

Abstract
Keratins 8 and 18 (K8/K18) are heteropolymeric intermediate filament phospho-glycoproteins of simple-type epithelia. K8/K18 protect hepatocytes from apoptosis and their mutations predispose to liver disease. K18 undergoes dynamic O-linked N-acetylglucosamine glycosylation at Ser30/31/49, the function of which is unknown. We addressed the function of K18 glycosylation by generating mice that overexpress human K18 S30/31/49A (Gly−), and compared their susceptibility to injury with wild-type and other keratin-mutant mice. Gly− mice are selectively more susceptible to liver and pancreas injury and apoptosis induced by streptozotocin or combined N-acetyl-D-glucosaminidase inhibition and Fas administration. The enhanced apoptosis in Gly− livers involves Akt1 and protein kinase Cθ inactivation due to their site-specific hypophosphorylation. Akt1 binds to K8 and this binding likely contributes to reciprocal Akt1 hyperglycosylation and hypophosphorylation upon K18 hypoglycosylation, with consequent decreased Akt1 kinase activity. Therefore, K18 glycosylation provides a unique protective role in epithelial injury by promoting the phosphorylation and activation of cell survival kinases.
Keywords: glycosylation, intermediate filaments, liver, pancreas, phosphorylation, streptozotocin
INTRODUCTION
Intermediate filaments (IFs), together with microfilaments and microtubules form the major cytoskeleton proteins that are expressed in a tissue and cell type-specific manner in mammalian cells1. Mutations in IFs cause or predispose to a wide range of human diseases that generally reflect the tissue specific expression of the mutant IF gene2, 3. In epithelial cells, keratin (K) 8 and 18 are the major IF proteins in simple-type epithelia4, 5 and, as such, are found in a broad range of organs including the digestive tract, lung and kidney. K8 and K18 are obligate noncovalent heterodimers whose individual mutations typically behave in a dominant manner and predispose their carriers to acute or chronic liver disease progression6–9. Keratins (including K8/K18) serve several mechanical and nonmechanical functions that include cytoprotection from cell stress and apoptosis4, regulation of epithelial polarity and protein targeting to subcellular compartments10, 11 and regulation of translation12.
Keratin functions are facilitated by the dynamic interaction with associated proteins and by posttranslational modifications with the best studied being K8/K18 serine phosphorylation13. The in vivo significance of phosphorylation at K8 S74 and K18 S34/S53 were demonstrated in studies of keratin-mutant transgenic mice that were susceptible to liver injury (K8 S74A and K18 S53A overexpressing mice) or that had altered mitotic progression and filament organization (K18 S34A mutant)13, 14. K8 and K18 also undergo dynamic single O-linked N-acetylglucosamine (O-GlcNAc) Ser/Thr glycosylation15. The three major human K18 O-GlcNAc glycosylation sites (Ser30/31/49) have been identified, which do not appear to be major K18 phosphorylation sites, though their function is not known16. K8/K18 glycosylation and phosphorylation occur concurrently17, likely at distinct residues, unlike some of the other O-GlcNAcylated proteins that have been characterized in detail18, 19. For example, K8/K18 hyperglycosylation occurs in the context of mitotic arrest or viral infection in cultured cells which also result in keratin hyperphosphorylation17, 20, while inhibition of protein phosphorylation inhibits stimulus-induced K8/K18 hyperglycosylation21.
O-GlcNAcylated proteins are modulated on Ser/Thr by O-GlcNAc transferase (OGT) and N-acetyl-D-glucosaminidase (O-GlcNAcase) via GlcNAc addition or removal, respectively18, 22. O-GlcNAcylation modifies numerous nuclear/cytoplasmic proteins, and regulates several functions including protein turnover, transcription and stress responses18, 22–27. O-GlcNAcylation and phosphorylation frequently occur at adjacent or identical Ser/Thr, and each modification can interfere with the other18, 22, 23, 28. Exposure of animal models to streptozotocin (STZ), an O-GlcNAcase inhibitor that also has other non-O-GlcNAcase effects29, results in accumulation of O-GlcNAc-modified proteins, diabetes and neurodegeneration18. Similarly, adipocyte exposure to O-(2-acetamido-2-deoxy-D-glucopyranosylidene)amino-N-phenylcarbamate (PUGNAc), another O-GlcNAcase inhibitor, leads to insulin resistance30 but no reported studies tested the function of site-specific glycosylation in animal models.
We addressed the function of K18 glycosylation by generating mice that overexpress human K18 S30/31/49→A (K18 Gly−), and compared their susceptibility to STZ- or PUGNAc/Fas-mediated injury with nontransgenic mice and three other control mouse lines that overexpress human wild-type K1831, phospho-mutant S53A K1813, or R90C K18 which disrupts keratin filaments and predisposes hepatocytes to apoptosis32. In contrast with the other control mouse lines, K18-Gly− mice are significantly more susceptible to STZ- or combined PUGNAc/Fas-induced liver and pancreatic injury, including prominent hepatocyte apoptosis. K18-null mice, which by definition lack K18 glycosylation, phenocopy the findings of K18 Gly− mice. The observed susceptibility to cell death is specific to O-GlcNAcase inhibition since Fas-ligand alone induces apoptosis similarly in K18 Gly− and control mice. The enhanced apoptosis in K18 Gly− mice involves Akt1 and protein kinase C (PKC)θ inactivation due to site-specific kinase hypophosphorylation in vivo and ex vivo. Akt1 binds to K8 and its inactivation by K18 hypoglycosylation is coupled by reciprocal Akt1 hyperglycosylation and hypophosphorylation. Our findings demonstrate a functional role for IF glycosylation by promoting cell survival kinase activation.
RESULTS
Expression of K18 Gly− mutant protein in transgenic mice
A genomic human (h)K18 construct, identical to that used to generate several other K18 overexpressing transgenic mice8 was used to introduce K18 point mutations to block glycosylation of its three major glycosylation sites (Fig 1a) that were previously identified16. The expression of hK18 S30/31/49A (Fig 1b) and ablation of K18 glycosylation were confirmed by in vitro galactosylation (Fig 1c), which labels endogenous terminal GlcNAcs, and by an epitope-specific antibody that recognizes K18 WT but not K18 Gly− (Fig 1b). One major advantage of the keratin-overexpression models is that endogenous mouse K18 (mK18) expression decreases in parallel to hK18 overexpression (Fig 1b). Given that K18 phosphorylation and glycosylation involve proximal residues (Fig 1a), we tested whether K18 Gly− affects K18 S34/S53 phosphorylation in transfected cells. K18 S34/S53 phosphorylation was assessed using anti-K18 pS53 antibody or 14-3-3 binding to K18 which is regulated by K18 S34 phosphorylation13. The K18 glyco-mutations have no effect on K18 S34/S53 phosphorylation (Supplemental Fig S1) which indicates that any observed phenotype of K18 Gly− mice is not due to alteration in K18 S34/S53 phosphorylation.
Figure 1. K18 Gly− predisposes transgenic mice to STZ-induced injury.

a, Posttranslational modifications of K18. All IFs consist of three structural motifs (not drawn to scale): a central relatively conserved α-helical “rod” domain that is flanked non-conserved “head” and “tail” domains. Three glyco-serines (gS30/31/49) and two phospho-serines (pS34/53) are located in the head domain, while two caspase digestion sites (D238/397) are located in the rod and tail domains. b, Livers were obtained from non-transgenic (N, lane 1), or transgenic mice that overexpress hK18 WT (lanes 2,3) or Gly− (Ser30/31/49→Ala) (lanes 4,5). Each lane represents separate livers that were used to generate a highly enriched keratin fraction by high salt extraction (HSE) or that were solubilized in SDS-containing sample buffer to prepare total lysates50. The samples were separated by SDS-PAGE then stained with Coomassie blue, or were blotted with antibodies specific to hK18 (DC10), mouse/human (m/h) K18 (EndoB), mK8 (Troma-I), or nonphospho-K18 S33 (antibody 4668) that recognizes the hK18 WT sequence 26RPVSSAASVYAGA, but not the K18 Gly− because of the underlined mutant serines50. c, In vitro galactosylation of K8/K18. K8/K18 immuno-precipitates from livers similar to those used in panel (b) were obtained by using the human K18-specific antibody L2A150 and then subjected to in vitro galactosylation using UDP-[4,5-3H]-galactose and galactosyltransferase to radiolabel K8/K18 Ser/Thr-linked O-GlcNAcs. Note that mutation of the three K18 S30/31/49 glycosylation sites abolished K18 glycosylation in the Gly− livers. d, In vitro [3H]-galactosylation of detergent-free cytosolic extracts isolated from transgenic K18-WT and K18-Gly− livers 2 days after STZ administration. Equal amounts of protein were separated by SDS-PAGE followed by Coomassie staining or fluorography to assess the extent of protein O-GlcNAc modification. e, f, The indicated mouse genotypes [total number of mice/genotype are shown in panel (f); all in an FVB background] were injected with STZ then monitored daily for survival. Most deaths occurred within 10 days and no deaths were observed beyond 24 days. Mortality was significantly increased in the Gly− mice as compared with other mice strains (~70% Gly− versus ~20–40% in the other listed control strains, p=0.0002).
K18 Gly− predisposes transgenic mice to STZ-induced injury
K18 Gly− mice are viable, breed normally, and have normal lifespan and histological phenotypes under basal conditions. Given that K18 glycosylation increases during few stress cell culture conditions such as mitotic arrest or rotavirus infection17, 20, we first tested the susceptibility of K18 Gly− mice to STZ. The overall protein O-GlcNAc content increases similarly (~40%) in WT and Gly− cytosolic liver homogenates after STZ (Fig 1d). However, K18 Gly− mice are markedly more susceptible to STZ-induced lethality (Fig 1e,f), which is accompanied by significantly higher serum alanine aminotransferase and lower serum insulin as compared with controls (Supplemental Table S1). In contrast, no statistically significant differences were noted after STZ administration when comparing control mice versus K18 S53A or K18 R90C mice (Fig 1f). Notably, K18 S53A or K18 R90C mice have increased susceptibility to hepatotoxins (versus control mice) including microcystin-LR (MLR, a phosphatase inhibitor) or Fas-ligand, respectively8, 32. However, K18 Gly− mice had similar injury when compared to non-transgenic and hK18 WT mice after Fas-alone injection (Supplemental Fig S2) or MLR (not shown). These results indicate that K18 Gly− mice are selectively and highly susceptible to specific types (e.g., STZ but not Fas) of apoptosis-associated injury.
The increased lethality in K18 Gly− mice was likely caused by multi-organ failure since several abdominal organs (liver/pancreas/kidney; Fig 2a) were clearly paler in K18 Gly− versus K18 WT mice. These findings were confirmed by histological analysis which showed extensive liver hemorrhage and cell drop-off, pancreatic edema and islet-cell necrosis in K18 Gly− animals (Fig 2b; Supplemental Fig S3). Also, there was significant hepatic steatosis and glycogen depletion in K18 Gly− mice as confirmed by oil red-O and periodic acid-Schiff staining, respectively (Fig 2c). Pancreatic endocrine damage was corroborated by the dramatically decreased insulin staining (Fig 3a) and lower serum insulin (Supplemental Table S1) in K18 Gly− pancreata. The predisposition to STZ-mediated pancreatic damage is not related to differences in glucose or insulin tolerance (Supplementary Fig S4). Collectively, these findings indicate that the dramatic STZ-mediated injury in K18 Gly− mice is not related to selective glucose homeostasis changes but to generalized epithelial tissue injury in several K18-expressing organs.
Figure 2. Comparison of the STZ-induced tissue injury in K18 WT versus K18 Gly−mice.

a, b, Gross appearance (a) and histopathology (b) of mouse organs were compared before and 2 days after STZ injection. E, edema; H, hemorrhage; I, islet; K, kidney; L, liver; P, pancreas; S, spleen. Bars = 200 μm (panel b). See supplemental Fig S3 for higher magnification of panel b. c, Liver sections from the indicated mice were stained with Oil red O for neutral lipids or with PAS for glycogen. The increased steatosis (Oil red O) and the effect on glycogen levels (PAS staining) in the STZ-treated Gly− mice both corroborate the diabetes associated changes (low insulin and islet damage). Bar = 100 μm.
Figure 3. Immostaining of pancreata from K18 WT and Gly− mice and biochemical assessment of apoptosis in liver and pancreas tissues.

a, Pancreas sections were triple-stained for K8 (green), insulin (red) and nuclei (blue). Note that the K8 staining in Gly−pancreata after STZ treatment is lower because of severe edema, islet cell necrosis and blood cell infiltration. Arrows indicate infiltrating red blood cells in islet that express neither K8 nor insulin. Bar = 50 μm. b, c, Total lysates were prepared from mouse livers (b) and pancreata (c). The lysates were separated by SDS-PAGE then blotted with the indicated antibodies. A duplicate gel was stained with Coomassie blue to verify equal protein loading. Each lane represents the analysis of one independent organ.
We examined the cause of cell death in K18 Gly− mice. No effect on liver and pancreas keratin filament organization under basal conditions was noted (Supplemental Fig S5a), but immunostaining of K18 Gly− liver and pancreas after STZ showed dramatic increased K18 apoptotic fragment formation and more prominent K8 S80 phosphorylation (a stress sensor13) as compared with K18 WT tissues (Supplemental Fig S5b,c). Increased apoptosis in K18 Gly− tissues (liver>pancreas) was supported by blotting with antibodies to cleaved caspase-3 and the K18 apoptotic fragment (Fig 3b,c). Hence, the liver injury involves extensive apoptosis while the pancreatic injury includes limited apoptosis which is consistent with experimental pancreatitis models where apoptosis (rather than necrosis) is typically inversely proportional to the severity of pancreatitis33.
K18 Gly− predisposes transgenic mice to PUGNAc/Fas-induced injury
The protective role of K18 glycosylation is further confirmed by using a more selective O-GlcNAcase inhibitor, PUGNAc29, combined with Fas antibody. We first tested various doses of PUGNAc in nontransgenic mice given that the effect of PUGNAc is not well-studied in mice. Although all mice appeared normal and had no liver damage, PUGNAc caused liver accumulation of O-GlcNAc-proteins (Fig 4a). Fas-antibody administration 2-days after PUGNAc treatment showed that the K18 Gly− mice are significantly more susceptible to lethality as compared with controls (Fig 4b,c). PUGNAc/Fas phenocopies the severe liver hemorrhage, apoptosis, hepatic steatosis and glycogen depletion (Fig 4d,e) seen with STZ (Fig 2,3). Given that the K18 Gly− mutation has no additional effect on Fas-alone mediated liver injury (Supplemental Fig S2), the phenotype of K18 Gly− mice after PUGNAc/Fas administration is likely related to the accumulation of O-GlcNAc proteins after PUGNAc treatment.
Figure 4. K18 Gly− predisposes transgenic mice to PUGNAc/Fas-induced injury.
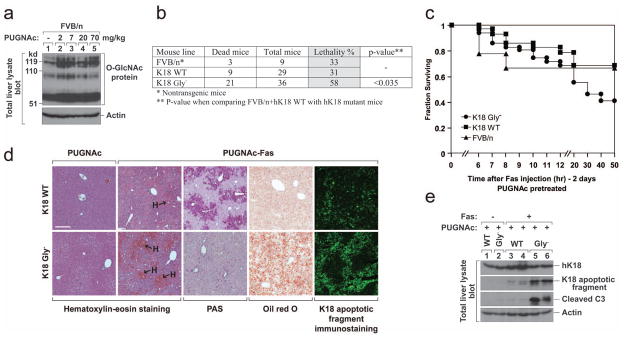
a, Nontransgenic FVB/n mice were injected intraperitoneally with the indicated doses of PUGNAc or vehicle (lane 1, PBS). Mice were then euthanized by CO2 inhalation 48 hr after PUGNAc administration. Total liver lysates were prepared and immunoblotted with anti-O-GlcNAc antibody (Ab 110.6). An actin blot is shown as a loading control. b, c, FVB/n, K18 WT or K18 Gly− mice were pretreated with PUGNAc (7 mg/kg body weight) for 48 hr to accumulate O-GlcNAc proteins, and then injected with Fas antibody (0.15 mg/kg mouse body weight). The mice were observed for 3 days (no deaths occurred after 3 days). Lethality (b) and survival curves (c) are shown. d, e, K18 WT or K18 Gly−mice were treated with PUGNAc ±Fas as in panels b and c. Livers were isolated 5 hr after Fas injection and used for the indicated stainings (d) and immunoblots (e). Bar in panel d = 200 μm.
K18 Gly− alters protein kinase phosphorylation in mouse hepatocyte primary cultures treated with STZ
The phosphoinositide 3-kinase (PI3K) pathway is central to hepatic glucose, lipid and insulin metabolism34. Phosphorylation of the PI3K effectors Akt and glycogen synthase kinase (GSK) is inhibited in adipocytes using a GlcNAcase inhibitor30. Hence, we tested if K18 Gly− expression affected Akt and GSK phosphorylation after STZ treatment in primary hepatocyte cultures, and also assessed hepatocyte susceptibility to apoptosis after STZ exposure in vivo. Phosphorylation of Akt1 T308 and its substrate GSK3α S21 were dramatically inhibited in K18 Gly− hepatocytes, while cleaved caspase-3 was more prominent in Gly− hepatocytes (Fig 5a), which parallels the profound apoptosis that is seen in intact livers (Fig 4e). The effects on Akt1 pT308 and GSK3α pS21 were selective since phosphorylation of GSK3β S9, PTEN S380 and MAPK p38 T180/Y182 were similar in all three mouse genotypes (Fig 5a). K18 WT and non-transgenic hepatocyte Akt1 T308 and GSK3α S21 were highly phosphorylated, even during basal conditions (Fig 5a; lanes 1,6), and their phosphorylations were minimally affected after STZ treatment (Fig 5a) which is likely due to the stress induced during hepatocyte isolation and culture.
Figure 5. K18 Gly− alters protein kinase phosphorylation and promotes apoptosis in response to STZ.

a, Hepatocytes were isolated from nontransgenic, K18-WT or K18-Gly− mice and treated ex vivo with 5 mM STZ for the indicated times. Total cell lysates were then prepared, separated by SDS-PAGE, stained with Coomassie blue (to verify equal protein loading) and blotted with antibodies to the indicated antigens. b, K18 WT or Gly− mice were injected i.p. with STZ or vehicle. Total liver lysates were prepared and analyzed as described in panel (a). Note that Akt1 T308 phosphorylation rose 12 hr after STZ in K18-WT livers (2.2 fold as determined by densitometric scanning) but remained flat up to 70 hr in K18-Gly− livers. c, The phosphoinositide-dependent protein kinase 1 (PDK1) consensus motif of the catalytic domain of several protein kinases. The first Thr of the motif (shown in light font) is phosphorylated by PDK1. d, Total liver lysates similar to those used in panel b were blotted with antibodies to the indicated phospho and non-phospho PKC sites. PKCθ T538, PKCδ T505 and PKCξ/λ T410/402 are PDK1 substrates (panel c), whereas PKCθ/δ S676/643, PKCβII S660 and PKCα/βII T637/641 are not. e, Pancreas and liver lysates were isolated from K18 WT or Gly− mice at the indicated times (+/− STZ administration), then analyzed by blotting with antibodies to the indicated antigens (actin is used as a loading control).
K18 Gly− alters protein kinase phosphorylation upon exposure to STZ in vivo
We then examined Akt1 T308 and GSK3α S21 in-situ phosphorylation in K18 WT and K18 Gly− mice after STZ administration. In contrast to primary hepatocyte cultures (Fig 5a), induction of GSK3α S21 phosphorylation was similar in STZ-injected K18 WT and K18 Gly− livers (Fig 5b). However, Akt1 T308 phosphorylation rose 12 hr after STZ in K18 WT livers but remained flat up to 70 hr in K18 Gly− livers (Fig 5b). Both ex vivo and in vivo results show enhanced susceptibility of K18 Gly− hepatocytes to apoptosis in association with site-specific blunting of Akt1 T308 but not Akt1 S473 phosphorylation (Fig 5a,b). Consistent with this finding, Akt activation leads to cell survival, proliferation and growth35, 36 while disruption of mouse Akt1 causes growth retardation and increased apoptosis37. Notably, Akt1 T308 (the activation loop phospho-site) mutation abolishes its kinase activity whereas S473 mutation results in partial inactivation38.
The AGC family of kinases (Akt/PKA/PKC) share a conserved (TFCGT) activation-loop phospho-motif (T308 in Akt1) that is phosphorylated by PDK139 (Fig 5c). We tested whether phosphorylation of the comparable threonine in PKC isoforms is inhibited in STZ-treated Gly− livers. Similar to Akt, PKCθ T538 but not S676 hyperphosphorylation after STZ exposure is markedly blunted in K18 Gly− livers (Fig 5d). Phosphorylation of the PDK1 consensus threonines of other tested PKC isoforms (α/β/δ/ξ/λ) showed no, or limited, changes in phosphorylation differences when comparing STZ-treated Gly− versus WT livers (Fig 5d). Notably, PKCθ T538A disrupts PKCθ activity while S676A results in limited inactivation40. In T cells, PKCθ is critical for cell activation and promotes survival by antagonizing apoptotic signals41. Therefore, blocking K18 glycosylation leads to site-specific inhibition of Akt T308/PKCθ T538 phosphorylation thereby providing an additional potential explanation for the observed accelerated STZ-induced apoptosis in K18 Gly− livers.
The selective site-specific differences in PDK1 substrate phosphorylation of Akt1/PKCθ are not related to differences in PDK1 activation since its activity based on PDK1 S241 phosphorylation is similar in WT/Gly− livers (Fig 5b). The effects that are seen in Akt 308/PKCθ T538 phosphorylations are selective to the liver and are not present in the pancreas (Fig 5e), which suggests that the mechanism of liver injury is distinct from the pancreas. This is supported by the limited apoptosis in K18 Gly−pancreas versus liver after STZ exposure (Fig 3b,c; Supplemental Fig S5b) despite extensive injury of both organs.
Effect of PUGNAc/Fas-induced injury on protein kinase phosphorylation
We then compared kinase phosphorylation in K18 WT and K18 Gly− mice after PUGNAc or PUGNAc/Fas treatments. PUGNAc alone causes hypophosphorylation of Akt T308 in K18 WT mouse livers but does so more prominent in Gly− livers, with a minimal effect on PKCθ T538 phosphorylation (Fig 6a,b). After PUGNAc/Fas treatment, Akt1 T308 phosphorylation and expression of Hsp70 were dramatically inhibited in K18 Gly− livers in association with more prominent cleaved caspase-3 (Fig 6c). Akt1 is a known modulator of HSF1 which, in turn, leads to transcriptional upregulation of Hsp7042. Hence, inhibition of K18 glycosylation inactivates Akt and blocks its downstream regulation.
Figure 6. K18 Gly− inhibits Akt T308 phosphorylation and Hsp70 expression, companying with enhanced apoptosis in response to PUNAc/Fas.

a, b, FVB/n mice (a), or K18 WT and K18 Gly− mice (b) were given PUGNAc (7 mg/kg body weight) or vehicle. Livers were harvested at the indicated times after PUGNAc injection and total liver lysates were blotted with antibodies to the indicated antigens. c, K18 WT or K18 Gly− mice were pretreated with PUGNAc for 48 hr then injected with Fas Ab. Livers were harvested at 2, 5, 7, 9 hr after Fas injection and total liver lysates were immunoblotted with antibodies to the indicated antigens. Akt1 T308 phosphorylation rose 2 hr after PUGNAc+Fas in K18-WT livers (4.9 fold; compare lanes 1 and 2) but the increase in K18-Gly− livers was limited. d, BHK cells were transfected with vector alone, Akt1 WT or T308A mutant. After 2 days, the transfected cells were treated with 100 μM PUGNAc for 18 hrs. Total cell lysates were prepared and blotted with antibodies to the indicated antigens. Note that O-GlcNAc proteins accumulated after PUGNAc treatment (lanes 4 and 5). The expression of Akt WT and T308A mutant were confirmed using Akt or phospho-specific Akt antibody. e, Transfected BHK cells (with Akt1 WT or T308A mutant) were treated with 100 μM PUGNAc for 18 hr. O-GlcNAc proteins were immunoprecipitated with two different O-GlcNAc antibodies, Ab110.6 or Ab RL2. The immunoprecipitates were analyzed by blotting with antibodies to Akt or vimentin (as a loading control). Note that similar patterns were obtained by using two different O-GlcNAc antibodies. The Akt1 T308A mutant was not efficiently immunoprecipitated with the O-GlcNAc antibodies as compared with Akt1 WT, which indicates that mutation of T308 inhibits Akt glycosylation and suggests that the O-GlcNAc modification occurs at or near Akt T308.
Effect of PUGNAc on Akt1 glycosylation
Given the potential reciprocity between Ser/Thr phosphorylation and glycosylation, we tested if Akt T308 mutation affects Akt O-GlcNAclyation. PUGNAc causes accumulation of O-GlcNAc proteins in BHK cells transfected with Akt1 WT or T308A (Fig 6d). Notably, PUGNAc treatment results in Akt T308 hypophosphorylation (Fig 6d). In order to test the effect of Akt1 T308 phosphorylation on Akt1 glycosylation, the O-GlcNAc proteins were immunoprecipitated from transfected cells using two independent anti-O-GlcNAc antibodies then blotted with anti-Akt antibody. As compared with Akt1 WT, Akt1 T308A was much less efficiently immunoprecipitated using both O-GlcNAc antibodies under conditions that immunoprecipitated similar levels of endogenous O-GlcNAc-vimentin (Fig 6e). These findings indicate that the O-GlcNAc modification occurs at or near Akt1 T308 though an effect of T308 mutation on other Akt1 Ser/Thr modifications is also possible.
Akt1 associates with K8 but not with K18
The relationship between K18 glycosylation and Akt glycosylation/phosphorylation was investigated by asking whether Akt binds to K8/K18 and, if so, whether this binding depends on K18 glycosylation or Akt phosphorylation. There is already precedence for binding of the N-terminal region of K10 (which is expressed in keratinocytes) with Akt and this physical interaction causes sequestration of the kinase within cytoskeleton43. As shown in Fig 7, K8/K18 co-immunoprecipitate with Akt, using anti-K8/K18 or anti-Akt antibodies, when isolated from transfected cells (panel a) or from the livers of K18 WT or K18 Gly− mice (panel b). However, the K8/K18-Akt interaction is independent of keratin glycosylation, when tested using K18 Gly− transfectants or transgenic mice (Fig 7a,b), and is also independent of Akt T308 phosphorylation (Fig 7a). Transfection of BHK cells with K8, K18, K8+K18 or individual keratin deletion mutants showed that both K8 and K18 co-immunoprecipitated with Akt (Fig 7c, lane 2) due to the known obligate heterodimeric nature of K8/K18, but single keratin transfections showed Akt association with K8 (Fig 7c, lane 3) but not K18 (Fig 7c, lane 5) (Fig 7d for an independent experiment). The Akt-K8 binding does not involve the N-terminal domains of K8 or K18 (Fig 7d), thereby suggesting that Akt association with K8 is likely to occur via the K8 C-terminal domain within amino acids 254–483.
Figure 7. K8/K18 and Akt association is independent of PUGNAc/Fas treatment and Akt associates with keratin 8.

a, BHK cells were transfected with the indicated cDNA constructs then treated with PUGNAc (100 μM, 18 hr). K8/K18 or Akt immunoprecipitates were obtained then analyzed by SDS-PAGE and Coomassie blue staining or were blotted with the indicated antibodies. b, K18 WT or K18 Gly− mice were pretreated with PUGNAc (7 mg/kg body weight, 48 hr) and either injected with Fas antibody (0.15 mg/kg) or with saline (lanes 1,5). Livers were harvested 5, 7, 9 hr after Fas injection. K8/K18 and Akt immunoprecipitates were obtained then analyzed as in panel a. c, BHK cells were co-transfected with Akt WT and the indicated keratin constructs. K8 (1–253) and K18 (1–244) represent the N-terminal 253 or 244 amino acids, respectively. High salt extraction (HSE) was performed to generate a cytoskeletal preparation that contains primarily vimentin (the endogenous IF of BHK cells) and the transfected keratins followed by Coomassie blue staining (vimentin and K8 have similar migration under the analyzed conditions). The transfected cells undergo apoptosis due to the transfection stress conditions (Lipofectamine) and generate a stable 238 amino acid fragment as reported51. Total cell lysates were also prepared from the transfected cells and blotted with antibody 8592 which recognizes K8 and K18. K8* and K18* represent major bands in the blot, while other weak bands in the same positions represent degraded K8 or background bands also found in Akt-transfectants (lane 1). Akt immunoprecipitates were also prepared and blotted with the indicated antibodies. d, BHK cells were transfected as in panel c. Akt immunoprecipitates were prepared and blotted with antibodies to K8/K18 or Akt. Expression of K8 WT and K8 (1–253) were similar as noted in the blot of the total lysates (panel c; lanes 3,4) (not shown). The displayed K8/K18 blot shows the details of the antibody reactive species in the range of 28–53 kd. Note that intact K8 WT (483 amino acids) co-precipitated with Akt (lane 2), but the K8 N-terminal fragment (1–253) did not (lane 3). The band marked with white arrow represents secondary antibody crossreactivity.
DISCUSSION
Keratin glycosylation protects from protein over-O-GlcNAcylation related injury
Our findings demonstrate that site-specific ablation of K18 O-GlcNAc glycosylation in mice leads to susceptibility to injury that is related to STZ or a combined effect of PUGNAc and Fas. Blocking K18 glycosylation does not predispose to apoptosis or tissue injury per se (Fig 2, Supplemental Fig S2) but requires another insult that inhibits O-GlcNAcase and thereby raises the intracellular levels of protein O-GlcNAcylation (Fig 1d, Fig 6d). Therefore, K18 glycosylation appears to serve as a cytoprotective and anti-apoptosis buffer during conditions that promote increased protein O-GlcNAcylation. The cytoprotective role of K18 glycosylation appears to be different in the liver versus the pancreas since the site-specific hypophosphorylation of Akt and PKC that was observed in the liver was not observed in the pancreas (Fig 5e), thereby invoking organ-specific effects of K18 glycosylation.
The stoichiometry of K18 glycosylation is estimated to be ~2 molecules of O-GlcNAc per keratin molecule15, which provides a potential robust buffering capacity due to K18 abundance as a major cytoskeletal protein of simple epithelia (e.g., K8/K18 make up ~0.3% of total mouse liver protein32). The buffering capacity of K18 glycosylation is related to specific proteins that include signaling kinases (e.g., Akt1 as demonstrated herein) and is not a global effect since not all proteins became hyperglycosylated in K18-glycosylation deficient liver (Fig 1b).
The K18 glycosylation protective effect in mice is highly specific to this posttranslational modification since mice that overexpress either the K18 S53A phospho-mutant44 or the K18 R90C mutant that disrupts the keratin filament network of hepatocytes and renders them markedly susceptible to Fas-alone mediated apoptosis45 behave similar to WT mice when exposed to STZ. Notably, K18 R90C in vivo expression results in hyperglycosylation (and hyperphosphorylation) of K8/K1846 which, given the findings herein, raises the possibility that K18 hyperglycosylation may serve a protective role that can partially compensate for the effect of the R90C mutation. In addition, the similar susceptibility of K18 WT and K18 Gly− mice to Fas-alone mediated injury, in contrast with the disruptive structural effect of K18 R90C on keratin cytoplasmic filament organization and consequent predisposition to Fas-alone mediated apoptosis, suggests that the K18 Gly− mice cytoprotective phenotype is unlikely to be due to a structural effect of the three K18 Ser→Ala mutations that were introduced to inhibit K18 glycosylation. Further support for the specificity of our findings is based on the observation that K18-null mice are also predisposed to STZ-induced liver/pancreatic injury (Supplemental Fig S6).
How does K18 glycosylation provide a cytoprotective effect?
O-GlcNAcase inhibition unmasks a new K18 glycosylation function that protects K18-expressing epithelial tissues from injury, and links glycosylation of the keratin cytoskeleton to activation of cell survival kinases. Our findings support a model (Fig 8) whereby K18 glycosylation promotes a phospho-Akt pT308 active state that inhibits cell death. Phospho-Akt T308 is likely to be hypoglycosylated since O-GlcNAcase inhibition by PUGNAc or STZ leads to Akt hypophosphorylation (Fig 5b, Fig 6d). Also, Akt T308A mutation decreases Akt glycosylation as measured using two independent anti-O-GlcNAc antibodies (Fig 6e). Hence, these results implicate reciprocal phosphorylation and glycosylation at or near the Akt T308 residue, whereby each type of modification interferes with the other. The importance of O-GlcNAcase in regulating Akt phosphorylation and activity is also supported by the previously reported increase in Akt phosphorylation at T308 upon overexpression of O-GlcNAcase in mouse liver47. Therefore, K18 glycosylation provides its protective effect likely via promoting Akt phosphorylation at the critical T308 residue that regulates its kinase activity and possibly via PKCθ phosphorylation at T538, though additional regulatory phospho/glyco-proteins may be involved. Such crosstalk between O-GlcNAcylation and phosphorylation is an emerging theme in the regulation of several signaling cascades related to coping with stress including NFκB and other transcription factors19.
Figure 8. Model of K8 association with Akt and the effect of K18 glycosylation on reciprocal Akt phosphorylation and glycosylation.
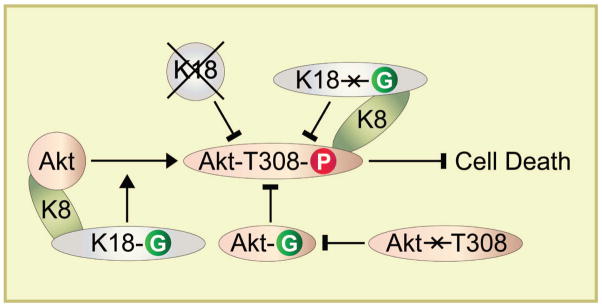
The schematic summarizes the results of the genetic animal models whereby ablation of K18 glycosylation or absence of the entire K18 protein in K18-null mice leads to Akt hypophosphorylation and inactivation which in turn promotes cell death. Inhibition of Akt T308 phosphorylation (by mutation to Akt T308A) inhibits Akt glycosylation (which may occur at T308 or an adjacent site), while hyperglycosylation of Akt (presumably at T308) leads to Akt hypophosphorylation at its regulatory T308.
The relationship of reciprocal Akt glycosylation and phosphorylation to K18 glycosylation does not appear to be mediated by upstream kinase regulation of Akt such as PDK1 activation (Fig 5,6). The binding of K8 to Akt, albeit in a K18 glycosylation and Akt T308 independent manner (Fig 7), suggests that the K8/K18 obligate heterodimeric complex scaffolds Akt akin to K10 association with Akt43. It remains to be determined whether K18 glycosylation helps recruit Akt kinase(s) or O-GlcNAcase given the general property of O-GlcNAcylated proteins to form multimeric complexes19. Such potential recruitment could promote Akt hyperphosphorylation and hypoglycosylation which in the absence of K18 glycosylation (e.g., in the K18 Gly− mice) could lead to the opposite effect of Akt hypophosphorylation and hyperglycosylation. Similar effects can occur if glyco-K18 is to recruit Akt phosphatase(s) and parallel paradigms might account for the observed PKCθ T538 hypophosphorylation as a consequence of K18 hypoglycosylation. However, PKCθ T538 phosphorylation does not appear to be significantly affected in the presence of PUGNAc as compared with Akt T308 phosphorylation which raises the possibility that more than one mechanism may be involved in transmitting the K18 glycosylation signal to a kinase inactivation effect.
In summary, our findings provide a link between keratin glycosylation and critical reciprocal kinase regulation by phosphorylation and glycosylation, which leads to anti-apoptotic and cytoprotective effects, as shown herein for Akt in the liver. These findings may extend to other IFs that are known to have the O-GlcNAc modification such as neurofilaments48 and vimentin49.
METHODS
Reagents
The primary reagents we used included: streptozotocin and galactosyl-transferase (Sigma-Aldrich); UDP-[4,5-3H]-galactose (PerkinElmer); collagenase type II (Worthington Biochemical Corp.); PUGNAc (Carbogen); antibodies to cleaved caspase 3, phospho-PTEN, phospho-protein kinases (Cell Signaling Technology), insulin (Abcam), Fas (BD Biosciences), O-GlcNAc (Covance), vimentin (Sigma) and keratins and phospho-keratins 50.
Generation of transgenic lines
The 10 kb human (h) K18 genomic DNA was mutated at three glycosylation sites (Ser30/31/49→Ala) using a Transformer Mutagenesis kit (Clontech). This genomic construct contains all the regulatory elements needed to retain the normal tissue-specific expression31. The mutated genomic DNA (Gly−) was used for injection into pronuclei of fertilized FVB/n mouse eggs. Progeny mice were screened by PCR amplification of mouse tail genomic DNA for the presence of hK18. Two independent heterozygous mouse lines were established and used for the studies herein, and both lines gave similar results (not shown). The two K18 Gly− mutant lines had comparable expression of hK18 protein to the hK18-WT mouse line. Of note, the human and mouse K18 amino acid sequences show 86% sequence identity and 92% sequence similarity [accession numbers: NP_000215.1 (human K18) and NP_034794.2 (mouse K18)].
The other experimental lines included the hK18 Ser53Ala13 (which ablates a major K18 phosphorylation site), the hK18 Arg90Cys that causes keratin filament disruption and predisposes to apoptosis32, and K18-null mice (originally kindly provided by Dr. Thomas Magin, University of Bonn; now available through The Jackson Laboratory). All mouse transgenic lines were in an FVB/n background. The K18-null mouse (originally in a mixed strain background) were backcrossed to an FVB/n background for 6 generations.
Drug administration
Mice were fasted overnight before each drug administration. Mice were injected intraperitoneally with STZ (200 mg/kg body weight) or vehicle (50 mM sodium citrate; pH 4.5). Alternately, mice were injected intraperitoneally with PUGNAc (7 mg/kg body weight) or vehicle (PBS), and after 48 hours the mice were retreated with Fas antibody (0.15 mg/kg mouse body weight). Mice were then euthanized by CO2 inhalation. Blood was collected by intracardiac puncture, and tissues were: (i) fixed in 10% formalin for hematoxylin/eosin staining, (ii) fixed in acetone for immunofluorescence staining, or (iii) frozen in liquid nitrogen for subsequent biochemical analysis50. Blood glucose was measured from tail blood using an automated glucose monitor (One Touch Ultra, Lifescan). Insulin levels were determined by ELISA (Insulin ELISA kit, Linco Research Inc).
In vitro galactosylation, immune staining and blot analysis
In vitro galactosylation was carried out using keratin immunoprecipitates or total tissue detergent-free cytosol fractions that were incubated with UDP-[4,5-3H]-galactose and galactosyltransferase which labels terminal GlcNAcs50. Immunofluorescence staining was performed as described50. Keratin enriched fractions were prepared by high salt extraction, separated by SDS-PAGE then stained with Coomassie blue to compare protein levels of endogenous mouse K18 and ectopic expressed human K18. Total lysates were obtained by solubilizing in SDS-containing sample buffer, separated by SDS-PAGE, transferred to membranes and then immunoblotted.
Cell transfections
BHK-21 (hamster kidney) cells were transfected by using LipofectAMINE (Invitrogen) as recommended by the supplier. After 2 days transfection, the cells were harvested for further experiments.
Statistical analysis
Statistical comparisons were carried out using Fisher’s exact test performed with StatView software.
Supplementary Material
Acknowledgments
We are grateful to Drs. Robert Oshima (The Sanford-Burnham Medical Research Institute) and Thomas Magin (University of Bonn) for making the K18 WT or K18-null mice available to us, Dr. Yanru Chen-Tsai and the Stanford University Transgenic Facility for helping generate the transgenic mice, Evelyn Resurreccion for assistance with immune staining and Pauline Chu for assistance with hematoxylin and eosin staining. This work was supported by NIH grants DK47918 and the Department of Veterans Affairs (M.B.O.), NIH Digestive Disease Center grant DK56339 to Stanford University, and WCU project (R31-2008-000-10086-0) from the Korean Ministry of Education, Science and Technology (N-O.K.).
Abbreviations
- ALT
alanine aminotransferase
- GSK
glycogen synthase kinase
- h
human
- IF
intermediate filament
- H&E
hematoxylin and eosin
- i.p
immunoprecipitate
- K
keratin
- O-GlcNAc
O-linked N-acetylglucosamine
- OGT
O-GlcNAc transferase
- O-GlcNAcase
N-acetyl-D-glucosaminidase
- p
phospho
- PDK1
phosphoinositide-dependent protein kinase 1
- PI3K
phosphoinositide 3-kinase
- PKC
protein kinase C
- WT
wild type
Footnotes
AUTHOR CONTRIBUTIONS: N.-O.K. and M.B.O. conceived and designed the study. N.-O.K., D.M.T. and P.S. performed the experiments and analyzed the data. N.-O.K., D.M.T., P.S. and M.B.O. interpreted the data. N.-O.K. and M.B.O. wrote the manuscript with comments by D.M.T. and P.S.
COMPETING FINANCIAL INTERESTS: The authors declare no competing financial interests.
References
- 1.Ku NO, Zhou X, Toivola DM, Omary MB. The cytoskeleton of digestive epithelia in health and disease. Am J Physiol. 1999;277:G1108–1137. doi: 10.1152/ajpgi.1999.277.6.G1108. [DOI] [PubMed] [Google Scholar]
- 2.Fuchs E, Cleveland DW. A structural scaffolding of intermediate filaments in health and disease. Science. 1998;279:514–519. doi: 10.1126/science.279.5350.514. [DOI] [PubMed] [Google Scholar]
- 3.Omary MB, Coulombe PA, McLean WH. Intermediate filament proteins and their associated diseases. N Engl J Med. 2004;351:2087–2100. doi: 10.1056/NEJMra040319. [DOI] [PubMed] [Google Scholar]
- 4.Coulombe PA, Omary MB. “Hard” and “Soft” principles defining the strucuture, function and regulation of keratin intermediate filaments. Curr Opin Cell Biol. 2002;14:110–122. doi: 10.1016/s0955-0674(01)00301-5. [DOI] [PubMed] [Google Scholar]
- 5.Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell. 1982;31:11–24. doi: 10.1016/0092-8674(82)90400-7. [DOI] [PubMed] [Google Scholar]
- 6.Ku NO, Gish R, Wright TL, Omary MB. Keratin 8 mutations in patients with cryptogenic liver disease. N Engl J Med. 2001;344:1580–1587. doi: 10.1056/NEJM200105243442103. [DOI] [PubMed] [Google Scholar]
- 7.Ku NO, et al. Keratins as susceptibility genes for end-stage liver disease. Gastroenterology. 2005;129:885–893. doi: 10.1053/j.gastro.2005.06.065. [DOI] [PubMed] [Google Scholar]
- 8.Omary MB, Ku NO, Strnad P, Hanada S. Towards unraveling the complexity of ‘simple’ epithelial keratins in human disease. J Clin Invest. 2009;119:1794–1805. doi: 10.1172/JCI37762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strnad P, et al. Keratin variants predispose to adverse outcomes from acute liver failure: Race and ethnic associations. Gastroenterology. 2010 doi: 10.1053/j.gastro.2010.06.007. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oriolo AS, Wald FA, Ramsauer VP, Salas PJ. Intermediate filaments: a role in epithelial polarity. Exp Cell Res. 2007;313:2255–2264. doi: 10.1016/j.yexcr.2007.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toivola DM, Tao GZ, Habtezion A, Liao J, Omary MB. Beyond cellular integrity: organelle-related and protein-targeting functions of intermediate filaments. Trends Cell Biol. 2005;15:608–617. doi: 10.1016/j.tcb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 12.Kim S, Coulombe PA. Emerging role for the cytoskeleton as an organizer and regulator of translation. Nat Rev Mol Cell Biol. 2010;11:75–81. doi: 10.1038/nrm2818. [DOI] [PubMed] [Google Scholar]
- 13.Omary MB, Ku NO, Tao GZ, Toivola DM, Liao J. “Heads and tails” of intermediate filament phosphorylation: multiple sites and functional insights. Trends Biochem Sci. 2006;31:383–394. doi: 10.1016/j.tibs.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 14.Ku NO, Omary MB. A disease and phosphorylation related non-mechanical function for keratin 8. J Cell Biol. 2006;174:115–125. doi: 10.1083/jcb.200602146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chou CF, Smith AJ, Omary MB. Characterization and dynamics of O-linked glycosylation of human cytokeratin 8 and 18. J Biol Chem. 1992;267:3901–3906. [PubMed] [Google Scholar]
- 16.Ku NO, Omary MB. Identification and mutational analysis of the glycosylation sites of human keratin 18. J Biol Chem. 1995;270:11820–11827. doi: 10.1074/jbc.270.20.11820. [DOI] [PubMed] [Google Scholar]
- 17.Omary MB, Ku NO, Liao J, Price D. Keratin modifications and solubility properties in epithelial cells and in vitro. Subcell Biochem. 1998;31:105–140. [PubMed] [Google Scholar]
- 18.Kudlow JE. Post-translational modification by O-GlcNAc: another way to change protein function. J Cell Biochem. 2006;98:1062–1075. doi: 10.1002/jcb.20926. [DOI] [PubMed] [Google Scholar]
- 19.Zeidan Q, Hart GW. The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J Cell Sci. 2010;123:13–22. doi: 10.1242/jcs.053678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haltiwanger RS, Philipsberg GA. Mitotic arrest with nocodazole induces selective changes in the level of O-linked N-acetylglucosamine and accumulation of incompletely processed N-glycans on proteins from HT29 cells. J Biol Chem. 1997;272:8752–8758. doi: 10.1074/jbc.272.13.8752. [DOI] [PubMed] [Google Scholar]
- 21.Chou CF, Omary MB. Mitotic arrest with anti-microtubule agents or okadaic acid is associated with increased glycoprotein terminal GlcNAc’s. J Cell Sci. 1994;107:1833–1843. doi: 10.1242/jcs.107.7.1833. [DOI] [PubMed] [Google Scholar]
- 22.Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–1022. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 23.Yang WH, et al. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol. 2006;8:1074–1083. doi: 10.1038/ncb1470. [DOI] [PubMed] [Google Scholar]
- 24.Love DC, Hanover JA. The hexosamine signaling pathway: deciphering the “O-GlcNAc code”. Sci STKE. 2005:re13. doi: 10.1126/stke.3122005re13. [DOI] [PubMed] [Google Scholar]
- 25.Zachara NE, et al. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem. 2004;279:30133–30142. doi: 10.1074/jbc.M403773200. [DOI] [PubMed] [Google Scholar]
- 26.Ngoh GA, et al. Unique hexosaminidase reduces metabolic survival signal and sensitizes cardiac myocytes to hypoxia/reoxygenation injury. Circ Res. 2009;104:41–49. doi: 10.1161/CIRCRESAHA.108.189431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Slawson C, Copeland RJ, Hart GW. O-GlcNAc signaling: a metabolic link between diabetes and cancer? Trends Biochem Sci. 2010 doi: 10.1016/j.tibs.2010.04.005. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Z, Gucek M, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc Natl Acad Sci U S A. 2008;105:13793–13798. doi: 10.1073/pnas.0806216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao Y, Parker GJ, Hart GW. Streptozotocin-induced beta-cell death is independent of its inhibition of O-GlcNAcase in pancreatic Min6 cells. Arch Biochem Biophys. 2000;383:296–302. doi: 10.1006/abbi.2000.2094. [DOI] [PubMed] [Google Scholar]
- 30.Vosseller K, Wells L, Lane MD, Hart GW. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci U S A. 2002;99:5313–5318. doi: 10.1073/pnas.072072399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abe M, Oshima RG. A single human keratin 18 gene is expressed in diverse epithelial cells of transgenic mice. J Cell Biol. 1990;111:1197–1206. doi: 10.1083/jcb.111.3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ku NO, Strnad P, Zhong BH, Tao GZ, Omary MB. Keratins let liver live: mutations predispose to liver disease and crosslinking generates Mallory-Denk bodies. Hepatology. 2007;46:1639–1649. doi: 10.1002/hep.21976. [DOI] [PubMed] [Google Scholar]
- 33.Gukovskaya AS, Gukovsky I, Jung Y, Mouria M, Pandol SJ. Cholecystokinin induces caspase activation and mitochondrial dysfunction in pancreatic acinar cells. Roles in cell injury processes of pancreatitis. J Biol Chem. 2002;277:22595–22604. doi: 10.1074/jbc.M202929200. [DOI] [PubMed] [Google Scholar]
- 34.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 35.Stambolic V, Woodgett JR. Functional distinctions of protein kinase B/Akt isoforms defined by their influence on cell migration. Trends Cell Biol. 2006;16:461–466. doi: 10.1016/j.tcb.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 36.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 37.Chen WS, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andjelkovic M, et al. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- 39.Parker PJ, Murray-Rust J. PKC at a glance. J Cell Sci. 2004;117:131–132. doi: 10.1242/jcs.00982. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, Graham C, Li A, Fisher RJ, Shaw S. Phosphorylation of the protein kinase C-theta activation loop and hydrophobic motif regulates its kinase activity, but only activation loop phosphorylation is critical to in vivo nuclear-factor-kappaB induction. Biochem J. 2002;361:255–265. doi: 10.1042/bj3610255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hayashi K, Altman A. Protein kinase C theta (PKCtheta): a key player in T cell life and death. Pharmacol Res. 2007;55:537–544. doi: 10.1016/j.phrs.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nadeau SI, Landry J. Mechanisms of activation and regulation of the heat shock-sensitive signaling pathways. Adv Exp Med Biol. 2007;594:100–113. doi: 10.1007/978-0-387-39975-1_10. [DOI] [PubMed] [Google Scholar]
- 43.Paramio JM, Segrelles C, Ruiz S, Jorcano JL. Inhibition of protein kinase B (PKB) and PKCzeta mediates keratin K10-induced cell cycle arrest. Mol Cell Biol. 2001;21:7449–7459. doi: 10.1128/MCB.21.21.7449-7459.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ku NO, et al. Mutation of a major keratin phosphorylation site predisposes to hepatotoxic injury in transgenic mice. J Cell Biol. 1998;143:2023–2032. doi: 10.1083/jcb.143.7.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ku NO, Soetikno RM, Omary MB. Keratin mutation in transgenic mice predisposes to Fas but not TNF-induced apoptosis and massive liver injury. Hepatology. 2003;37:1006–1014. doi: 10.1053/jhep.2003.50181. [DOI] [PubMed] [Google Scholar]
- 46.Ku NO, Michie S, Oshima RG, Omary MB. Chronic hepatitis, hepatocyte fragility, and increased soluble phosphoglycokeratins in transgenic mice expressing a keratin 18 conserved arginine mutant. J Cell Biol. 1995;131:1303–1314. doi: 10.1083/jcb.131.5.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soesanto YA, et al. Regulation of Akt signaling by O-GlcNAc in euglycemia. Am J Physiol Endocrinol Metab. 2008;295:E974–980. doi: 10.1152/ajpendo.90366.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong DL, Xu ZS, Hart GW, Cleveland DW. Cytoplasmic O-GlcNAc modification of the head domain and the KSP repeat motif of the neurofilament protein neurofilament-H. J Biol Chem. 1996;271:20845–20852. doi: 10.1074/jbc.271.34.20845. [DOI] [PubMed] [Google Scholar]
- 49.Slawson C, Lakshmanan T, Knapp S, Hart GW. A mitotic GlcNAcylation/phosphorylation signaling complex alters the posttranslational state of the cytoskeletal protein vimentin. Mol Biol Cell. 2008;19:4130–4140. doi: 10.1091/mbc.E07-11-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ku NO, et al. Studying simple epithelial keratins in cells and tissues. Methods Cell Biol. 2004;78:489–517. doi: 10.1016/s0091-679x(04)78017-6. [DOI] [PubMed] [Google Scholar]
- 51.Ku NO, Liao J, Omary MB. Apoptosis generates stable fragments of human type I keratins. J Biol Chem. 1997;272:33197–33203. doi: 10.1074/jbc.272.52.33197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.