, Leeds General Infirmary, for their assistance throughout this work. This publication made use of the Clostridium difficile Multilocus Sequence Typing Web site (available at: http://pubmlst.org/cdifficile/sited) at the Department of Zoology, University of Oxford. The development of this Web site has been funded by the Wellcome Trust.
Financial support. This work was supported by the Oxford NIHR Biomedical Research Centre (Senior Investigator Award to T. E. P. and D. W. C. and doctoral research fellowship to D. W. E.); by the UKCRC Modernising Medical Microbiology Consortium, which is funded under the UKCRC Translational Infection Research Initiative and supported by the Medical Research Council, the Biotechnology and Biological Sciences Research Council, and the National Institute for Health Research on behalf of the United Kingdom Department of Health (grant G0800778) and The Wellcome Trust (grant 087646/Z/08/Z); and by the Engineering and Physical Sciences Research Council (to M. A. A.).
Potential conflicts of interest. D. W. C. and T. E. P. have received per-patient grant funding from Optimer Pharmaceuticals. M. H. W. has received honoraria for consultancy work, financial support to attend meetings, and research funding from Actelion, Astellas, Astra-Zeneca, bioMerieux, Cerexa, Cubist, Merck, Novacta, Optima, Pfizer, Summit, and The Medicines Company. All other authors report no potential conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed
References
- 1.Croucher NJ, Harris SR, Fraser C, et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 2011;331:430–34. doi: 10.1126/science.1198545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayer LW, Reeves MW, Al-Hamdan N, et al. Outbreak of W135 meningococcal disease in 2000: not emergence of a new W135 strain but clonal expansion within the electophoretic type-37 complex. J Infect Dis. 2002;185:1596–605. doi: 10.1086/340414. [DOI] [PubMed] [Google Scholar]
- 3.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–9. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pallen MJ, Loman NJ, Penn CW. High-throughput sequencing and clinical microbiology: progress, opportunities and challenges. Curr Opin Microbiol. 2010;13:625–31. doi: 10.1016/j.mib.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Bauer MP, Notermans DW, van Benthem BH, et al. Clostridium difficile infection in Europe: a hospital-based survey. Lancet. 2011;377:63–73. doi: 10.1016/S0140-6736(10)61266-4. [DOI] [PubMed] [Google Scholar]
- 6.Calabi E, Ward S, Wren B, et al. Molecular characterization of the surface layer proteins from Clostridium difficile. Mol Microbiol. 2001;40:1187–99. doi: 10.1046/j.1365-2958.2001.02461.x. [DOI] [PubMed] [Google Scholar]
- 7.Waligora AJ, Hennequin C, Mullany P, Bourlioux P, Collignon A, Karjalainen T. Characterization of a cell surface protein of Clostridium difficile with adhesive properties. Infect Immun. 2001;69:2144–53. doi: 10.1128/IAI.69.4.2144-2153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calabi E, Fairweather N. Patterns of sequence conservation in the S-Layer proteins and related sequences in Clostridium difficile. J Bacteriol. 2002;184:3886–97. doi: 10.1128/JB.184.14.3886-3897.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McGowan AP, Lalayiannis LC, Sarma JB, Marshall B, Martin KE, Welfare MR. Thirty-day mortality of Clostridium difficile infection in a UK National Health Service Foundation Trust between 2002 and 2008. J Hosp Infect. 2011;77:11–5. doi: 10.1016/j.jhin.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 10.Merrigan M, Venugopal A, Mallozzi M, et al. Human hypervirulent Clostridium difficile strains exhibit increased sporulation as well as robust toxin production. J Bacteriol. 2010;192:4904–11. doi: 10.1128/JB.00445-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonald LC, Killgore GE, Thompson A, et al. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med. 2005;353:2433–41. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 12.Goorhuis A, Bakker D, Corver J, et al. Emergence of Clostridium difficile infection due to a new hypervirulent strain, polymerase chain reaction ribotype 078. Clin Infect Dis. 2008;47:1162–70. doi: 10.1086/592257. [DOI] [PubMed] [Google Scholar]
- 13.Cerquetti M, Molinari A, Sebastianelli A, et al. Characterization of surface layer proteins from different Clostridium difficile clinical isolates. Microb Pathog. 2000;28:363–72. doi: 10.1006/mpat.2000.0356. [DOI] [PubMed] [Google Scholar]
- 14.Sebaihia M, Wren BW, Mullany P, et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 2006;38:779–86. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 15.Ristl R, Steiner K, Zarschler K, Zayni S, Messner P, Schäffer C. The S-layer glycome-adding to the sugar coat of bacteria. Int J Microbiol. 2011;pii:127870. doi: 10.1155/2011/127870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qazi O, Hitchen P, Tissot B, et al. Mass spectrometric analysis of the S-layer proteins from Clostridium difficile demonstrates the absence of glycosylation. J Mass Spectrom. 2009;44:368–74. doi: 10.1002/jms.1514. [DOI] [PubMed] [Google Scholar]
- 17.Wright A, Drudy D, Kyne L, Brown K, Fairweather NF. Immunoreactive cell wall proteins of Clostridium difficile identified by human sera. J Med Microbiol. 2008;57:750–56. doi: 10.1099/jmm.0.47532-0. [DOI] [PubMed] [Google Scholar]
- 18.Ryan A, Lynch M, Smith SM, et al. A role for TLR4 in Clostridium difficile infection and the recognition of surface layer proteins. PLoS Pathog. 2011;7:e1002076. doi: 10.1371/journal.ppat.1002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karjalainen T, Saumier N, Barc MC, Delmee M, Collignon A. Clostridium difficile genotyping based on slpA variable region in S-layer gene sequence: an alternative to serotyping. J Clin Microbiol. 2002;40:2452–58. doi: 10.1128/JCM.40.7.2452-2458.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta S, Maiden MC. Exploring the evolution of diversity in pathogen populations. Trends Microbiol. 2001;9:181–85. doi: 10.1016/s0966-842x(01)01986-2. [DOI] [PubMed] [Google Scholar]
- 21.Péchiné S, Gleizes A, Janoir C, et al. Immunological properties of surface proteins of Clostridium difficile. J Med Microbiol. 2005;54:193–96. doi: 10.1099/jmm.0.45800-0. [DOI] [PubMed] [Google Scholar]
- 22.Fagan RP, Fairweather NF. Clostridium difficile has two parallel and essential Sec secretion systems. J Biol Chem. 2011;286:27483–93. doi: 10.1074/jbc.M111.263889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dingle KE, Griffiths D, Didelot X, et al. Clinical Clostridium difficile: clonality and pathogenicity locus diversity. PLoS One. 2011;6:e19993. doi: 10.1371/journal.pone.0019993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griffiths D, Fawley W, Kachrimanidou M, et al. Multilocus sequence typing of Clostridium difficile. J Clin Microbiol. 2010;48:770–78. doi: 10.1128/JCM.01796-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–29. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jolley KA, Maiden MC. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li KB. ClustalW-MPI: ClustalW analysis using distributed and parallel computing. Bioinformatics. 2003;19:1585–86. doi: 10.1093/bioinformatics/btg192. [DOI] [PubMed] [Google Scholar]
- 28.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–99. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 29.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- 30.Rutherford K, Parkhill J, Crook J, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–45. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 31.Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–23. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- 32.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carver T, Thomson N, Bleasby A, Berriman M, Parkhill J. DNA Plotter: circular and linear interactive genome visualization. Bioinformatics. 2009;25:119–20. doi: 10.1093/bioinformatics/btn578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Molecular Biology and Evolution. 1986;3:418–26. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 35.Health Protection Agency. Clostridium difficile Ribotyping Network (CDRN) for England and Northern Ireland 2009/2010 report. http://www.hpa.org.uk/webc/HPAwebFile/HPAweb_C/1296681523205. Accessed 14 June 2012. [Google Scholar]
- 36.Spigaglia P, Galeotti CL, Barbanti F, Scarselli M, Van Broeck J, Mastrantonio P. The LMW surface-layer proteins of Clostridium difficile PCR ribotypes 027 and 001 share common immunogenic properties. J Med Microbiol. 2011;60:1168–73. doi: 10.1099/jmm.0.029710-0. [DOI] [PubMed] [Google Scholar]
- 37.He M, Sebaihia M, Lawley TD, et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci U S A. 2010;107:7527–32. doi: 10.1073/pnas.0914322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brockhurst MA, Buckling A, Rainey PB. The effect of a bacteriophage on diversification of the opportunistic bacterial pathogen, Pseudomonas aeruginosa. Proc Biol Sci. 2005;272:1385–91. doi: 10.1098/rspb.2005.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brazier JS. The epidemiology and typing of Clostridium difficile. J Antimicrob Chemother. 1998;41:47–57. doi: 10.1093/jac/41.suppl_3.47. [DOI] [PubMed] [Google Scholar]
- 40.Kato H, Kato H, Ito Y, et al. Typing of Clostridium difficile isolates endemic in Japan by sequencing of slpA and its application to direct typing. J Med Microbiol. 2010;59:556–62. doi: 10.1099/jmm.0.016147-0. [DOI] [PubMed] [Google Scholar]
- 41.Meader E, Mayer MJ, Gasson MJ, Steverding D, Carding SR, Narbad A. Bacteriophage treatment significantly reduces viable Clostridium difficile and prevents toxin production in an in vitro model system. Anaerobe. 2010;16:549–54. doi: 10.1016/j.anaerobe.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 42.Giraud MF, Naismith JH. The rhamnose pathway. Curr Opin Struct Biol. 2000;10:687–96. doi: 10.1016/s0959-440x(00)00145-7. [DOI] [PubMed] [Google Scholar]
- 43.Belmares J, Johnson S, Parada JP, et al. Molecular epidemiology of Clostridium difficile over the course of 10 years in a tertiary care hospital. Clin Infect Dis. 2009;49:1141–47. doi: 10.1086/605638. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.