

Abstract
Confocal fluorescence microscopy is a broadly used imaging technique that enhances the signal-to-noise ratio by removing out of focal plane fluorescence. Confocal microscopes come with a variety of modifications depending on the particular experimental goals. Microscopes, illumination pathways, and light collection were originally focused upon obtaining the highest resolution image possible, typically on fixed tissue. More recently, live-cell confocal imaging has gained importance. Since measured signals are often rapid or transient, thus requiring higher sampling rates, specializations are included to enhance spatial and temporal resolution while maintaining tissue viability. Thus, a balance between image quality, temporal resolution, and tissue viability is needed. A subtype of confocal imaging, termed swept field confocal (SFC) microscopy, can image live cells at high rates while maintaining confocality. SFC systems can use a pinhole array to obtain high spatial resolution, similar to spinning disc systems. In addition, SFC imaging can achieve faster rates by using a slit to sweep the light across the entire image plane, thus requiring a single scan to generate an image. Coupled to a high-speed charge-coupled device camera and a laser illumination source, images can be obtained at greater than 1,000 frames per second while maintaining confocality.
Keywords: swept field confocal microscopy, prairie technology, RedShirt imaging, live-cell imaging
Introduction
Fluorescence microscopy is based on the principle that a fluorophore absorbs a photon of a particular wavelength, moves to an excited state where energy loss leads to emission of a photon of a longer wavelength (lower energy), and thereby returns to the starting energy level (Pawley, 2006). Separation of the excitation photons from the emitted photons and capture of the emitted photons allows for the generation of an image that provides information as to the location and relative number of these emitted photons (Pawley, 2006). A variety of specializations have been developed over the past decades to enhance the quality of the resulting image. For example, better filter sets are available to better separate excitation and emission wavelengths. Lasers are often used as excitation sources to provide coherent narrowband light with higher intensities (Gratton & vande Ven, 2006). Additionally, higher resolution cameras with more and smaller pixels also contribute to enhance image quality. Higher sensitivity detectors reduce the noise floor and allow for single photon detection.
First described more than 50 years ago, confocal microscopy was a major technical advancement by which out of focus light was removed from the image plane, resulting in better resolution and cleaner optical sections (Minsky, 1988). The basic principle is that a pinhole is placed in the image plane before the detector, consequently blocking any emitted photons not in the image plane (Inoue, 2006). Depending on the excitation light source, a pinhole can also be placed in the excitation path, providing the required point illumination. The pinhole diameter, which is typically set to match the diameter of the Airy disc of a diffraction-limited optical system, is directly proportional to the magnification between the pinhole plane and object as well as to the wavelength of light and inversely proportional to the numerical aperture of the objective (Toomre & Pawley, 2006). This creates a point source system that requires scanning of the image by the excitation light that is synchronized with the light collection and rasterization of an image. The hardware used for generating the excitation light source, scanning the beam across the tissue, and the collection of the emitted light varies among systems, which are typically specialized to application. Swept field confocal (SFC)microscopy is one of these specializations and the focus of this brief review.
The SFC microscope was designed as a tool that allows for live-cell imaging at high spatial and temporal resolutions. To achieve these goals, several important features are incorporated into the system’s design. First, the system allows for flexibility by using an aperture plate that contains either a set of pinholes or a slit. In the pinhole mode, parallel line scanning set by a galvanometer module must be synchronized with a perpendicular piezoelectric movement to form a complete frame using 16 horizontal scans. This pinhole mode allows for maximizing resolution. In the slit mode, the use of a single slit for sweeping the sample achieves higher rates with low intensity light for excitation. The trade-off here is slightly diminished spatial resolution and optical sectioning due to the increase of fluorescence detected from out of focus light. A second feature for the high-speed SFC microscope is the addition of a high-speed camera with larger pixel sizes that allow for more efficient light capture, which increases the signal-to-noise ratio (SNR) at the expense of decreased spatial resolution. A third specialization is the use of cameras with smaller pixel arrays such that an 80 × 80 pixel image can be transferred much faster than one that is 512 × 512. The final feature is the use of higher power excitation lasers. Although lasers are used in many confocal systems, the higher power lasers serve to reduce the excitation time required to generate an image.
Methods
Hardware
Figure 1 presents a photograph of the electrophysiological recording station coupled to the SFC imaging system (Prairie Technologies, Middlefield, WI, USA). An upright fixed stage microscope (BX51 Olympus, Center Valley, PA, USA) was used with a dual port for either bright-field imaging or SFC imaging (note the second camera in Fig. 1B). This station was modified for low vibration to investigate the mechanosensitive sensory cells of the inner ear. To this end the stage as well as mounting components were produced in house. Here, optical rail was used for both the microscope stage and to mount the SFC system. The mounting system is critical for accurate and stable alignment of the SFC system. For live-cell imaging high magnification was used: a 100× 1.0 NA dipping lens (LUMPlan Fl, Olympus), further magnified 2× and projected to a RedShirt camera (80 × 80 pixels) (RedShirtImaging, Decatur, GA, USA) resulting in a 10 × 10 μm field of view with a pixel size of 120 nm. This small field of view required accurate alignment of all components for optimal sensitivity. A fiber optic cable (Figs. 1A, 1B) was used to direct the excitation light into the light path (Fig. 1C). Dichroic and emission filters were individually selected and placed by hand into the SFC head. We typically use multipeaked filters that allow imaging of two fluorophores without a need to change filters. One weakness of the system is the need to manually change these filters and to fine-tune the alignment based on which the filter set is mounted.
Figure 1.

SFC and electrophysiological recording station. A: Presents a photograph of the recording station with key elements highlighted. The SFC system is integrated into an electrophysiology recording station using an optical rail based support system. B: Presents a higher power view of the assembly, again highlighting the major components. A separate camera is used for the bright-field image needed for electrophysiological recordings. C: Presents the optical light path for excitation (blue) and emission (green) pathways. Again key features are labeled. Significant is the lack of moving parts that ensures stability while at the same time enhancing speed of imaging.
Light Path
Confocal microscopy can be performed with a variety of light sources. Mercury lamps or metal halide can be used depending on wavelength and power requirements. Mercury lamps tend to be better for histological samples due to their high power. However, the tendency for these bulbs to flicker makes temporal measurements more difficult for live-cell imaging. Xenon lamps are more stable and therefore better for live-cell imaging, but also deliver less power. The ability to deliver greater power at specific wavelengths makes lasers particularly useful for high-speed live-cell imaging. We used a laser launch system supplied by Prairie Technologies that incorporates five laser lines, which are selected through an acoustic optical tunable filter (AOTF). The AOTF selects wavelengths via RF oscillation frequency of the crystal and intensity via RF oscillation amplitude, thus serving for both laser selection and intensity control in our system. There is a user-friendly simple interface in the imaging software for control of the AOTF. A hard shutter can also be remotely controlled for both safety and to ensure no low-level excitation of tissue. The efficiency of light transfer from the launch to the SFC is quite high and largely dependent upon the length of the fiber optic. Appropriate alignment of the fiber is critical to maximize sensitivity.
Figure 1C provides a view of the optical path (or layout) of the SFC system with the excitation path shown in blue and the emission path shown in green. The light enters via the fiber and passes through the aperture plate (slit or pinholes). A unique feature of this system is that the aperture plate is set up in duplicate so that the excitation light will pass through a slit or a set of pinholes, while the emission will pass through a second set. The pinhole or slit is selected prior to imaging, and no plate movement is required during imaging. This speeds imaging and eliminates vibration that can degrade the image or, as in our work, activate mechanoreceptive hair cells. The light is then beamed onto a set of galvanometers with mirrors that are used to scan the excitation light across the tissue and the emission light across the camera. In fact, the mirrors are used three times. They scan the excitation light onto the specimen, then de-scan the emitted light through the emission apertures, and finally scan this emission onto the camera. A focusing lens was present in front of the camera to ensure parfocality.
Camera Selection
We have selected the NeuroCCD-SMQ (RedShirtImaging) camera as our detector for several reasons. First, it can acquire images up to 2,000 frames per second (fps) full frame (80 × 80 pixels). It is cooled for low dark noise and does not use an internal fan to avoid vibration. It has a large well size (215,000 e−) and digitizes at 14 bits, thus enhancing both speed and SNR. The software control was programmed in IDL (Exelis, Visual Information Solutions, Fort Wayne, IN, USA), a user-friendly modifiable interface. The standard software is well integrated with electrophysiological measurements so timing between measurements was well controlled. Both the laser launch system and the SFC system can be controlled via the same software interface. The pixel size was 24 × 24 μm. At this pixel size we used a 100× objective with an additional 2× magnification to produce a 120 nm pixel at the specimen. The images presented in this article show stereocilia, the mechanosensitive structures characteristic of auditory cells of the inner ear. The minimum sampling frequency (Nyquist limit) requires sampling at least two times the maximal spatial frequency to be reconstructed. As the stereocilia being imaged range between 250–500 nm in diameter, this pixel size satisfies the Nyquist limit to resolve these structures.
Results
Phalloidin labels filamentous actin, the core component of the stereocilia of hair cells. Figure 2 presents images of hair bundles obtained with the same objective [Plan-Apochromat 100× 1.4NA, Zeiss (Carl Zeiss, Oberkochen, Germany)] using our three different confocal systems: SFC (pinhole mode), spinning disc, and laser-scanning confocal (LSCM). The images are shown under conditions that allow the best quality image from each system to demonstrate the compromise provided in image quality to gain speed. The spinning disc images were taken using a Yokogawa CSU10 scan head with the identical 100× oil immersion objective, but used a Photometrics QuantEM512SC (Tucson, AZ, USA) camera (pixel size 16 × 16 μm). After magnification with a 2.5× optovar, the spinning disc achieves 46 nm/pixel at the specimen, compared to 120 nm/pixel for the SFC. The spinning disc uses a 50 μm pinhole. The LSCM images were obtained in a LSM 5 Exciter laser system (Carl Zeiss). These images look better mainly due to oversampling and signal averaging with longer pixel dwell times. These results showed that the SFC microscopy allows levels of sharpness and confocality appropriate for resolving subcellular structures.
Figure 2.

Comparison of SFC with spinning disc and laser scanning confocal systems. Images show stereocilia from rat inner hair cell bundles labeled with Alexa Fluor 488-phalloidin using a 100× 1.4 NA oil immersion objective. The SFC image is the average of 40 frames obtained at 40 fps using a 60 μm pinhole at 120 nm/pixel. The spinning disc image was obtained in a Yokogawa CSU10 using a 50 μm pinhole with a 2 s exposure at 46 nm/pixel. The LSCM image was obtained with a Zeiss LSM 5 Exciter for a total of 120 s at 10 nm/pixel. Scale = 1 μm.
That spatial resolution can be maintained in the SFC at higher sampling rates is demonstrated in Figure 3, where sampling was increased from 40 to 1,000 fps. Figure 3A presents a hair cell hair bundle, stained with phalloidin-Alexa Fluor 488 and imaged with a 35 μm slit. Single frames obtained at different frequency rates (120 nm/pixel) show that single stereocilia can be resolved, although the SNR was reduced at higher sampling rates due to lower signal levels. For higher frequencies the laser intensities had to be increased and the dynamic range adjusted to obtain equivalent images. Moreover, sampling at 1,000 fps at these high intensities produced a strong photobleaching effect. Figure 3B shows single frame images of another bundle obtained with a CSU10 Yokogawa spinning disc confocal microscope (114 nm/pixel), having a maximal rate of 360 fps. New spinning disc microscopes can, however, reach higher rates. Figure 3C contains examples of laser scanning confocal images obtained with the same pixel size as the SFC images in Figure 3A (120 nm/pixel), where the maximum acquisition rate was 16 fps. In general this figure demonstrates that the SFC system using a slit scanner is able to maintain resolution at high frequencies.
Figure 3.

Comparison of different sampling rates in three confocal systems. A: A hair bundle was imaged with the SFC system at different acquisition rates (2 to 1,000 fps) using a 50 μm slit. The laser intensity, depicted in the lower right corner of each panel, had to be increased for higher frequencies to obtain equivalent illumination intensities. B: Another hair bundle was imaged with the CSU10 Yokogawa spinning disc system at different sampling rates (4 to 333 fps) using a 50 μm pinhole. The maximal rate in our spinning disc microscope was 360 fps. Scan line inhomogeneity observed at higher rates is due to the inability of our spinning disc head to change its rotation speed to have integer scans of the image during our camera exposure time. C: Another hair bundle was imaged with the LSM 5 Exciter laser scanning confocal system at two acquisition rates. The image was set to 80 × 80 pixels for the sake of comparison. The fastest frequency allowed by the LSCM was 16 fps using 4 μs dwelling time. Scale = 1 μm.
Figure 4 demonstrates the importance of slit selection regarding optical sectioning and resolution. Here, images were obtained by averaging 20 frames acquired at 40 fps. Slits with a width close to 37 μm, the optimal diameter according to the Fraunhofer formula (equal to the full-width at half-maximum of the Airy figure), provided better resolution than larger slit widths. Image resolution obtained with a smaller diameter slit was no better and required more illumination than the 35 μm slit. The reduced out-of-focus fluorescence from the optical sections using a slit close to 37 μm allowed individual stereocilia to be resolved better than the 70 μm slit. This demonstrates that the same principles for pinhole confocality apply for the slit and, therefore, a slit width selection matching the Airy unit is preferred. Pinholes provided slightly better resolution, at the expense of lower SNRs and higher laser intensities as predicted. The major difference in light discrepancy is due to light passing through 32 pinholes versus 1 slit and the fact that 16 sweeps are done to collect an image in pinhole mode as compared to 1 sweep in slit mode. For live-cell imaging, where photodamage is an issue as well as for bleaching, this could be a major obstacle. A similar type of problem would exist for spinning disc type confocal microscopes at high acquisition rates.
Figure 4.

Comparison of a hair bundle SFC section imaged with different slits and pinhole sizes. Rat hair cell bundle was fixed and stained with phallodin-Alexa Fluor 488. Images were acquired at 40 fps during 500 ms using different slit or pinhole sizes. Slit widths closer to the Airy limit improved resolution. Contrast was improved by using pinholes to the detriment of light exposure. The laser intensity is depicted in the lower right corner of each panel. Scale = 1 μm.
The SFC system was developed to maintain spatial resolution at higher speeds in live-cell high-speed imaging. Auditory hair cells have calcium channels clustered at synapses (Tucker & Fettiplace, 1995; Ricci et al., 2000), which open and close rapidly with changes in membrane voltage, allowing for calcium entry. Figure 5 presents a hair cell imaged with the SFC system revealing a complex distribution of calcium changes. In this case, each position shows multiple kinetic components to the calcium increase. In addition there is a spatial difference where the calcium signal near the synapse rises much faster than the signal at a distance. The confocality allows for individual synapses to be resolved. The temporal resolution allows for differences in local calcium changes to be observed. Slower sampling would allow for more calcium diffusion, blurring the differences between cellular locations. Additionally, the signal at the synapse changes quite rapidly. Slower integration would miss this rapid change.
Figure 5.
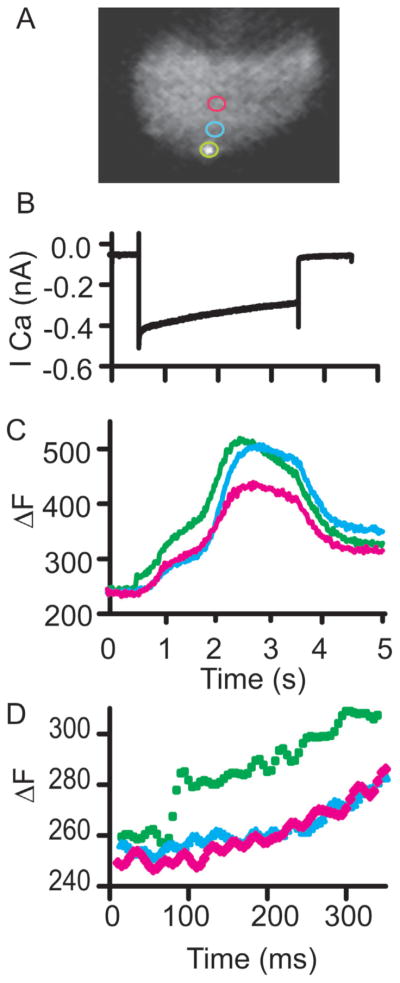
Calcium imaging of depolarization induced calcium entry using SFC imaging. A: Presents images with colored rings indicating regions sampled. The SFC image is a single frame of a hair cell with a synapse labeling peptide where a single synapse is in focus. B: Presents the calcium current. Cesium based intracellular solutions were used with apamin (100 nM) external to isolate the calcium currents. C: Provides the change in fluorescence observed at the color coded spots indicated in image A. Note the multiple components in the SFC time course. D: Provides an expanded time view to illustrate the value of temporal resolution as seen in the green trace.
To further demonstrate the capacity of the SFC system, calcium imaging of individual stereocilia is presented in Figure 6. Figure 6A shows a bright-field image taken with the secondary camera and also an Alexa Fluor 594-filled stereocilia. This bright-field image was taken just prior to the calcium time lapse to identify the image plane through the hair bundle. Using the AOTF to select the 594 laser and using a dichroic mirror and barrier filter that allows imaging at wavelengths for Alexa Fluor 594 as well as the calcium indicator fluo4FF (Invitrogen/Life Technologies, Carlsbad, CA, USA) greatly simplifies this experiment. Figure 5B presents the stimulus used to elicit mechanically activated calcium signals in the sensory hair bundle stereocilia. To avoid moving artifacts while imaging calcium, the hair cell is first depolarized to +80 mV to reduce calcium entry. At this point the hair bundle is moved to elicit outward mechanically activated currents. Once the bundle has stabilized mechanically, with channels open, the calcium driving force was returned by hyperpolarizing the cell to −84 mV. The current traces in Figure 5B show the results of this stimulus protocol, with first voltage dependent currents activating, followed by mechanically gated channel activation. Hyper-polarization then gives inward mechanically gated currents. The arrows in the figure indicate where the images in Figure 6C were taken. Individual stereocilia are resolvable such that each stereocilium with active channels can be identified. The confocality was important for providing spatial resolution. Images were collected at 500 fps. Plotting fluorescence change against time allowed for the use of the kinetic change to aid in localizing the position of the channels. The faster the rise time, the closer is the source of the calcium. In this way the spatial and temporal components of the system work together. Slower sampling would allow calcium to spread more, thereby dissipating the gradient from the source.
Figure 6.
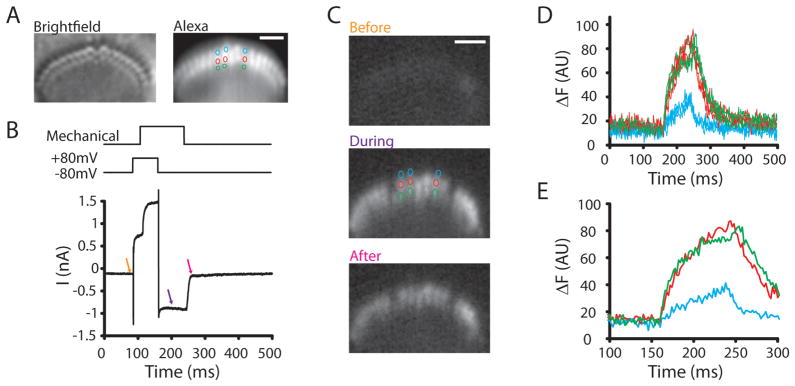
Method used to localize the site of mechanotransduction channels in inner hair cell stereocilia. A: Presents the bright-field hair bundle image and the Alexa Fluor 594-filled stereocilia for identification of all stereocilia. B: Provides the stimulus above and the hair cell current responses below. The stimulus protocol first depolarizes hair bundles to reduce inward driving force for calcium while the hair bundle is moving. The hair bundle is deflected with a picospritzer to activate mechanotranducer channels, and then the cell is hyperpolarized while channels are open in order to image calcium entry. Arrows indicate time points in image C where images were obtained. C: Presents calcium images, Fluo4ff was the calcium indicator, at time points indicated in image B. The colored circles are the stereocilia where fluorescence was tracked in time (D). Scale bar represents 2 μm. D: Plots the fluorescence against time to illustrate the rapid increase in calcium levels for the second and third row but not the first. E: An expanded time view of the onset to better represent the different time courses.
Discussion
The purpose of this review was to describe the fundamental features of swept field confocal microscopy. We used as an example a system that couples a Prairie SFC to a RedShirt neuro CCD-SMQ camera. There are several advantages to this system. First, the few moving parts are simply the galvanometers for positioning the light, thus limiting vibration and maintaining confocality by having the apertures (i.e., slits and pinholes) in fixed positions. Second, it has a small footprint and can be easily incorporated into an electrophysiological station. Third, the software is compatible with electrophysiological measurements. Fourth, the SFC microscope, by using a slit scanning system, can image at high speeds (>1,000 fps) while maintaining confocality.
Laser scanning confocal microscopy of fixed tissue was developed to enhance resolution and allow imaging in thick tissue samples by eliminating or reducing out of focus fluorescence. Further enhancements consist of using cameras with a large number of small pixels, increasing the dwell times per pixel, using high numerical aperture oil immersion objectives and higher-powered illumination sources. Many of these enhancements are not compatible with live-cell imaging. For example, dipping lenses with lower numerical aperture are required for many tissue preparations because of the need for longer working distances. Smaller pixel size limits photons per pixel, requiring longer sampling. The need for higher sampling rates and more limited light exposure to work with live cells with rapidly changing signals has led to compromises in imaging. One of these is the swept field slit scanning system, which enables imaging with confocal resolution to work at frequencies greater than 1,000 fps.
We have demonstrated that the use of a slit as compared to a pinhole is justified when sampling at higher rates. The loss of resolution is offset by the additional light and lower phototoxicity, allowing for reduced camera gain. Under comparable conditions the pinhole generates a better section than the slit, but this comes at the cost of higher intensity excitation laser, which for live-cell imaging is a major problem for tissue viability. Newer spinning disc confocal microscopes can sample at higher rates compared to the initial types and have made efforts to enhance light passage through the pinholes using microlenses. Whether this can offset the requirement of higher laser power intensities needs to be determined. Also, mechanical noise introduced by the spinning disc into the system could be a problem at least for mechanoreceptor measurements.
We have also shown that when imaging rapidly changing systems, such as calcium imaging in stereocilia or at synapses, the higher temporal and spatial resolution provided by the SFC system enables the tracking of these small and rapidly occurring events. It also to some degree makes possible experiments such as those presented in Figures 5 and 6 (Beurg et al., 2009; Schnee et al., 2011), by providing the necessary optical sectioning and therefore resolving these spatially important sites. The higher sensitivity allows for higher sampling rates, limiting diffusion from the source and allowing the kinetics of the response to provide the necessary spatial information. SFC imaging has also been used for imaging hippocampal slices containing dendrites too small to record (Gasparini et al., 2007). Calcium sparklets, a transient and low intensity signal from smooth muscle, have also been characterized using the SFC system (Santana et al., 2008).
Imaging at higher rates usually requires exposure of living samples to high intensity laser illumination. This may cause both photodamage and photobleaching. Confocal microscopy in any of its present forms does not completely circumvent this problem. Alternative imaging techniques such as multiphoton microscopy are much less damaging to live tissue and allow even deeper tissue penetration than does confocal microscopy. The combined use of the different microscopy technologies available is happening at a fast pace, yielding important new insights into how to obtain high resolution at high speed in living cells without increasing toxicity or bleaching.
In summary, the SFC system provides the ability to image in living tissue at high rates. Confocality can be maintained and illumination requirements are reduced as compared to pinhole systems. For experiments that need spatial and temporal resolution in living systems, the SFC system provides these abilities.
Acknowledgments
This work was supported by NIDCD RO1 grants DC003896 and DC009913 to A.J.R., by core grant P30 44992, by a Dean’s postdoctoral fellowship, and a Cajamadrid Foundation fellowship to M.C.M., and an NRSA F32 from NIDCD to A.W.P.
References
- Beurg M, Fettiplace R, Nam JH, Ricci AJ. Localization of inner hair cell mechanotransducer channels using high-speed calcium imaging. Nat Neurosci. 2009;12:553–558. doi: 10.1038/nn.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini S, Losonczy A, Chen X, Johnston D, Magee JC. Associative pairing enhances action potential backpropagation in radial oblique branches of CA1 pyramidal neurons. J Physiol. 2007;580:787–800. doi: 10.1113/jphysiol.2006.121343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratton E, vandeVen MJ. Chapter 5: Laser sources for confocal microscopy. In: Pawley JB, editor. Handbook of Biological Confocal Microscopy. 3. New York: Springer; 2006. pp. 80–125. [Google Scholar]
- Inoue S. Chapter 1: Foundations of confocal scanned imaging in light microscopy. In: Pawley JB, editor. Handbook of Biological Confocal Microscopy. 3. New York: Springer; 2006. pp. 1–16. [Google Scholar]
- Minsky M. Memoir on inventing the confocal microscope. Scanning. 1988;10:128–138. [Google Scholar]
- Pawley JB. Fundamental limits in confocal microscopy. In: Pawley JB, editor. Handbook of Biological Confocal Microscopy. 3. New York: Springer; 2006. pp. 20–41. [Google Scholar]
- Ricci AJ, Gray-Keller M, Fettiplace R. Tonotopic variations of calcium signaling in turtle auditory hair cells. J Physiol (Lond) 2000;524(Pt 2):423–436. doi: 10.1111/j.1469-7793.2000.00423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana LF, Navedo MF, Amberg GC, Nieves-Cintron M, Votaw VS, Ufret-Vincenty CA. Calcium sparklets in arterial smooth muscle. Clin Exp Pharmacol Physiol. 2008;35:1121–1126. doi: 10.1111/j.1440-1681.2007.04867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnee ME, Santos-Sacchi J, Castellano-Munoz M, Kong JH, Ricci AJ. Calcium-dependent synaptic vesicle trafficking underlies indefatigable release at the hair cell afferent fiber synapse. Neuron. 2011;70:326–338. doi: 10.1016/j.neuron.2011.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toomre D, Pawley JB. Chapter 10: Disc scanning confocal microscopy. In: Pawley JB, editor. Handbook of Biological Confocal Microscopy. 3. New York: Springer; 2006. pp. 221–237. [Google Scholar]
- Tucker T, Fettiplace R. Confocal imaging of calcium microdomains and calcium extrusion in turtle hair cells. Neuron. 1995;15:1323–1335. doi: 10.1016/0896-6273(95)90011-x. [DOI] [PubMed] [Google Scholar]