

Abstract
Aims
Coagulopathy is often present after resuscitation from cardiac arrest but plays an undefined role in the post cardiac arrest syndrome. The aim of this study was to characterize coagulation changes during cardiac arrest and post-resuscitation care in order to direct further focused study.
Methods
Ventricular fibrillation (VF) was induced electrically in immature male swine, followed by normothermic American Heart Association Advanced Cardiac Life Support and a uniform post-resuscitation goal-directed resuscitation protocol. PT, aPTT, fibrinogen, Thrombelastography (TEG), platelet contractile force (PCF), clot elastic modulus (CEM), and collagen-induced platelet aggregation were compared at baseline, at 8 minutes of VF, during the 3rd round of chest compressions (CPR), and at 15, 90, 180, and 360 minutes after return of circulation using repeated measures ANOVA.
Results
8/18 (44%) animals were resuscitated after 10.9+/− 0.9 minutes of VF and 7.6+/−3.4 minutes of CPR. TEG revealed a significant impairment in clot strength (MA) and clot formation kinetics (K, Alpha Angle) arising during CPR, followed by a brief prolongation of clot onset times (R) after return of circulation. Both PCF and CEM fell significantly during CPR (PCF by 50%, CEM by 47% of baseline) and platelet aggregation was significantly decreased during CPR. Coagulation changes were partially recovered by 3 hours of post-resuscitation care.
Conclusions
Whole blood coagulation was rapidly impaired during CPR after electrically-induced VF in this swine model by impaired platelet aggregation/contractile function and clotting kinetics. Further platelet-specific study is indicated.
Introduction
Only 7.9% of the estimated 295,000 out of hospital cardiac arrest patients with initial shockable rhythms survive to hospital discharge in the United States.1 Furthermore, hospital mortality remains high (55-71%) in those who are resuscitated initially. 2,3 A primary contributor to the high post-resuscitation mortality is the post-cardiac arrest syndrome (PCAS), consisting of impaired hemodynamic function, neurologic injury, and uncontrolled inflammation. PCAS can emerge rapidly following return of spontaneous circulation (ROSC) and there is a growing body of evidence that global ischemia/reperfusion injury contributes to the pathology and high mortality of PCAS.4,5
The coagulation system may play a key role in PCAS. Anticoagulation has been identified in cardiac arrest survivors by early prolongation in prothrombin and activated partial thromboplastin times.6,7 These coagulation changes are more prominent in those with refractory hemodynamic shock and are associated with early in-hospital mortality.7 However, our current understanding is incomplete, being limited to plasma-based assays only.
The purpose of this study was to improve our understanding of the coagulation system during cardiac arrest and resuscitation by characterizing the the whole blood response. We hypothesize that tests of whole blood coagulation function will be significantly altered during cardiac arrest and the post-resuscitation period when compared to pre-arrest values.
Methods
Subjects and Instrumentation
Immature 40-50kg domestic male swine were fed ad-libitum with freely available drinking water except that food was withheld the night before the planned experiment to decrease gastric distention. Animals were sedated by intramuscular ketamine/xylazine (20mg/kg and 2mg/kg respectively) and induction was by intravenous pentathol injection (25 mg/kg). Intravenous alpha chloralose bolus (50 mg/kg) and continuous infusion (10 mg/kg/hr) provided continuous anesthesia. Subjects were intubated and ventilated with FiO2 = 21% (iVent201, VersaMed Medical Systems, Inc., Pearl River, NY). The ventilator rate was adjusted for normal pH and pCO2 = 35-45 mmHg and was not altered for the rest of the protocol other than during cardiac arrest when the ventilator was disconnected.
Electrodes were placed on the precordium for continuous ECG monitoring and the right carotid artery was cannulated for continuous recording of mean arterial pressure (MAP), arterial blood gas, and coagulation sampling. A tongue oximeter measured arterial oxygen saturation. The right internal jugular vein was cannulated using a central venous catheter (Edwards Life Sciences, Irvine, CA) with the tip placed at the junction of the superior vena cava and right atrium for monitoring of central venous pressure (CVP) and administration of medications and fluids. An equilibration period of 10 minutes followed, during which time baseline measurements were recorded. Body temperature was maintained in the normal porcine range 38+/− 1 deg C by a heating blanket and monitored by rectal probe.
Cardiac Arrest Protocol
Ventricular fibrillation (VF) was induced by passage of 100 mA 60 Hz alternating current for 3-5 seconds using a rheostat via electrodes at the precordium. Cardiac arrest was confirmed by the presence VF rhythm on ECG and disappearance of the arterial blood pressure waveform. The ventilator was then disconnected and all infusions except anesthesia stopped to simulate an untreated cardiac arrest scenario for 10-12 minutes. Previous model development indicated a ROSC rate of 50% with this arrest duration in preparation for future testing of therapeutic interventions.
Cardiopulmonary resuscitation (CPR) was performed according to the American Heart Association Advanced Cardiac Life Support guidelines (2005 revision), but a mechanical device was used for chest compressions.8 Mechanical CPR was initiated using LUCAS™ Chest Compression System (Physio Control Inc., Redmond, WA) providing 100 chest compressions per minute with at least 2 inches of precordial chest wall collapse with full rebound between each stroke. At the same time, asynchronous mechanical ventilations were started with FiO2 at 100%. Chest compressions were performed uninterrupted for a period of 2 minutes with rhythm checks performed every 2 minutes. If VF or pulseless Ventricular Tachycardia (VT) was present, biphasic defibrillation was attempted once at 120 Joules using a manual defibrillator (Zoll M Series, Zoll Medical Corporation, Chelmsford, MA) via paddles placed at the precordium. Intravenous Vasopressin (40IU), epinephrine (0.01mg/kg repeated every 4 minutes), atropine (0.5mg repeated till a maximum dose of 0.04mg/kg for asystole or PEA) and amiodarone 300mg were given according to ACLS guidelines. Resuscitation was continued until ROSC or 16 minutes (8th rhythm check). Without ROSC the animal was euthanized with an intravenous injection of 2mmol/L of potassium chloride under anesthesia. Successful ROSC was defined as an initial return of a sinus ECG rhythm with an obvious arterial wave form and mean arterial pressure >30 mmHg lasting at least 5 minutes in order to exclude unstable animals requiring further rounds of CPR after initial ROSC.
A simplified post-ROSC goal-directed treatment protocol was used in order to standardize post-resuscitation physiology and care due to the influence of refractory shock on coagulation status.7 The protocol was adapted from clinical septic shock and cardiac arrest protocols as reported by Rivers et al, and Gaieski et al, respectively.9,10 Our chosen goals were CVP >8 (achieved by normal saline bolus and infusion), MAP > 65 mmHg (achieved by titration of norepinephrine infusion), and tongue oxygen saturation >92% by supplemental oxygen titration. Of note, we chose a lower mean MAP goal of >65 mmHg rather than the MAP > 80 mmHg suggested by Gaieski et al, because we felt that the large volumes of vasopressor and crystalloid required to maintain this higher blood pressure in the face of significant capillary leakage and cardiac dysfunction. These volumes would have likely impacted coagulation function, and possibly confounded our results. FiO2 was decreased incrementally after ROSC to achieve adequate oxygen saturation using the lowest possible oxygen concentration. Surviving animals were maintained up to 6 hours following ROSC then euthanized under anesthesia using 2mmol/kg intravenous potassium chloride. Therapeutic hypothermia was not induced due to lack of evidence for an acute effect on coagulation function and the need for large volumes of iced intravenous fluids to rapidly produce hypothermia, causing plasma dilution and obscuring the native coagulation response.11
Measurements
Hemodynamic measurements, cell counts, chemistry, and coagulation studies were measured from the arterial circulation at baseline (BL), at 8 minutes of VF (VF), at the start of the third round of chest compressions (CPR), and 15, 90, 180, and 360 minutes post-resuscitation (PR). Arterial samples were required due to collapse of veins during VF which prevented adequate blood sampling. Systemic blood gases and lacate measurements were made using the Stat Profile Critical Care Xpress bedside analyzer (Nova Biomedical Corp., Waltham, MA). Hematocrit (Hct), hemoglobin (Hgb), white blood cell (WBC) and platelet counts (PLT) were obtained using the VetScan HM2 Hematology System, bedside analyzer (Abaxid, Union City, CA).
Coagulation Studies
Coagulation studies included; Prothrombin time (PT), Activated Partial Thromboplastin Time (aPTT), and fibrinogen concentration in platelet poor plasma using the STart-4 coagulation analyzer (Diagnostica Stago, Asnières, France). Whole-blood coagulation function was determined by Thrombelastography™ (TEG) (Haemoscope Corporation, Niles, IL).
The Hemostasis Analysis System (HAS) (Hemodyne Inc. Richmond, VA, USA) was also used to clarify the platelet contribution to clot formation. The device and its applications have been summarized previously.12 In brief; Platelet Contractile Force (PCF) represents platelet-induced clot contraction and is measured in Kilodynes once per minute for 20 minutes. A separate estimate of Clot Elastic Modulus (CEM) is made by recording stress/strain ratio after application of a brief constant force to the surface of the clot once per minute and is reported in Kilodynes/cm2 after 700 μL of whole citrated blood is mixed with 50 μL of calcium chloride (10mmol/l final concentration) to initiate clotting. The terminal values of each measurement taken at 20 minutes of clot formation time were used for analysis after being performed in duplicate at 37°C.
Platelet aggregation was measured to clarify the role of platelet membrane receptors using collagen (4ug/ml) as the platelet agonist due to its selectivity for activating platelets via surface membrane receptors (glycoprotein Ia/IIa and glycoprotein VI).13 Aggregation was measured in whole blood by impedance at 37°C in reference to platelet-poor plasma using the Chrono-log aggregometer (Chrono-log Corp, Havertown, PA).
Effect of Vasoactive Medications
Coagulation function and platelet activity are altered in response to circulating epinephrine and vasopressin.14,15 Therefore, a separate ex vivo experiment examining the contribution of vasoactive drugs administered during CPR to coagulopathy. Whole blood was sampled at baseline prior to cardiac arrest and incubated with the same estimated concentration that the animal would have received upon 3 rounds of CPR of; 1. epinephrine (6×10−4 mg/ml), 2. vasopressin (0.01 U/ml), or 3. epinephrine + vasopressin. TEG and HAS were then used to evaluate for any drug-specific effects on coagulation function compared to baseline controls.
Statistical Analysis
Measurements were summarized as means with standard deviation and standard error. One way repeated-measure ANOVA was used to identify changes in individual variables over time. One way ANOVA evaluated for a significant effect of epinephrine, vasopressin, or epinephrine+vasopressin on coagulation function. Tukey Kramer adjustment for multiple comparisons was made to maintain overall significance at p=0.05 in all cases. Statistical analysis was made using JMP8.0 software (SAS Inc., Durham, NC).
Ethical Issues
This study was approved by the Virginia Commonwealth University Institutional Animal Use Committee and abided by international standards for the ethical treatment of animals.
Results
Eighteen immature male swine (weight (Mean/SD) 48 +/− 7.6 kg) were studied. VF was induced for an average of 10.9 +/− 0.9 minutes. Eight swine (44%) achieved ROSC after 7.6+/−3.4 minutes or approximately 3 rounds of cardiopulmonary resuscitation. All 8 of the successfully resuscitated animals achieved the post-resuscitation hemodynamic goals and survived at least six hours following ROSC. Core temperature averaged 37.7 ± 1.6 deg C during the protocol. Repeated-measure ANOVA revealed no significant change in core temperature between the baseline, arrest, and post-resuscitation periods (p=0.53).
Table 1 summarizes the changes in standard coagulation tests, chemistries, and cell counts. There were no changes in plasma-phase coagulation tests, other than a briefly prolonged PT and decreased fibrinogen during CPR. Blood pH was decreased for the period of 15-90 minutes post-ROSC. Arterial PO2 was only minimally lowered during VF and increased significantly during CPR. Lactate increased significantly during CPR compared to baseline, continued to increase during the first 15 minutes post-ROSC, and then declined during the remainder of the post-ROSC period. Hematocrit increased with CPR and after ROSC. Platelet count decreased during CPR but rebounded to near baseline levels by 15 minutes post-ROSC.
Table 1.
Summary statistics of coagulation tests, cell counts, and metabolic measurements compared over time.
| Protocol Time | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BL (n=18) | VF (n=18) | CPR (n=18) | PR15 (n=8) | PR90 (n=8) | PR180 (n=8) | PR360 (n=8) | ||||||||
| Coagulation | Mean | SE | Mean | SE | Mean | SE | Mean | SE | Mean | SE | Mean | SE | Mean | SE |
| PT (sec) | 14.7 | 0.6 | 14.3 | 0.6 | 16.8 | 0.9 | 13.4 | 0.8 | 13.8 | 0.8 | 12.5 | 0.8 | 13.4 | 0.9 |
| aPTT (sec) | 27.3 | 1.4 | 25.6 | 1.4 | 27.2 | 2.2 | 27.2 | 2.0 | 26.9 | 2.2 | 24.1 | 2.0 | 28.5 | 2.2 |
| Fibrinogen (mg/dl) | 138.9 | 17.9 | 135.4 | 17.9 | 74.1 | 27.9 | 159.3 | 26.1 | 172.0 | 28.0 | 173.3 | 26.1 | 180.0 | 30.2 |
| Cell Counts | ||||||||||||||
| Hgb (mg/dl) | 9.2 | 0.3 | 9.3 | 0.5 | 11.7* | 0.6 | 12.6* | 0.6 | 12.5* | 0.5 | 12.4* | 0.4 | 12.2* | 0.5 |
| Hct (%) | 27.9 | 1.1 | 26.3 | 1.7 | 31.9 | 2.1 | 35.8* | 2.1 | 35.2* | 1.6 | 32.7 | 1.5 | 32.1 | 1.8 |
| plt (×10^3) | 273.5 | 17.3 | 255.8 | 27.3 | 147.3* | 33.5 | 237.0 | 33.5 | 306.7 | 25.3 | 313.0 | 23.7 | 268.2 | 29.9 |
| Metabolism | ||||||||||||||
| pH | 7.454 | 0.026 | 7.513 | 0.026 | 7.318 | 0.042 | 7.12 9* | 0.039 | 7.29 4* | 0.042 | 7.352 | 0.039 | 7.339 | 0.045 |
| pO2 (mmHg) | 115.2 | 27.5 | 73.4 | 22.4 | 282.7* | 35.9 | 350.7* | 33.5 | 268.3* | 35.9 | 264.6* | 33.5 | 262.0* | 38.7 |
| Lac (mmol/L) | 1.0 | 0.5 | 2.0 | 0.5 | 6.2* | 0.8 | 11.0* | 0.8 | 5.4* | 0.8 | 4.4* | 0.8 | 2.7* | 0.9 |
Plasma-phase coagulation tests, arterial blood cell counts, and markers of metabolism at baseline (BL), after 8 minutes of ventricular fibrillation (VF), at 3rd round of cardiopulmonary resuscitation (CPR), and post-resuscitation (PR) 15, 90, 180, and 360 minutes.
= significantly different than baseline value by repeated measure ANOVA with Tukey HSD.
Thrombelastography
Results of whole blood TEG parameters are summarized in Figures 2 and 3. Clot initiation time (R) was shortened and strength (MA) was increased during VF, followed by rapid inhibition of clot formation (K, Alpha Angle) and MA during CPR which partially recovered by 180 minutes post-ROSC. Changes in K, Alpha Angle, and MA tended to precede prolongation of R time (Figure 3.).
Figure 2.
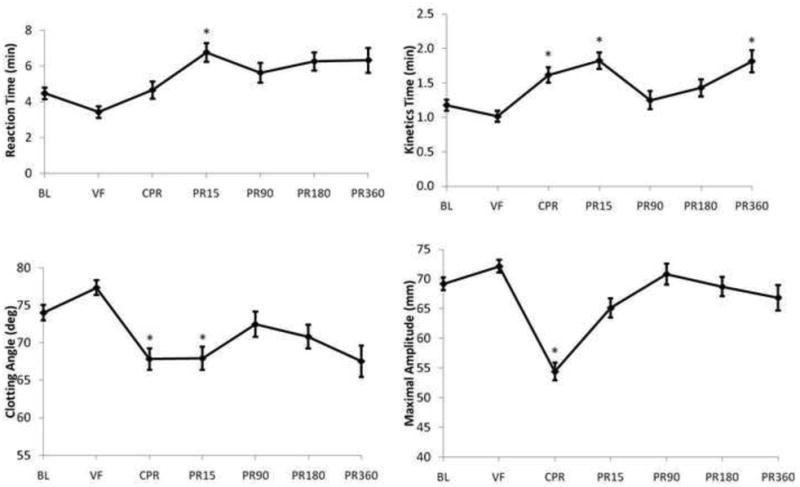
Summary of Thrombelastography (TEG) results in whole blood sampled as described under Methods. An aliquot of 340 μL of citrated whole blood was recalcified with 20 μL of 0.2M calcium chloride to initiate coagulation. Reaction time= time to onset of clot formation, Kinetics time= estimate of speed of clot buildup/polymerization, Clotting Angle= estimate of clot polymerization rate, Maximal Amplitude= estimate of clot strength. *= significantly different than baseline value by repeated measure ANOVA with Tukey HSD.
Figure 3.
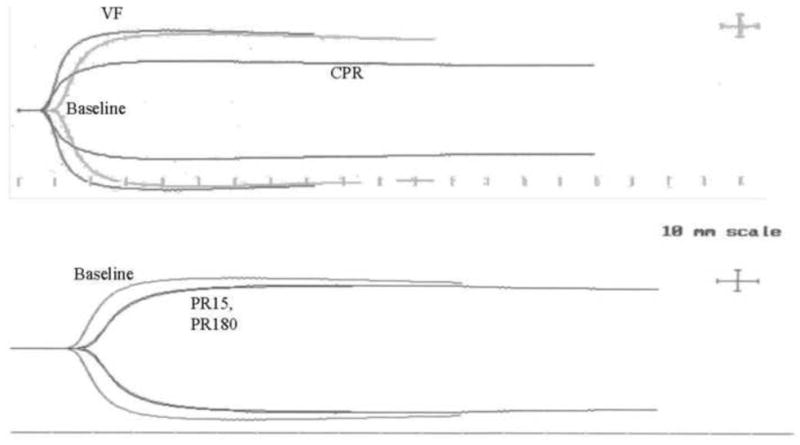
Representative TEG clot formation curves for a single subject at various times during the experimental protocol as described under Methods.
Hemostasis Analysis System
Both PCF and CEM were slightly increased during VF and then fell significantly during CPR (PCF by 50%, CEM by 47% of baseline) and remained so for up to 180 minutes post-resuscitation (Figure 4).
Figure 4.
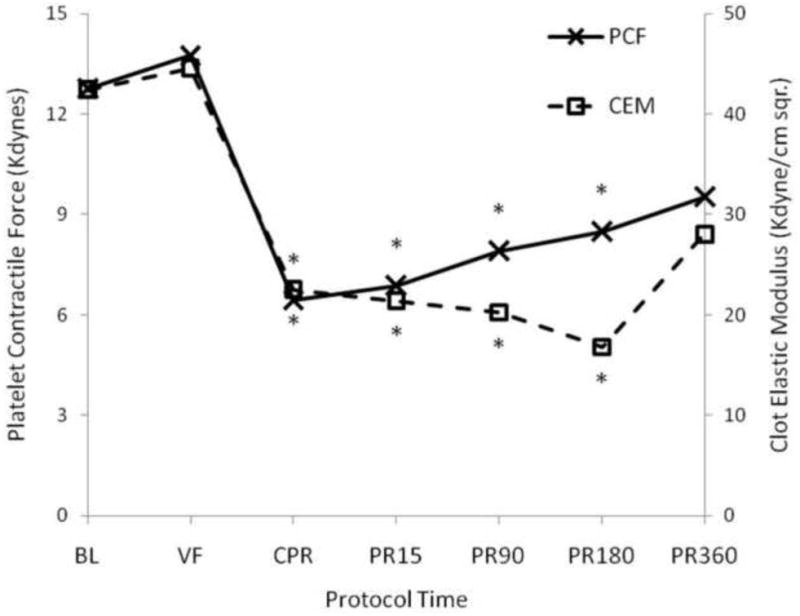
Summary of platelet-induced clot contraction and clot elastic modulus during cardiac arrest and resuscitation. Mean platelet contractile force (PCF) on the left axis and clot elastic modulus (CEM) on the right axis measured before, during and after cardiac arrest and resuscitation as described in Methods. *= significantly different than baseline value by one way ANOVA with Tukey HSD.
Platelet Aggregation
Figure 5 summarizes changes in collagen-induced platelet aggregation which tended to increase during VF, significantly decrease during CPR, and partially recover post-ROSC.
Figure 5.
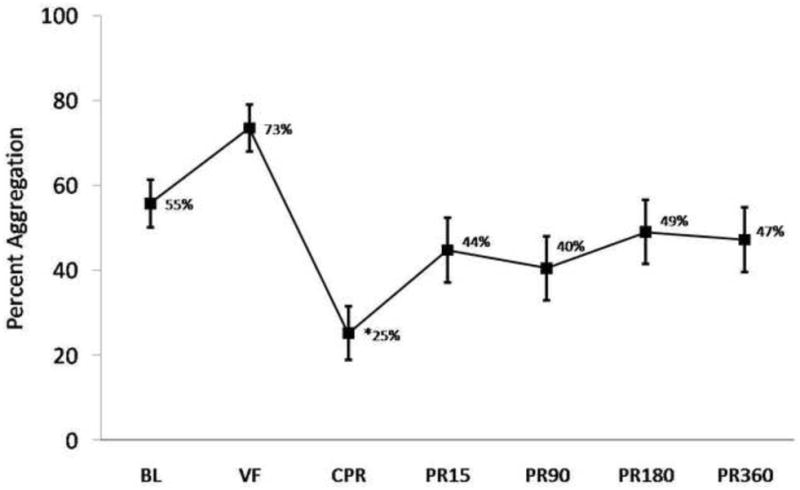
Average Collagen-induced platelet aggregation in whole blood by impedance performed as described in Methods. *= significantly different than baseline value by one way ANOVA with Tukey HSD. Error bars = SE.
Effect of Vasoactive Medications
Incubation of blood samples drawn at baseline with vasoactive medications used during CPR singularly or in combination (N=10, each group) had no significant effect on TEG or HAS values compared to controls (all p > 0.32, data not shown).
Discussion
In this study whole-blood clot formation became impaired during CPR, lasting up to 3 hours post-ROSC. The impaired clot formation was associated with reduced platelet contraction and aggregation, suggesting an important impact of cardiac arrest and resuscitation on platelet function. Our result differs from current clinical literature which has described platelet activation and disseminated intravascular coagulation in cardiac arrest survivors. The specific mechanism of platelet inhibition in our study remains unclear.
The effect of electrically-induced VF on platelet function in our model differs from clinical reports describing platelet hyperfunction in ischemia-induced cardiac arrest and disseminated intravascular coagulation (DIC) in hypoxia-driven experimental arrest models.16,17 Spiel et al, used collagen/ADP-induced high shear platelet aggregation (PFA-100®) to study platelet function in cardiac arrest survivors classified as having either a “cardiac” or “noncardiac” cause of arrest. Those patients with cardiac causes demonstrated faster platelet-dependent clotting times. All patients in the cardiac group experienced VF as the presenting rhythm. No patients in the noncardiac group experienced VF and demonstrated normal platelet function despite a similar increase in plasma vonWillebrand factor. These results suggest a platelet-activating effect of pre-arrest cardiac ischemia or an effect of VF itself16 To the contrary, we clearly demonstrated evidence for platelet inhibition using multiple assays making it less likely that VF itself was the primary cause of platelet activation observed by Spiel et al. We also used presumably healthy and cardiovascular disease-free animals, thus avoiding pre-arrest platelet activation from cardiac ischemia. This may be the reason why our results differ. In addition, Spiel et al, used a mixed Collagen/ADP agonist in their study while we used collagen as a single agonist. Platelets stimulated by ADP may be activated even in the face of collagen receptor inhibition because these two agonists can activate platelets by distinct signaling mechanisms. In the setting of hypoxia-induced cardiac arrest, Gaszynski used a rabbit model to report prolonged TEG clot onset times (R), reduced fibrinogen and platelet concentration, and increased fibrin monomers starting before CPR.17 These changes were consistent with consumption and DIC taking place during pre-arrest hypoxia. Increased thrombin generation and consumption during CPR have also been found in resuscitated cardiac arrest patients.18,19 We found only isolated platelet-specific inhibition during CPR without persistent consumption of fibrinogen that might indicate DIC. The PO2 in blood sampled for coagulation studies during VF was also maintained at nearly normal levels in our model (mean PO2 >70 mmHG) and may not have reached hypoxic levels needed to trigger DIC. Therefore, aside from the species difference, the lack of a significant component of pre-arrest hypoxia or cardiac ischemia in our model may explain our contrary results.
Inhibited platelet aggregation and availability was likely responsible for the reduced MA, PCF, and CEM noted during CPR. The mechanism for impaired aggregation remains unknown. There was no discernable independent effect of vasoactive medications on clot formation when added to blood obtained at baseline. In-vitro studies using human blood support this conclusion.20 Platelet adhesion and aggregation are required for proper clot contraction and direct inhibitors of aggregation can reduce MA, PCF, and CEM.21 Collagen receptor-dependent platelet adhesion/aggregation has been linked to Na+/Ca2+ membrane exchanger activity, integrin α2β1, andglycoproteins VI/IV making these potential targets for future mechanistic studies.22,23 Recent results from Padosch et al, using a glycoprotein IIb/IIIa inhibitor given after ROSC in a rat model of electrically-induced cardiac arrest did not impact post resuscitation microcirculatory dysfunction.24 Our results suggest that perhaps earlier administration of platelet-targeted therapies prior to ROSC may be required. Alternatively, specific targeting of collagen-mediated platelet adhesion upstream of glycoprotein IIb/IIIa activation, or the Na+/Ca2+ membrane exchanger may be important potential avenues for future research.
Impaired collagen-induced platelet aggregation likely explains the initial decrease in MA, PCF, and CEM noted during CPR, but does not explain why clot formation was reduced for several hours during the post-resuscitation period after recovery of aggregation to near baseline levels.
TEG R and K times were first prolonged at 15 minutes after ROSC, reflecting impaired clot initiation and buildup. This was presumably a direct effect of lactic acidosis. The level of blood acidosis achieved shortly after ROSC (average pH = 7.129) was consistent with previously published coagulopathy induced by blood acidification in swine.25
The CPR measurement also curiously demonstrated a transient loss of platelets from the arterial sample which may have contributed to the noted decreased in overall platelet function. The decrease in platelet count was not due to plasma dilution or sampling error because hemoglobin and hematocrit were both increased in the same sample. The reason for this transient reduction in circulating platelets during CPR remains unknown and warrants further investigation.
Limitations
Our conclusions are limited for several reasons including our lack of direct measurement of the concentrations of vasoactive drugs (Vasopressin, Epinephrine) administered during CPR. Instead, we calculated average doses administered after the third round of CPR for our in-vitro studies and found no significant changes in clot formation. This conclusion should be further verified by directly measuring drug concentrations in future studies. The effect of anesthesia was also not tested in our model and deserves further study. Ketamine can display antiplatelet activity in humans.26 However, no reports of significant ketamine or chloralose-induced hemostatic abnormalities have been reported in swine. Our baseline PT was also similar to other heavily instrumented experimental swine models using different anesthesia.27 Significant hyperoxia was also induced during CPR and during the 6 hour post-resuscitation period. Hyperoxia may have contributed to changes in platelet function because platelet integrin activity can be affected by oxygen radicals.28 This relationship may be important and deserves further study because prolonged hyperoxia after resuscitation from cardiac arrest has also been associated with increased mortality.29 Future studies of coagulation function during cardiac arrest and resuscitation should include a normoxic control group for comparison. Moreover, the lack of an instrumented control group without cardiac arrest is an acknowledged limitation of our study. Future mechanistic studies would require instrumented control groups to achieve validity. It is also unclear if the same coagulation changes would manifest with chest compression-only CPR.30,31 Overall, this study provides an initial characterization of the whole blood coagulation response to VF arrest and is intended to spur further hypothesis generation and guide experimental model development.
Conclusion
Whole blood clot formation was impaired during CPR in this electrically-induced VF model. Platelets were first affected and demonstrated impairment of aggregation, contractile function, and clotting kinetics, persisting up to 3 hours after return of circulation.
Figure 1.

Schematic representation of cardiac arrest protocol. CPR= cardiopulmonary resuscitation, ACLS= Advanced Cardiac Life Support, ROSC= return of spontaneous circulation. Blood was sampled described under Methods.
Acknowledgments
The authors acknowledge the efforts of the VCURES laboratory team. NJW was supported by NIH grant GM008695-09. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Footnotes
This data was presented in preliminary form at the 2009 American Heart Association Resuscitation Science Symposium, Orlando, FL.
Conflict of interest statement: None to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Nathan J. White, Department of Medicine/Division of Emergency Medicine, University of Washington, Seattle, WA.
Benjamin Sieu-Hon Leong, Department of Emergency Medicine, National University Hospital, Singapore.
Jessica Brueckner, Virginia Commonwealth University School of Medicine, Richmond, VA.
Erika J. Martin, Department of Pharmacy, Virgnia Commonwealth University, Richmond, VA.
Donald F. Brophy, Department of Pharmacy, Virginia Commonwealth University, Richmond, VA.
Mary A. Peberdy, Department of Medicine, Division of Cardiology, Virginia Commonwealth University, Richmond VA.
Joseph Ornato, Department of Emergency Medicine, Virginia Commonwealth University, Richmond VA.
Kevin R. Ward, Department of Emergency Medicine, Virginia Commonwealth University, Richmond VA.
References
- 1.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Executive summary: heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121(7):948–54. doi: 10.1161/CIRCULATIONAHA.109.192666. [DOI] [PubMed] [Google Scholar]
- 2.Nadkarni VM, Larkin GL, Peberdy MA, et al. First documented rhythm and clinical outcome from in-hospital cardiac arrest among children and adults. JAMA. 2006;295:50–7. doi: 10.1001/jama.295.1.50. [DOI] [PubMed] [Google Scholar]
- 3.Nolan JP, Laver SR, Welch CA, Harrison DA, Gupta V, Rowan K. Outcome following admission to UK intensive care units after cardiac arrest: a secondary analysis of the ICNARC Case Mix Programme Database. Anaesthesia. 2007;62:1207–16. doi: 10.1111/j.1365-2044.2007.05232.x. [DOI] [PubMed] [Google Scholar]
- 4.Neumar RW, Nolan JP, Adrie C, Aibiki M, Berg RA, Bottiger BW, et al. Post-Cardiac arrest syndrome Epidemiology, pathophysiology, treatment, and prognostication. Circulation. 2008;118:2452–2483. doi: 10.1161/CIRCULATIONAHA.108.190652. [DOI] [PubMed] [Google Scholar]
- 5.Ayoub IM, Radhakrishnan J, Gazmuri RJ. Targeting mitochondria for resuscitation from cardiac arrest. Crit Care Med. 2008;36:S440–S446. doi: 10.1097/ccm.0b013e31818a89f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adrie C, Adib-Conquy M, Laurent I, Monchi M, Vinsonneau C, Fitting C, et al. Successful cardiopulmonary resuscitation after cardiac arrest as a “sepsis-like” syndrome. Circulation. 2002;106(5):562–568. doi: 10.1161/01.cir.0000023891.80661.ad. [DOI] [PubMed] [Google Scholar]
- 7.Adrie C, Monchi M, Laurent I, Um S, Yan SB, Thuong M, et al. Coagulopathy after successful cardiopulmonary resuscitation following cardiac arrest: implication of the protein C anticoagulant pathway. Journal of the American College of Cardiology. 2005;46(1):21–28. doi: 10.1016/j.jacc.2005.03.046. [DOI] [PubMed] [Google Scholar]
- 8.2005 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Part 7.2: Management of Cardiac Arrest. Circulation. 2005;112:IV-58–IV-66. doi: 10.1161/CIRCULATIONAHA.105.166550. [DOI] [PubMed] [Google Scholar]
- 9.Rivers E, Nguyen B, Havstad S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001;345:1368–77. doi: 10.1056/NEJMoa010307. [DOI] [PubMed] [Google Scholar]
- 10.Gaieski DF, Band RA, Abella BS, Neumar RW, Fuchs BD, Kolansky DM, et al. Early goal-directed hemodynamic optimization combined with therapeutic hypothermia in comatose survivors of out-of-hospital cardiac arrest. Resuscitation. 2009;80(4):418–424. doi: 10.1016/j.resuscitation.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 11.Spiel AO, Kliegel A, Janata A, Uray T, Mayr FB, Laggner AN, et al. Hemostasis in cardiac arrest patients treated with mild hypothermia initiated by cold fluids. Resuscitation. 2009;7:762–5. doi: 10.1016/j.resuscitation.2009.03.026. [DOI] [PubMed] [Google Scholar]
- 12.Carr ME. Development of Platelet Contractile Force as a Research and Clinical Measure of Platelet Function. Cell Biochem Biophys. 2003;38:55–78. doi: 10.1385/CBB:38:1:55. [DOI] [PubMed] [Google Scholar]
- 13.Surin WR, Barthwal MK, Dikshit M. Platelet collagen receptors, signaling and antagonism: emerging approaches for the prevention of intravascular thrombosis. Thromb Res. 2008;122(6):786–803. doi: 10.1016/j.thromres.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 14.Kjeldsen SE, Weder AB, Egan B, Neubig R, Zweifler AJ, Julius S. Effect of circulating epinephrine on platelet function and hematocrit. Hypertension. 1995;25(5):1096–105. doi: 10.1161/01.hyp.25.5.1096. [DOI] [PubMed] [Google Scholar]
- 15.Filep J, Rosenkranz B. Mechanism of vasopressin-induced platelet aggregation. Thromb Res. 1987 Jan 1;45(1):7–15. doi: 10.1016/0049-3848(87)90252-0. [DOI] [PubMed] [Google Scholar]
- 16.Spiel AO, Frossard M, Mayr FB, et al. Pronounced platelet hyperfunction in patients with cardiac arrest achieving restoration of spontaneous circulation. Crit Care Med. 2009;37:975–9. doi: 10.1097/CCM.0b013e3181962cb9. [DOI] [PubMed] [Google Scholar]
- 17.Gaszynski W. Research Work on Blood Clotting System during Cardiorespiratory Resuscitation. Anasth Resus Inten Therap. 1974;2(4):303–316. [PubMed] [Google Scholar]
- 18.Bottiger BW, Motsch J, Bohrer H, et al. Activation of blood coagulation after cardiac arrest is not balanced adequately by activation of endogenous fibrinolysis. Circulation. 1995;92:2572–2578. doi: 10.1161/01.cir.92.9.2572. [DOI] [PubMed] [Google Scholar]
- 19.Gando S, Kameue T, Nanzaki S, et al. Massive fibrin formation with consecutive impairment of fibrinolysis in patients with out-of hospital cardiac arrest. Thromb Haemost. 1997;77:278–282. [PubMed] [Google Scholar]
- 20.Kawasaki J, Katori N, Taketomi T, Terui K, Tanaka KA. The effects of vasoactive agents, platelet agonists and anticoagulation on thrombelastography. Acta Anaesthesiol Scand. 2007;51(9):1237–44. doi: 10.1111/j.1399-6576.2007.01434.x. [DOI] [PubMed] [Google Scholar]
- 21.Greilich PE, Alving BM, Longnecker D, Carr ME, Jr, Whitten CW, Chang AS, Reid TJ. Near-site monitoring of the antiplatelet drug abciximab using the Hemodyne analyzer and modified thrombelastograph. J Cardiothorac Vasc Anesth. 1999;13(1):58–64. doi: 10.1016/s1053-0770(99)90175-1. [DOI] [PubMed] [Google Scholar]
- 22.Roberts DE, McNicol A, Bose R. Mechanism of collagen activation in human platelets. J Biol Chem. 2004;279(19):19421–30. doi: 10.1074/jbc.M308864200. [DOI] [PubMed] [Google Scholar]
- 23.Ruggeri ZM, Mendolicchio GL. Adhesion Mechanisms in Platelet Function. Circ Res. 2007;100:1673–1685. doi: 10.1161/01.RES.0000267878.97021.ab. [DOI] [PubMed] [Google Scholar]
- 24.Padosch SA, Teschendorf P, Fuchs A, del Valle y Fuentes D, Peter C, Popp E, Schneider A, Böttiger BW, Walther A. Effects of abciximab on postresuscitation microcirculatory dysfunction after experimental cardiac arrest in rats. Resuscitation. 2010;81(2):255–9. doi: 10.1016/j.resuscitation.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 25.Martini WZ, Pusateri AE, Uscilowicz JM, Delgado AV, Holcomb JB. Independent Contributions of Hypothermia and Acidosis to Coagulopathy in Swine. J Trauma. 2005;58:1002–1010. doi: 10.1097/01.ta.0000156246.53383.9f. [DOI] [PubMed] [Google Scholar]
- 26.Yi C, Ta-Liang C, Joen-Rong S, et al. Mechanisms Involved in the Antiplatelet Activity of Ketamine in Human Platelets. Journal of Biomedical Science. 2004;11(6):764–772. doi: 10.1007/BF02254361. [DOI] [PubMed] [Google Scholar]
- 27.Tomori T, Hupalo D, Teranishi K, Michaud S, Hammett M, Freilich D, McCarron R, Arnaud F. Evaluation of coagulation stages of hemorrhaged swine: comparison of thromboelastography and rotational elastometry. Blood Coagul Fibrinolysis. 2010 Jan;21(1):20–7. doi: 10.1097/MBC.0b013e32833113e9. [DOI] [PubMed] [Google Scholar]
- 28.Gregg D, De Carvalho DD, Kovacic H. Integrins and Coagulation: A Role for ROS/Redox Signaling? Antioxid Redox Signal. 2004;6:757–765. doi: 10.1089/1523086041361604. [DOI] [PubMed] [Google Scholar]
- 29.Kilgannon JH, Jones AE, Shapiro NI, Angelos MG, Milcarek B, Hunter K, et al. Association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. JAMA. 2010 Jun 2;303(21):2165–71. doi: 10.1001/jama.2010.707. [DOI] [PubMed] [Google Scholar]
- 30.Svensson L, Bohm K, Castrèn M, Pettersson H, Engerström L, Herlitz J, Rosenqvist M. Compression-only CPR or standard CPR in out-of-hospital cardiac arrest. N Engl J Med. 2010;363(5):434–42. doi: 10.1056/NEJMoa0908991. [DOI] [PubMed] [Google Scholar]
- 31.Rea TD, Fahrenbruch C, Culley L, Donohoe RT, Hambly C, Innes J, Bloomingdale M, Subido C, Romines S, Eisenberg MS. CPR with chest compression alone or with rescue breathing. N Engl J Med. 2010;363(5):423–33. doi: 10.1056/NEJMoa0908993. [DOI] [PubMed] [Google Scholar]