

SUMMARY
While transcriptional regulation of stem cell pluripotency and differentiation has been extensively studied, only a small number of studies have addressed the roles for post-translational modifications in these processes. A key mechanism of post-translational modification is ubiquitination by the ubiquitin-proteasome system (UPS). Here we map, using shotgun proteomics, the ubiquitinated protein landscape during ES cell differentiation and induced pluripotency. Moreover, using UPS-targeted RNAi screens, we identify novel regulators of pluripotency and differentiation. We focus on two of these proteins, the deubiquitinating enzyme, Psmd14, and the E3 ligase, Fbxw7, and characterize their importance in ES cell pluripotency and cellular reprogramming. This is the first global characterization of the UPS as a key regulator of stem cell pluripotency, opening the way for future studies that focus on specific UPS enzymes or ubiquitinated substrates.
INTRODUCTION
It is well established that embryonic stem (ES) cell self-renewal and differentiation are regulated by a network of transcription factors including Oct4, Nanog, and Sox2, while Sox2 and Oct4 are also key factors in cellular reprogramming (Avilion et al., 2003; Chambers et al., 2003; Nichols et al., 1998). Additional levels of ES cell-specific regulation have been characterized, including important roles for environmental cues, epigenetic regulators and miRNAs (Martinez and Gregory, 2010; Meissner, 2010; Wang et al., 2007; Ying et al., 2003). Recent studies suggested that transcription-independent regulation of proteins can also control ES cell fates (Lu et al., 2009); however, there is limited information on the role of post-translational modifications (PTMs) in ES cell differentiation and function. Although studies have addressed the role of phosphorylation (Li et al., 2011; Phanstiel et al., 2011) the role of protein turnover by the ubiquitin proteasome system (UPS) has not yet been explored.
The relative abundance as well as other functional modifications of proteins are regulated by a complex cellular machinery, the UPS, that specifically adds or removes ubiquitin to proteins (Pickart, 2001). The specificity of the reaction is provided by the E3 ligase complex, which conjugates activated ubiquitin to the substrate. Repetition of this action ensures poly-ubiquitination, and leads to recognition by the 26S proteasome followed by proteolysis. On the other hand, mono-ubiquitinated proteins have non-proteolytic fates. Moreover, apart from the function of distinct ligase complexes, the UPS is also regulated by a class of deubiquitinating enzymes (DUBs) responsible for removing ubiquitin conjugates from substrates (Kimura and Tanaka, 2010). The study of UPS-mediated regulation of stem cell function has been recently introduced by studies of hematopoietic stem cell (HSC) differentiation. Our laboratory and others have identified the F-box E3 ligase, Fbxw7, as a key component of hematopoietic and neural stem cell self-renewal and differentiation (Matsumoto et al., 2011; Matsuoka et al., 2008; Reavie et al., 2010; Thompson et al., 2008). Little is known on the function of UPS components in pluripotency. It has been suggested that the proteasome is important in sequestering transcription factor binding to regulatory regions of genes necessary for development, modulating their gene expression (Szutorisz et al., 2006). It was also suggested that some pluripotency regulators are ubiquitinated. However, the roles and significance of such modifications remain unclear (Gontan et al., 2012; Liao and Jin, 2010; Ramakrishna et al., 2011).
To study the role of the UPS in ES cell pluripotency and cellular reprogramming, we employed a combination of genomic and proteomic approaches. Initially, we applied quantitative mass spectrometry to map the global changes in protein expression and ubiquitination in pluripotent and differentiated ES cells and showed that a significant number of the members of the core pluripotency machinery are modified by ubiquitin. Secondly, we used RNAi-based screens, targeting the majority of known members of the UPS, and identified a significant number of putative novel regulators. Finally, we confirmed the role of selected UPS members in regulating pluripotency of ES cells. For example, in the current study, we showed that silencing of Fbxw7 expression inhibits ES cell differentiation and enhances cellular reprogramming through stabilization of c-Myc. Furthermore, we demonstrated that the DUB Psmd14 is essential for cellular reprogramming and maintenance of ES cell self-renewal due to its enzymatic activity within the proteasome lid. Finally, we directly compared the “ubiproteome” of ES and iPS cells and demonstrated a remarkable similarity of ubiquitin modifications in these two cell types. These are the first studies mapping protein ubiquination in ES cell pluripotentcy, differentiation and cellular reprogramming, introducing an additional level of regulation of these processes.
RESULTS
Mapping the ubiquitin-modified proteome during ES cell differentiation
To quantitatively assess ubiquitin modified proteins in pluripotent and differentiated mouse ES cells (Figure 1A, Figure S1), we labeled pluripotent ES cells with stable-isotope amino acids (SILAC). For direct comparison to differentiated cell populations, cells were maintained in light medium and differentiated for four days. We then mixed equal amount of labeled (heavy) and unlabeled lysates, digested the mixed lysate with trypsin, and enriched for ubiquitin-modified peptides using an affinity matrix coupled with antibodies against diGly-modified lysine (Kim et al., 2011; Xu et al., 2010). We analyzed the enriched peptides by LC-MS/MS (Figure 1A). We identified 2578 non-redundant ubiquitin-modified peptides with a false discovery rate of 1%, which were assigned to 1168 unique proteins (Table S1). We repeated these experiments using label-free detection to increase the efficiency of identification of ubiquitin-modified peptides (Table S1). We found a significant concordance between these two approaches, especially evident by the identification of ubiquitin-modified peptides from well-characterized members of the extended pluripotency network (Figure 1B, Table S2).
Figure 1. Mapping of the mouse ES cell Ubiproteome.
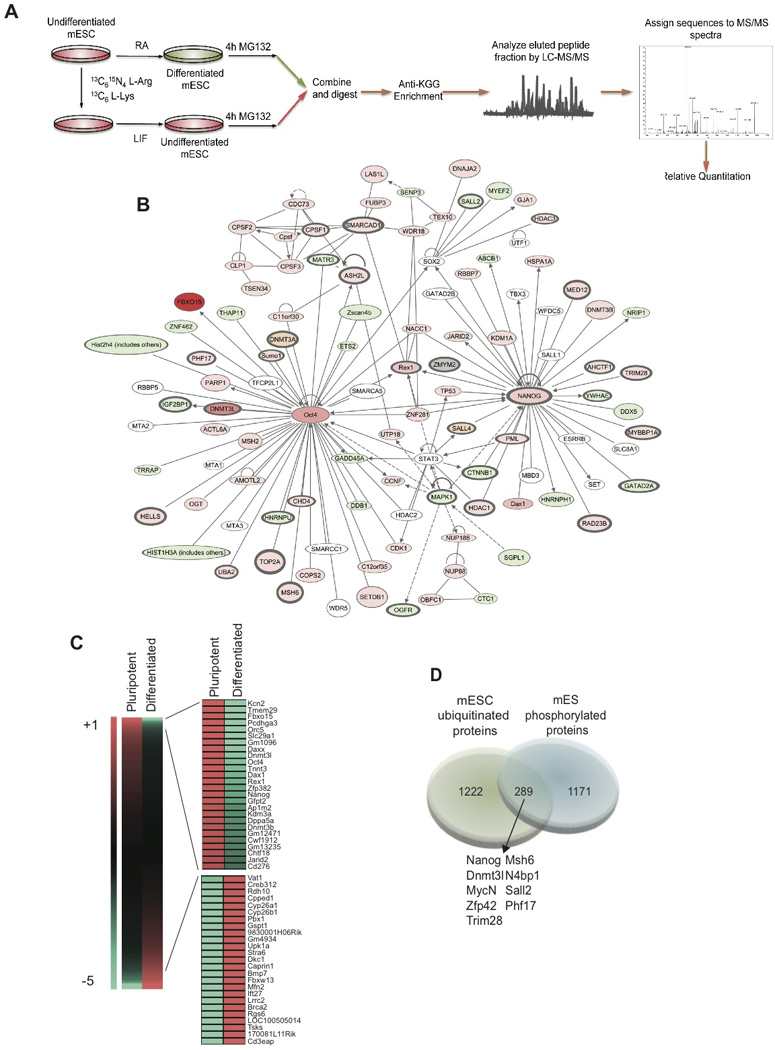
A) Scheme depicting the proteomic strategy followed using SILAC and MS/MS technology. B) Ingenuity Pathways-generated network with Nanog and Oct4 as nodes. Molecules in red show a higher ubiquitination rate in self-renewal conditions and in green in differentiated cells. Color intensities reflect ubiquitination detection values. White marks molecules that belong to the network and were not detected in our study. In orange are marked pluripotency factors only detected in the label-free experiments. Lines without arrows mean binding. Lines with arrows mean acting on. Dotted lines reflect an indirect interaction. C) Heat-map illustrating fold change of ubiquitinated peptides differentially detected in self-renewing ES cells and in differentiated cells. D) Venn diagram showing the overlap between the mouse phospho-proteome (Li et al, 2011) and the ubi-proteome obtained in this study. Common proteins are underlined in the pluripotency network in dark grey.
To understand the biological significance of the identified ubiquitin modified proteins, we classified these proteins using the Ingenuity Systems Pathway Analysis software (IPA) and created a pluripotency network centered around the ubiquitinated key regulator Nanog and Oct4. In pluripotent ES cells, we detected a significant number of ubiquitin-modified proteins that are key regulators of self-renewal in ES cells (Figure 1B, Figure S1). Notably, we were also able to identify novel sites of ubiquitination (Figure S1). We focused on ubiquitin-modified peptides representing proteins more highly detected in the pluripotent or in the differentiated ES cells. (Figure 1C, Table S1). DAVID functional annotation (Huang da et al., 2009) revealed enrichment of distinct biological classifications in both populations. The pluripotency-associated ubiquitin-modified proteins included several known pluripotency factors (e.g. Nanog, Oct4, Dax1, Rex1), epigenetic factors (e.g. Jarid2, Kdm3a, Hdac1), and translation-related proteins (e.g. eEF1A1, Rps10). On the contrary, in the differentiated-associated ubiquitin-modified proteins we detected proteins involved in retinoic acid metabolism (e.g. CYP26a, STRA8), protein scaffolding and cell adhesion (Figure S2).
Phosphorylation is the best-studied post-translational modification in ES cells in proteome-wide scale (Li et al., 2011). Since phosphorylation can prime proteins for ubiquitination (Deshaies and Joazeiro, 2009), we cross-referenced our databases to previously published phosphoproteome studies in ES cells and identified 289 (19.1%) proteins marked by both phosphorylation and ubiquitination (Figure 1D). Interestingly, many of those proteins are also part of the pluripotency network shown in Figure 1B (outlined), suggesting a potential crosstalk between ubiquitination and phosphorylation in the regulation of pluripotency in ES cells. These studies identified specific “ubiquitin signatures” for ES cell self-renewal and differentiation, strongly suggesting a connection between ubiquitination and ES cell pluripotency.
UPS-mediated protein half-life of key pluripotency factors
As the UPS can control protein half-life and degradation we set out to define such UPS-regulated pluripotency/differentiation factors. Using multi-dimensional protein identification technology (MudPIT), we identified 5440 proteins from the nuclear fraction of pluripotent ES cells in the presence or absence of the proteasome inhibitor, MG132. The fold changes of proteins with or without the MG132 treatment followed a Gaussian-like curve, suggesting that MG132 did not cause a systemic change of protein expression (Figure 2A). Ubiquitin-modified peptides identified using the diGly-lysine affinity matrix appeared to be found mostly in proteins with increased levels in the presence of MG132. From these identified proteins approximately 25% (1311) were found to show a >2-fold increase in expression in response to MG132, and ubiquitin-modified peptides were found in 145 among these proteins (Figure 2A, Table S3). Interestingly, several known stem cell transcription factors such as Nanog, Rex1, Dax1, or c-Myc were among these ubiquitin-modified proteins both ubiquitinated and stabilized. Next, we verified the ubiquitin modifications found on several pluripotency associated factors (Figure 2B and C; Figure S3) and confirmed the stabilization of selected proteins using western blot analysis (Figure S3). Moreover, we validated poly-ubiquitination of key pluripotency factors found ubiquitinated in pluripotent ES cells (Figure 2C and D, Figure S2). Both Nanog and Oct4 (but not Sox2) were specifically ubiquitinated in pluripotent cells, suggesting active regulation of their relative protein abundance in self-renewal conditions (Figure 2D, Figure S3). To test this hypothesis we quantified protein half-life at different stages of differentiation and were able to show that both factors were actively degraded in pluripotent but not differentiated ES cells (Figure 2E and F). This finding could have important consequences for the understanding of ES cell function, as it suggests that during pluripotency the UPS is actively restricting the protein abundance of such key regulators.
Figure 2. Half-life of key pluripotency factors is regulated by the UPS.
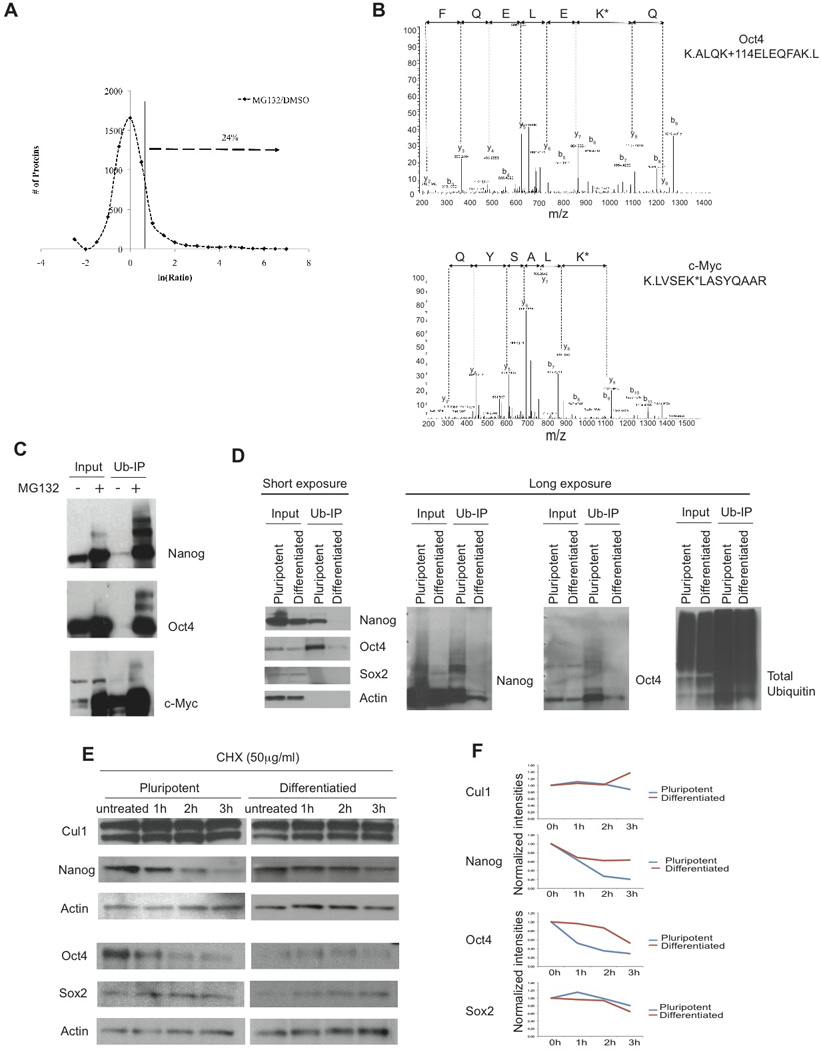
A) Differential expression of ES nuclear proteins in the presence of MG132. Proteins with increased expression in the presence of MG132 are indicated by an arrow (> 2 fold increase). X-axis shows the ratios of MS/MS spectra identified in the MG132 treatment over the MS/MS spectra identified in the DMSO treatment for each protein in the natural logarithm (ln). Y-axis shows the number of proteins collectively in each binned ratio. B) Representative annotated MS/MS spectra of the Oct4 and c-Myc remnant–containing peptides obtained by immunoprecipitation in pluripotent ES cells. The sequence of the ubiquitinated peptide and the diglycine-modified lysine (K*) are indicated. C) In vivo ubiquitination experiments of pluripotency factors in self-renewing conditions. D) In vivo ubiquitination of Nanog, Oct4 and Sox2 in self-renewal and 24 hours of differentiation with RA using ubiquitin pull-down enrichment. E) Western blot analysis of Nanog, Oct4, Sox2 and Cullin1 half-life in self-renewal and 24 hours of differentiation using 50µg/mL of cycloheximide. G) Quantification of the half-life of proteins depicted in E. Plots represent normalized intensity relative to actin.
Novel UPS members essential for the maintenance of ES cell self-renewal
To begin mapping the function of members of the UPS responsible for ubiquitination events identified by our mass spectrometry approach, we targeted the UPS using siRNA-mediated gene silencing in ES cells. We used siRNA libraries against E1 activating enzymes, E2 conjugating enzymes, E3 ligases, deubiquitinating enzymes, as well as, predicted E3 ligases. siRNA libraries contained a pool of 4 siRNAs targeting approximately 640 known and predicted UPS genes. Following transfection of siRNA, cells were maintained in cultures supplemented with LIF, maintaining pluripotency of ES cells. To monitor loss of self-renewal, we used a “knock-in” reporter ES cell line, in which GFP expression is driven by the Nanog regulatory elements (Schaniel et al., 2010) (Figure S4). Nanog-GFP expression was monitored by flow cytometry. Effects on differentiation were compared to silencing of known regulators of self-renewal (Oct4 and Sox2). To identify genes specifically affecting pluripotency we eliminated siRNAs that lead to cell death or cell cycle deregulation. The initial screen revealed 20 genes that when silenced lead to significant effects on ES cell self-renewal. Genes that were considered hits had >9% differential expression of GFP compared to the control non-target siRNA (differential expression= % GFP− cells in target gene - % GFP− cells in non-target control), and had a z-score > 1 (Figure 3A, Figure S4, Table S3).
Figure 3. UPS-targeted siRNA screen identifies genes required to maintain ES cell self-renewal and pluripotency.
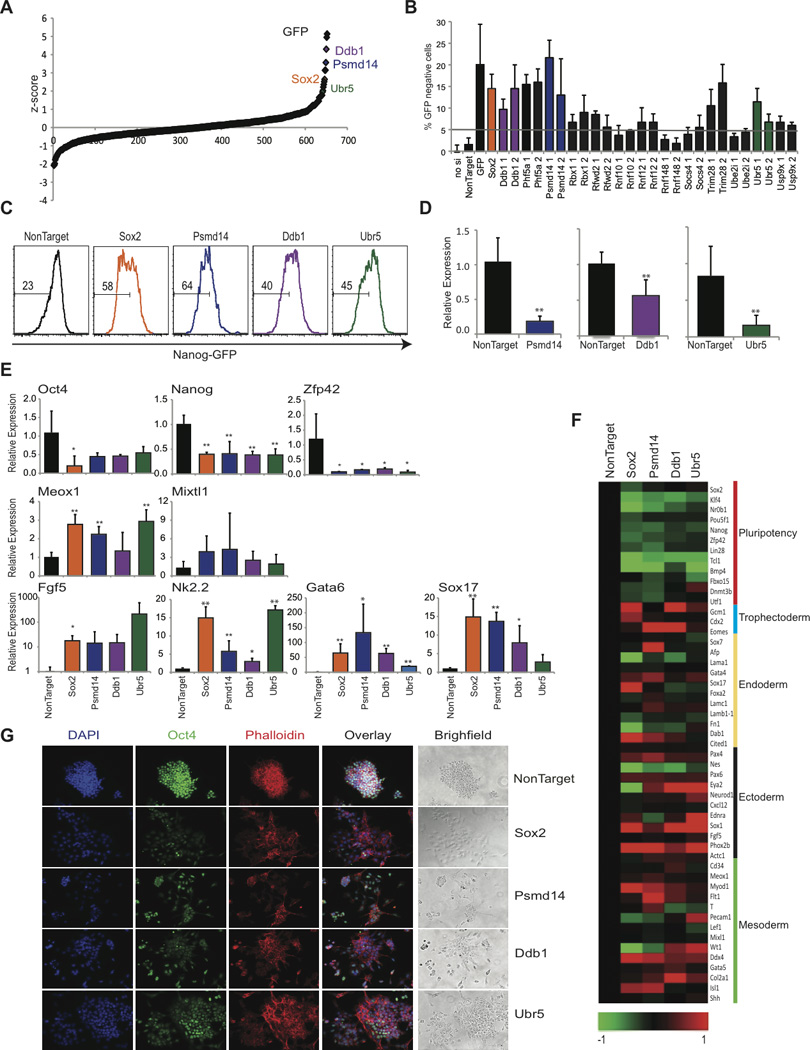
Nanog-GFP ES cells were transfected with pools of siRNAs under conditions of self-renewal and analyzed by FACS 48h later. A) Dot plot representing Z-score for all siRNAs. B) siRNA pool validation by individual siRNAs. C) FACS plots depicting loss of Nanog-GFP following siRNA transfection. D) Knockdown efficiency by qRT-PCR after siRNA transfection. E) Relative expression of pluripotency genes (Oct4, Nanog, Zfp42); endoderm genes (Gata6, Sox17); mesoderm (Meox1 and Mixtl1) and ectoderm genes (Nk2.2 and Fgf5) by qRT-PCR. Data represented as +SEM; N=3. *p-value <0.05, **p-value<0.01. F) Heat-map illustrating gene expression (log2). Selected pluripotency and differentiation genes are shown. G) Immunofluorescence and bright field images 48 hrs following siRNA knockdown. Staining for Oct4 (Green), Phalloidin (Red), and DAPI (Blue).
To rule out off-target effects and validate the utilized siRNAs, we screened 13 selected genes targeting distinct areas of the transcript. Nine out of the tested 13 genes validated (Figure 3B). These genes represented DUBs (Psmd14 and Usp9x), E3 ligases (Rbx1, Rfwd2, Rnf12, Ubr5, and Ddb1), and 2 putative E3 ligases based on RING motif homology (Trim28 and Phf5a). Three selected genes (Psmd14, Ubr5, and Ddb1) were further evaluated for their role in ES self-renewal and pluripotency (Figure 3C and D). We then performed assays to determine the role of these selected genes in ES cell identity. Knockdown of all three selected genes lead to a >50% decrease in the expression of the known pluripotency genes, Nanog, Oct4, and Zfp42 (Figure 3E). On the other hand, genes associated with early ES differentiation to distinct lineages showed marked (although heterogeneous) increase in expression (Figure 3E). The knockdown of all three genes showed moderate induction of early mesoderm markers (Meox1, Mixtl1), however, early ectoderm (Nk2.2 and Fgf5) and endoderm genes (Gata6 and Sox17) showed significant increase in gene expression (Figure 3E). To further support these initial expression studies, we used whole-transcriptome profiling. This analysis revealed a homogenous down-regulation of ES cell pluripotency-associated gene expression and concomitant activation of genes associated with early differentiation (Figure 3F). Although we observed a homogenous down-regulation of pluripotency genes, global expression profiling revealed that each siRNA had a unique set of differentially expressed genes suggesting a unique role in inhibiting specific lineage commitment (Figure S4). Immunofluorescence studies showed in response to siRNA-mediated knockdown of Psmd14, Ddb1, and Ubr5, ES cells had significant loss of Oct4 protein expression coupled to morphology changes, similar to ES cells lacking Sox2 (Figure 3G). These combined studies describe the first identification of UPS members playing roles in the regulation of ES cell self-renewal and identify Psmd14, Ddb1, and Ubr5 as novel regulators of ES cell pluripotency.
Novel UPS members essential for the regulation of ES cell differentiation
To study the influence of members of the UPS on early ES differentiation, we repeated our UPS-targeted siRNA screen, but this time we forced ES cell differentiation by LIF withdrawal and addition of retinoic acid (RA). As a “positive” control for known factors affecting ES differentiation, we used siRNA oligos against Sox17 (Niakan et al., 2010). Putative “hits” were defined as previously described. We identified 17 genes which when their expression was silenced lead to significant up-regulation of Nanog-GFP expression (Figure 4A, Figure S5, Table S5). To validate siRNAs, we selected 10 genes to screen using four individual siRNAs, distinct from the ones contained in the screen library. Knockdown of nine out of ten genes lead to a significant maintenance of Nanog-GFP expression when targeted with at least two siRNAs (Figure 4B). These genes included six E3 ligases (Fbxw7, Rnf152, Rnf31, Rnf8, Socs3, and Topors), 2 putative ligases based on ubiquitin-like domain analysis and conservation (Rnf36, and Tnfrsf25), and Ubl5, a ubiquitin-like protein (Figure 4B).
Figure 4. UPS-targeted siRNA screen identifies genes required for optimal ES cell differentiation.
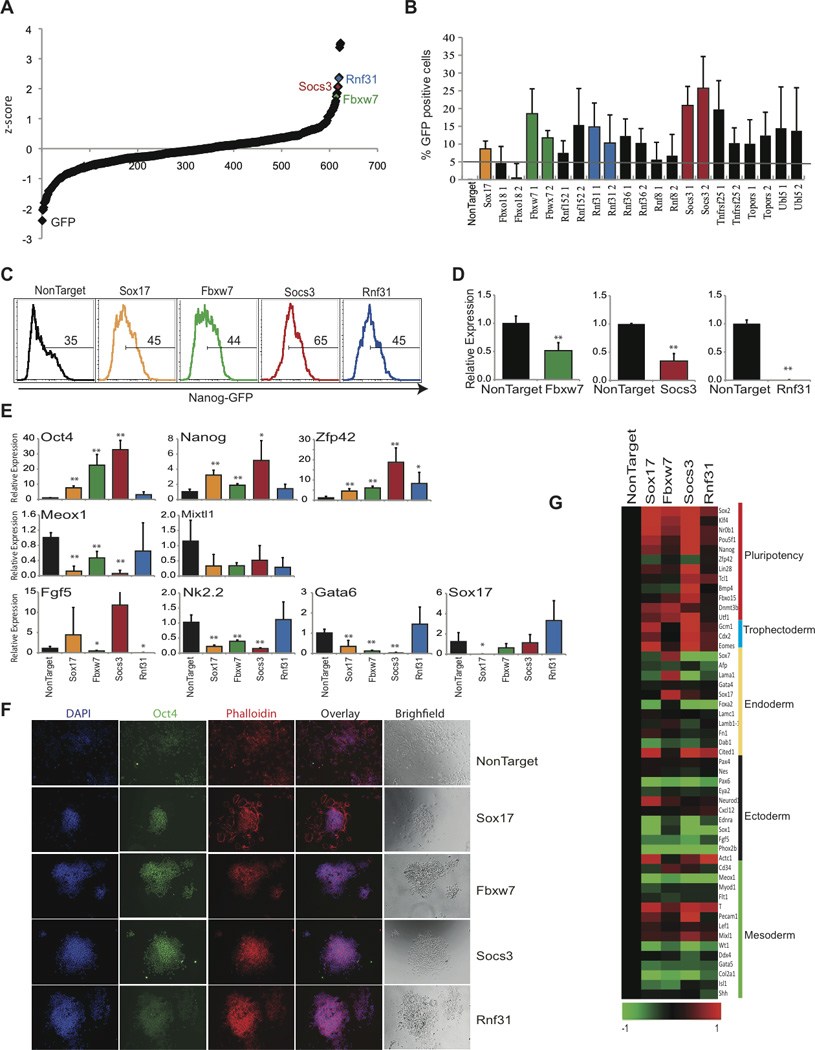
Nanog-GFP ES cells were transfected with pools of siRNAs and differentiated for 48hrs followed by FACS analysis. A) Dot plot representing z-score of all siRNAs. B) Differential expression of Nanog-GFP compared to NonTarget with individual siRNAs. C) Histogram FACS plots showing retention of GFP following siRNA transfection. D) Relative knockdown after siRNA transfection by qRT-PCR. E) Relative expression of pluripotency genes (Oct4, Nanog, Zfp42); endoderm genes (Gata6, Sox17); mesoderm (Meox1 and Mixtl1) and ectoderm genes (Nk2.2 and Fgf5), by qRT-PCR. Data represented as +SEM; N=3. *p-value<0.05, **p-value<0.01. F) Immunofluorescence and bright field images 72 hrs following siRNA knockdown. Staining for Oct4 (Green), Phalloidin (Red), and DAPI (Blue). G) Heat-map illustrating changes in gene expression (log2). Selected pluripotency and differentiation genes are shown.
Three of the validated genes (Fbxw7, Socs3, and Rnf31) were selected for further functional characterization (Figure 4C and D, Figure S5). All three genes showed an increase of RNA transcript levels in days 2 and 4 of differentiation when compared to self-renewing ES cells (Figure 6C, Figure S5). Silencing of these selected candidate genes revealed homogenous up-regulation of key pluripotency genes, including Oct4, Nanog, and Zfp42, whereas, early lineage differentiation genes revealed varied degrees of down-regulation (Figure 4E), reflecting putative specificity for distinct lineages. To further support this initial expression analysis we have used whole-transcriptome analysis in ES cells in which Fbxw7, Socs3, and Rnf31 expression was silenced. Gene silencing lead to a homogenous up-regulation of genes related to ES cell pluripotency, including Nanog, Zfp42, and Essrb (Figure 4G). However overall regulation of differentiation-specific gene expression was heterogeneous demonstrating differential roles of individual genes on inhibition of differentiation (Figure 4G, Figure S5). Finally, to gage morphological and protein expression changes of candidate cell populations in response to siRNA-mediated silencing, immunofluorescence was performed (Figure 4F). Consistent with the Nanog-GFP expression changes in our primary screen, the morphology varied between conditions, although all candidate genes showed more significant ES colony-like morphology, and these colonies consisted of more self-renewing (Oct4 positive) ES cells (Figure 4C and F). These combined studies presented the first identification of members of the UPS that play roles in the regulation of ES cell differentiation.
Figure 6. Fbxw7 targets c-Myc for degradation during ES cell differentiation.
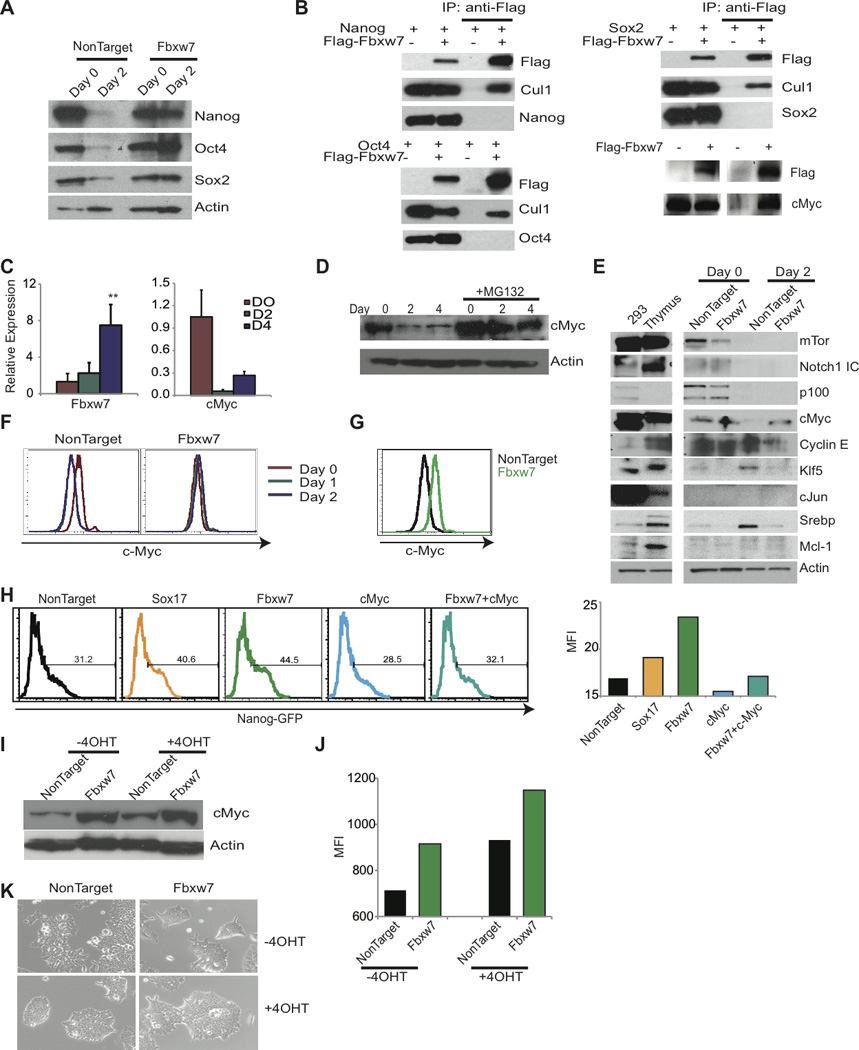
A) Western blot of pluripotency factors following Fbxw7 silencing in Day 0 and 2 of differentiation. B) Co-transfection of Fbxw7 and pluripotency factors in 293T cells followed by immunoprecipitation. Lane 1 and 2; whole cell extract and Lane 3 and 4; immunoprecipitates. C) qRT-PCR of Fbxw7 and c-Myc during differentiation. D) c-Myc protein expression during differentiation with or without 10µM MG132 treatment. E) Immunoblot of known Fbxw7 substrates at Day 0 and Day 2 of differentiation. F and G) Intracellular FACS staining with anti-c-Myc following shRNA transduction and differentiation with RA. H) Histogram representing retention of GFP 72hrs following siRNA transfection and 48hrs addition of differentiation media. Bar graph on the right represents MFI of ES cells in one representative experiment (n=3). I-J) MycT58AER ES cells following depletion of Fbxw7 with shRNA were differentiated for 2 days (−LIF+5µM RA) without or in the presence of 10nM 4-OHT, and analyzed by I) immunoblot for c-Myc, J) SSEA1 expression Bar graph represents MFI of SSEA1 in one representative experiment (n=2), and K) morphology of colonies. Data represented as +SEM; N=3. *p-value <0.05, **p-value<0.01.
Psmd14, a member of the proteasome lid is essential for ES cell pluripotency
We then decided to focus on selected screen “hits” and better understand their mechanisms of action. We initially focused on Psmd14, since it was identified in our RNAi screen as one of the most potent regulators of ES cell pluripotency (Figure 3, Figure S4). Pluripotent ES cells express significant amounts of Psmd14 and its expression is down-regulated as the cells differentiate, suggesting specific functions associated with pluripotency (Figure 5A and B). We sought to identify Psmd14 interacting partners by generating doxycycline (Dox)-inducible ES cell lines expressing StrepII/Flag tags. Psmd14 interacting partners were identified using liquid chromatography-tandem mass spectrometry (LC-MS/MS). In agreement to previous studies of the yeast proteasome, Psmd14 was shown to specifically associate with the majority of the members of the 19S proteasome lid, including Psmd3, Psmd6, Psmd7, Psmd11, Psmd12, and Psmd13 (Figure 5C, Table S6) (Lander et al., 2012). No such peptide was identified in control eGFP pulldowns, strongly suggesting the specificity of the interaction with the proteasome lid.
Figure 5. Psmd14, a component of the 19S proteasome lid, is required for optimal ES cell function.
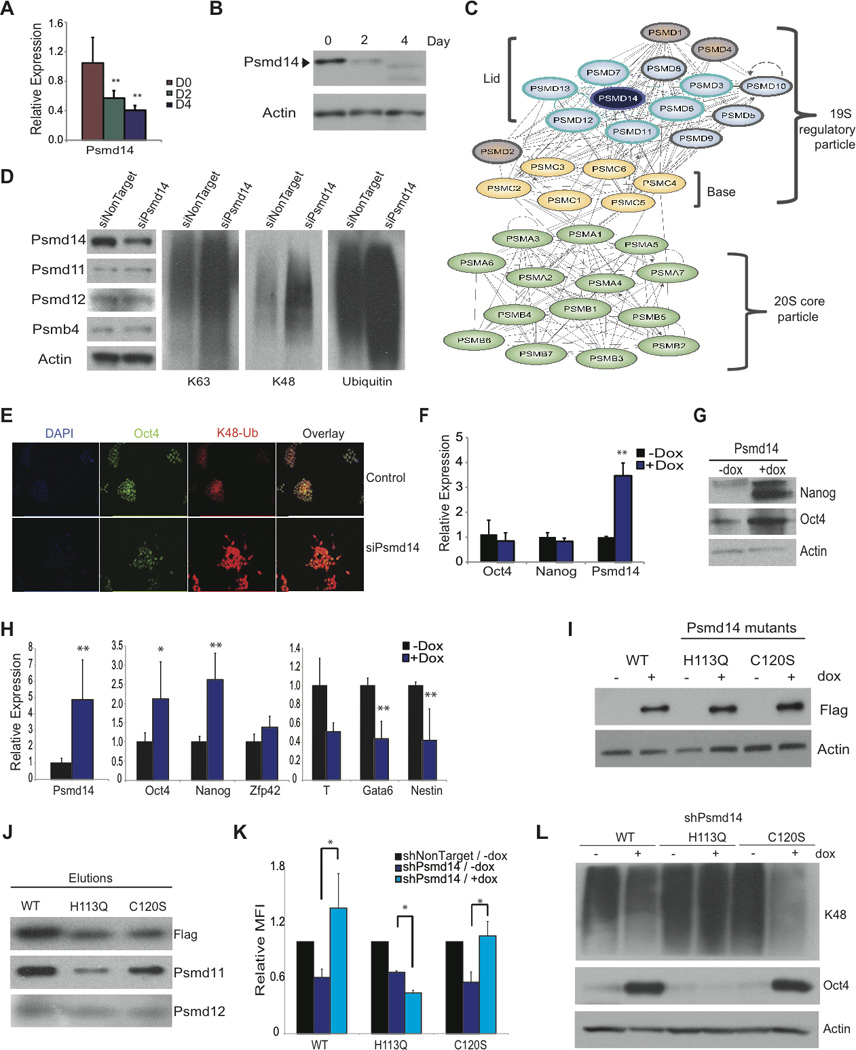
A-B) Relative expression of Psmd14 mRNA and protein during ES cell differentiation with RA. C) Scheme depicting 26S proteasome components and Psmd14 interacting partners. The bait, Psmd14 (purple), and interacting partners encircled in blue. D) Western blots with antibodies against Psmd14, Psmd11, Psmd12, Psmb4, K48- and K63-linkage specific linked, as well as pan-ubiquitin following Psmd14 knockdown in ES cells. E) Immunofluorescence showing K48-Ubiquitin accumulation in response to Psmd14 RNAi. F) Relative expression of genes following induction of exogenous Psmd14 in ES cells. G) Western blot analysis after 4 days of differentiation in the presence or absence of Dox. H) Relative expression of pluripotency genes Oct4, Nanog and Zfp42 (Rex1); and genes representing endoderm (Gata6), mesoderm (T) and ectoderm (Nestin) by qRT-PCR after 4 days of differentiation in the presence or absence of Dox. I) Western blot showing StrepII/Flag-tagged Psmd14WT, Psmd14H113Q, or Psmd14C120S mutant protein expression in targeted KH2 cells in the presence or absence of Dox. J) Western blot analysis with antibodies against Flag (Psmd14), Psmd11 and Psmd12 antibodies following elutions of StrepII-purified Flag-tagged Psmd14 proteins. K) Bar graph representing mean fluorescent intensity (MFI) of SSEA1-expressing cells following shRNA transfection against Psmd14 in the presence or absence of Dox and normalized to non-target control. L) Western blot analysis of Oct4 and K48-linkage specific polyubiquitinated proteins in ES cells following knockdown of endogenous Psmd14 and expression of Psmd14WT, Psmd14H113Q or Psmd14C120 proteins. Data represented as +SEM; N=3. *p-value <0.05, **p-value<0.01.
To start investigating Psmd14 functions we initially excluded significant effects on ES cell viability (Figure S6). Moreover, we noticed that upon Psmd14 downregulation, the expression of its interacting partners and components of the proteasome lid, Psmd11 and Psmd12 remained unaffected (Figure 5D). Similarly, the protein levels of the core component Psmb4 remained unaltered, suggesting that the overall stoichiometry of the 26S proteasome remained unchanged (Lander et al., 2012). Strikingly, we observed that both K48- and K63- polyubiquitinated proteins accumulated extensively by western blot analysis in response to Psmd14 downregulation, indicating defective proteasome activity (Figure 5D). Immunofluorescence revealed an increase in total K48-specific staining in ES cells, which corresponded to loss of Oct4 expression and morphology changes consistent with ES differentiation (Figure 3G and 5E). These results illustrate that Psmd14 is necessary for self-renewal due to proper deubiquitination and subsequent degradation of target proteins in ES cells. To further support this hypothesis we were able to show that Psmd14 overexpression could inhibit differentiation as illustrated in Figures 5F–G and S6. Interestingly, it appears that Psmd14-interacting proteins at the proteasome lid could play similar roles. Silencing of the Psmd14-interacting components Psmd13 and Psmd11 induced a significant loss of Nanog-GFP expression, and both are down-regulated during ES differentiation (Figure S6), suggesting a more global proteasome lid response during differentiation.
The DUB enzymatic activity of Psmd14 is essential for ES cell pluripotency
Psmd14 contains a JAMM/MPN+ motif sequence and its catalytic site resembles that of a zinc metalloprotease (Verma et al., 2002; Yao and Cohen, 2002). Two conserved histidines among different species from yeast to human, H113 and H115, are thought to play a key role in coordinating the zinc ion. To determine the role of the enzymatic activity of Psmd14, and exclude that the RNAi-induced effects are solely attributed to the structural requirement for Psmd14, we generated inducible ES lines expressing point mutants on the MPN+ domain (Figure 5I, Figure S6). Proper incorporation of mutant proteins into the proteasome lid (Gallery et al., 2007) was verified (Figure 5J).
Knockdown of the endogenous Psmd14 transcripts was achieved using short hairpins that target the 5' or the 3' UTR. Induced expression of Psmd14WT, Psmd14H113Q or Psmd14C120S coding region was utilized in order to investigate possible rescue of the differentiation phenotype. We observed that Psmd14WT-expressing ES cells were resistant to knockdown as measured by SSEA1 staining of transduced cells. In contrast the point mutant Psmd14H113Q failed to rescue the RNAi-induced phenotype. However, Psmd14C120S-expressing cells, similarly to Psmd14WT, were able to revert the differentiation phenotype measured by SSEA1 levels (Figure 5K). Moreover, Psmd14WT- and the Psmd14C120S- expressing cells retain elevated levels of Oct4 compared to uninduced cells after shRNA Psmd14 knockdown, but Psmd14H113Q- expressing ES cells did not (Figure 5L). In agreement with the above, only Psmd14 and Psmd14C120S, but not Psmd14H113Q - expressing ES cells were able to retain alkaline phosphatase (AP) activity after 4 days of differentiation with retinoic acid and under constant induction (Figure S6). Furthermore, western blots for K48- or K63- specific antibodies showed a marked decrease in both types of polyubiquitinated proteins following induction of Psmd14WT and Psmd14C120S but not Psmd14H113Q expression, indicating proper deubiquitinating activity that allows for efficient proteasome function (Figures 5L, Figure S6). The above combined results suggest that the enzymatic deubiquitinase activity of Psmd14 is necessary for proper ES cell identity.
The E3 ligase Fbxw7 controls ES cell pluripotency by regulating c-Myc protein stability
We then focused on the molecular function of Fbxw7, that when silenced leads to an inhibition of ES cell differentiation. We initially determined whether inhibition of differentiation following Fbxw7 silencing, was due to accumulation of core pluripotency factors. However, depletion of Fbxw7 did not lead to stabilization of Nanog, Oct4, or Sox2 (Figure 6A). Moreover, none of these factors co-immunoprecipitated with Fbxw7 (Figure 6B). Fbxw7 has been shown to ubiquitinate a number of substrates including mTor, Notch, p100, Cyclin E, Klf5, cJun, Srebp, Mcl-1, and c-Myc (Busino et al., 2012; Crusio et al., 2010; Welcker et al., 2004; Ye et al., 2004). c-Myc is an essential regulator of ES cell self-renewal and cellular reprogramming (Cartwright et al., 2005; Takahashi and Yamanaka, 2006) and is down-regulated during differentiation, whereas Fbxw7 is up-regulated at the transcriptional level as ES cells differentiate (Figure 6C). Ubiquitination of c-Myc was detected in ES cells in the mass-spectrometry experiments illustrated in Figure 1 and ubiquitination (as well as phosphorylation) sites were identified (Figure 1A, Figure S3). Also, during differentiation c-Myc accumulated upon proteasome inhibition (Figure 6D). Using shRNAs to silence Fbxw7, we evaluated accumulation of known substrates, including c-Myc during ES differentiation. These studies demonstrated that upon Fbxw7 silencing only c-Myc protein levels increased and remained equivalent to self-renewing ES cells (Figure 6E–G). We next sought to determine if the inhibition of ES cell differentiation phenotype resulting from Fbxw7 silencing could be rescued by decreasing c-Myc protein levels. Using the Nanog-GFP reporter cell line, and a combination of gene knock-down, we were able to show that c-Myc loss was indeed able to efficiently rescue Fbxw7-induced inhibition of differentiation phenotype (Figure 6H). Moreover, it was shown that degradation of c-Myc is preceded by a phosphorylation on threonine 58 (T58) by GSK3β (Gregory et al., 2003). To further correlate Fbxw7 silencing to c-Myc stability, we used an ES cell line that in the presence of 4-hydroxytamoxifen (4-OHT) expresses a form of c-Myc with a mutation in the T58 phosphorylation site (c-MycT58AER ES cells) (Cartwright et al., 2005). During differentiation, in the absence of 4-OHT, c-Myc protein is reduced, and cell morphology, and SSEA1 expression is consistent with differentiation, whereas either in the presence of 4-OHT or the depletion of Fbxw7 cells, c-Myc levels are maintained and cells maintain an ES morphology and SSEA-1 expression (Figure 6I–K). Silencing of Fbxw7 further enhances inhibition of differentiation in c-MycT58AER ES cells suggesting that it leads to accumulation of endogenous expressed c-Myc combined with mutant c-MycT58A (Figure 6I–K). These studies identify Fbxw7 as a novel key regulator of ES cell differentiation and demonstrate that c-Myc is its key substrate in ES cells.
The UPS members Psmd14 and Fbxw7 have key roles in cellular reprogramming
Based on our previous studies, we have hypothesized that Psmd14 and Fbxw7 could also play significant, although opposing, roles in cellular reprogramming. We first saw that Psmd14 is significantly higher expressed at the mRNA and protein level in both ES and iPS cells when compared to MEFs (Figure S7). On the other hand, Fbxw7 expression was similar in all three populations (Figure S7). We took advantage of a reprogrammable mouse model to determine the effect of silencing Psmd14 and Fbxw7 on iPS cell generation using siRNAs (Stadtfeld et al., 2010). These animals carry a Dox-inducible polycistronic cassette encoding the four reprogramming factors Oct4, Klf4, Sox2, and c-Myc (OKSM) downstream of the Collagen type I, alpha 1 gene (Col1a1) and a reverse tetracycline transactivator (rtTA) in the constitutively active ROSA26 locus. Using previously validated siRNAs against Psmd14 and Fbxw7 (Figure 3D and 4D), we silenced expression in “reprogrammable” MEFs at different time-points. MEFs were cultured in ES media containing Dox for 10 days at which point media was changed to ES media without Dox. Six days after the transfection cells were transfected a second time with siRNA. At this timepoint, morphology changes consistent with ongoing reprogramming were observed following silencing of Fbxw7, however, Psmd14-silenced MEFs maintained a fibroblast-like morphology (Figure 7A). At day 14 MEFs treated with siRNA against Fbxw7 produced ~60% more ES-like AP+ colonies when compared to MEFs treated with Dox alone (n=3, p<0.01). On the other hand, knock-down of Psmd14 led to significant reduction in iPS colony formation (n=3, p=0.01) (Figure 7B–D). These studies suggested that silencing of Fbxw7 enhances iPS generation, and that the expression of the self-renewal regulator Psmd14 is absolutely essential for cellular reprogramming.
Figure 7. Psmd14 and Fbxw7 regulate cellular reprogramming.
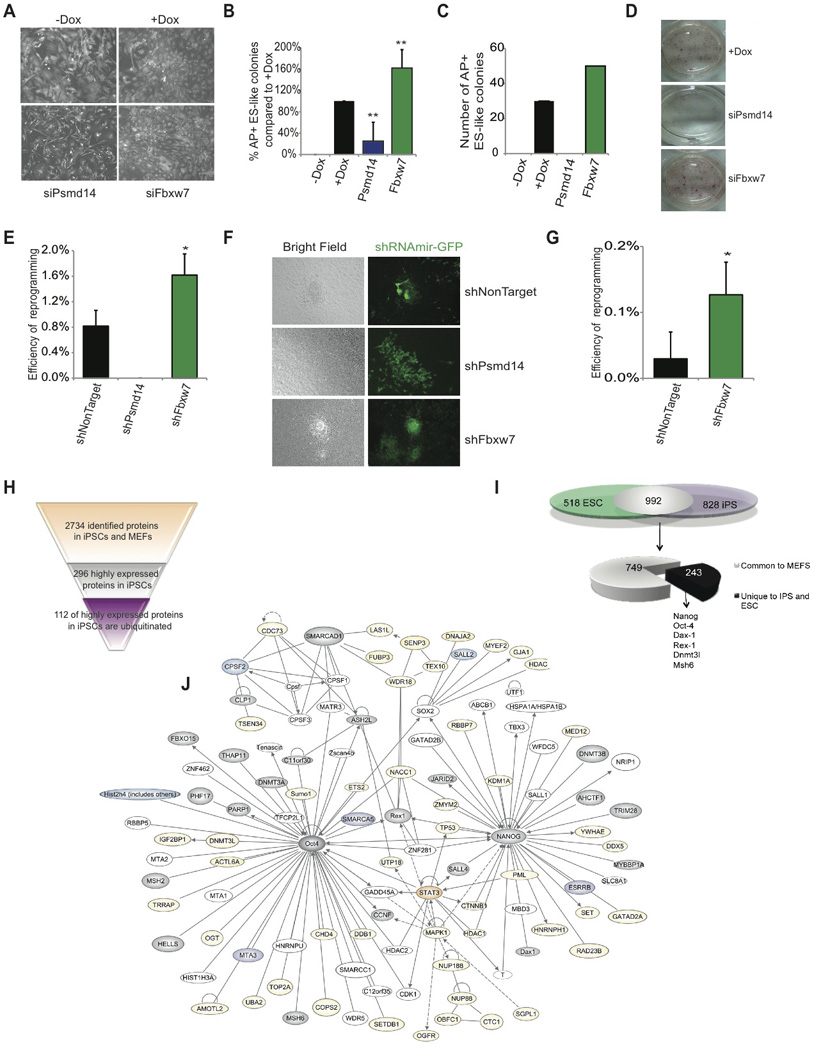
A) Images of ubiquitously expressed actin-RFP 6 days post siRNA transfection and Dox (OKSM) induction demonstrating morphology changes. B) AP+ ES-like colony count at Day 14 following siRNA knockdown of Fbxw7 and Psmd14 relative to +Dox control. C) Absolute number of AP+ ES-like colonies at day 14 following siRNA transfection and addition of Dox of MEFs. One representative experiment is shown (n=4). D) AP staining 14 days following siRNA transfection and addition of Dox. E) Reprogramming efficiency of OKSM MEFs expressing shRNAs against non-target, Psmd14, or Fbxw7 to generate AP+ ES-like colonies. Colonies were enumerated at day 14. F) Morphology of representative colonies following transduction of shRNAmir at day 14. Green; shRNAmir transduced cells. G) Efficiency of OSK MEFs expressing shRNAs against NonTarget, or Fbxw7 to generate AP+ ES-like colonies. Colonies were enumerated at day 14. H) Proteins identified and quantified in MEFs and iPS in previous studies, showing proteins highly expressed in iPS and the number of the ones that are ubiquitinated. I) Overlap between ubiquitinated proteins in ES and iPS and comparison with ubiquitinated proteins in MEFs. J) Self-Renewal network, as depicted in Figure 1D, demonstrating proteins that have been identified to be ubiquitinated in iPS and ES (grey). Proteins also ubiquitinated in MEFs are marked in yellow. Proteins in orange were found ubiquitinated in iPS and MEFs and proteins in purple were exclusively found in iPS. Data represented as +SEM; N=3. *p-value <0.05, **p-value<0.01.
To further evaluate the effects of stable silencing of UPS components on iPS formation, OKSM-MEFs were transduced with virus expressing shRNAs against either non-target control, Psmd14, or Fbxw7 (Figure S7). MEFs were sorted by GFP expression and plated on wild-type mitomycin C treated MEF feeder layer. MEFs expressing Psmd14 shRNAs failed to reprogram and generate iPS (Figure 7E and F). This data strongly suggested that Psmd14 is required for iPS generation. Once more, the absence of shPsmd14+ iPS colonies was not due to a general effect on cell viability, as shPsmd14+ MEFs were readily detectable (Figure 7F, Figure S7). This finding was consistent with the requirement of the full-function of the proteasome to induce pluripotency and even partial inhibition with low dose of MG132 completely inhibits reprogramming (Figure S7). In agreement with the experiments using siRNA, Fbxw7 knock-down promoted a two-fold increase in cellular reprogramming efficiency (Figure 7E). iPS colonies derived from MEFs expressing shRNA against Fbxw7 express pluripotency genes Nanog and Oct4 from the endogenous loci demonstrating reprogrammed state (Figure S7 and not shown).
Since our previous studies identified c-Myc as a substrate responsible for inhibition of differentiation following Fbxw7 silencing, we next asked if Fbxw7 silencing could replace exogenous c-Myc expression. Although exogenous expression of Oct4, Sox2, Klf4, and c-Myc together induce reprogramming, exogenous c-Myc can be omitted during reprogramming albeit low efficiency (Takahashi and Yamanaka, 2006). We thus determined whether silencing of Fbxw7 could augment reprogramming of MEFs that carry a Dox inducible polycistronic cassette encoding three reprogramming factors Oct4, Klf4, and Sox2 (OKS) (E.A. and K.H., unpublished). Silencing of Fbxw7 in OKS MEFs significantly enhanced efficiency from ~0.01% to ~0.15%, implicating once more Fbxw7 in the regulation of endogenous c-Myc protein abundance (Figure 7G). Together these studies identified, Psmd14 and Fbxw7, as novel regulators of cellular reprogramming.
Comparing ES and iPS cell “ubiproteomes”
Although ES and iPS cells are highly similar with respect to their morphology, behavior in culture, and developmental potential, several lines of evidence point towards important molecular and functional differences between these two pluripotent cell types (Stadtfeld and Hochedlinger, 2010). We therefore decided to study the iPS “ubiproteome” in a similar fashion as illustrated in Figure 1. Initially, we cross-referenced the ubiquitin-modified proteins identified in iPS cells with previously published large-scale protein expression analysis of iPS cells and MEFs (Huang et al., 2012) and found that 112 of the proteins highly expressed in iPS cells compared to MEFs were ubiquitinated (Figure 7H). Then, we compared the ubiquitin-modified proteins identified in pluripotent ES cells and iPS cells and found an overlap of 65.7% (992 proteins) between both populations (Figure 7I, Figure S7 Table S7). Interestingly, out of these 992 ubiquitin-modified proteins, 25% were exclusively ubiquitinated in ES and iPS cells but not in MEFs. Among the shared ubiquitinated proteins, we identified the majority of the pluripotency network proteins (Figure 7J). Our combined studies revealed that the UPS participates in the regulation of the complex process of cellular reprogramming and that both, ES and iPS cells, use ubiquitination in similar ways to regulate the abundance and activity of key factors of pluripotency.
DISCUSSION
This is the first study identifying the ubiquitin-proteasome system as a novel level of regulation of pluripotency and cellular reprogramming. Our work maps the ES cell “ubiproteome”, and identify a substantial amount of novel ubiquitinated proteins with important roles during ES cell differentiation. Moreover, using UPS-targeted RNAi screens, we have identified a significant number of novel ubiquitin enzymes essential for the regulation of pluripotency, ES cell differentiation and homeostasis. These genes are both ubiquitin ligases and deubiquitinases, suggesting that both modes of regulation could be important for ES cell function. We have focused on the function of Psmd14 and shown that this DUB is a part of the 19S proteasome lid, its expression is regulated during ES cell differentiation and its silencing affected ES pluripotency and abrogated cellular reprogramming. Additionally, we have studied a role of the ligase Fbxw7 in ES cell differentiation and induced pluripotency, and have identified c-Myc (within a large number of described substrates) as one of the key ubiquitinated protein substrate of Fbxw7 involved in ES cell differentiation. Finally, we have identified novel roles for UPS members in cellular reprogramming, along with characterizing the “ubiproteome” of fully reprogrammed somatic cells.
Our mass spectrometry-based mapping of the ES and iPS cell “ubiproteome” demonstrated that a significant number of known components of the ES cell pluripotency network, including members of the Nanog and Oct4 pathways, are ubiquitinated. A few of them, including Nanog, c-Myc and Oct4, have been previously suggested to be regulated by ubiquitination (Liao and Jin, 2010; Ramakrishna et al., 2011) however, there was little information on their sites of ubiquitination and the role of such ubiquitination events in ES pluripotency and differentiation. Moreover, our studies also identified novel sites of ubiquitination on additional known regulators of pluripotency (e.g. Setdb1, Jarid2, Fbxo15). These combined observations introduce the hypothesis that ubiquitin-mediated fine-tuning of the abundance of essential pluripotency regulators could support distinct states of pluripotency or predispose the cell for the initiation of a program of differentiation. Future studies will further detail the role of UPS and assign novel ligases and DUBs to the novel ES cell-specific substrates.
Our studies have identified a large number of UPS members, both ligases and deubiquitinases, that have essential roles in both ES cell self-renewal and differentiation. One of these proteins is the E3 ligase Fbxw7, the depletion of which lead to inhibition of differentiation, and more importantly, enhancement of iPS cell generation. c-Myc, one of the known Fbxw7 substrates, is known to play key roles in both ES cell homeostasis and in somatic cell reprogramming (Smith et al., 2010; Takahashi and Yamanaka, 2006). We were able to demonstrate that c-Myc depletion rescues inhibition of differentiation induced by silencing of Fbxw7, suggesting that the majority of the ES cell-specific Fbxw7 activity passes through c-Myc. In agreement with our findings, it was recently shown that self-renewing ES cells are characterized by active Akt signaling, leading to GSK3 translocation and inhibition of c-Myc phosphorylation (An et al., 2008), an event that leads to Fbxw7 recruitment. Our data provide the link between extracellular signaling controlling GSK3 activity and degradation of c-Myc during ES cell differentiation. However, we cannot exclude that additional, novel, Fbxw7 substrates are also important for optimal ES cell differentiation.
The DUB Psmd14 appears to be necessary for the maintenance of ES pluripotency. Our proteomic analysis revealed striking Psmd14 specificity for the 19S proteasome lid, in agreement with recent positional mapping of the yeast proteasome (Lander et al., 2012), suggesting that the structure of the mammalian proteasome lid adopts similar conformation. We have utilized mutagenesis of the Psmd14 active site and concluded that the requirement for Psmd14 at the proteasome lid is due to its specific DUB enzymatic activity. Indeed, a single point mutation on the conserved zinc-coordinating histidine 113 abolished DUB activity. These results are in agreement with mutational studies on Psmd14 function in human protein orthologues (Gallery et al., 2007). In contrast, we found cysteine 120 to be dispensable for Psmd14 function in ES cells, consistent with the fact that it belongs to a family distinct from the cysteine proteases. These results suggested that Psmd14 plays a crucial role in maintaining self-renewal and pluripotency by controlling deubiquitination - a key step in protein degradation. We suggest that active ubiquitination (and de-ubiquitination) and protein degradation is required for proper maintenance of pluripotency for two main reasons: to maintain expression threshold for members of the pluripotency network (i.e. Nanog, Oct4) and counter-act permissive expression of factors that could favor differentiation. Supporting this notion, Dillin and colleagues has recently identified PSMD11, an interacting partner of PSMD14, expression and function is essential for human ES cell pluripotency (Vilchez et al., 2012).
Recent systems biology-level studies suggested the existence of additional modes of regulation, responsible for changes in protein expression levels, controlling ES cell fate and homeostasis (Lu et al., 2009). Our study identifies the UPS as such a novel layer of regulation and suggests that dynamic ubiquitin tagging could be essential for physiological ES cell differentiation and early embryogenesis. The dynamic reversibility of the ubiquitin modification and the recent development of both general proteasome inhibitors (Adams and Kauffman, 2004) as well as specific antagonists of ligase function (Aghajan et al., 2010; Orlicky et al., 2010) open the way for future manipulation of the UPS in ES cells leading to more efficient induced pluripotency (iPS generation) and targeted differentiation towards pre-determined lineages.
Experimental Procedures
Isolation of ubiquitinated peptides and Mass Spectrometry
For SILAC experiments, undifferentiated ES cells were labeled with 13C615N4-arginine and 13C6-Lysine for five or more passages and treated with MG132. Cells were kept undifferentiated and were subjected to differentiation in the presence of 5 µM retinoic acid. Equal amounts of proteins of both populations were mixed, lysed together and the resulting protein extracts were reduced, carboxamidomethylated, and digested with trypsin. Peptides were separated from non-peptide material by solid-phase extraction with Sep-Pak C18 cartridges. Lyophilized peptides were re-dissolved, and peptides with the K- GG modification were purified using the K- GG motif antibody (Cell Signaling) conjugated to Protein-A Agarose (Roche). Eluted peptides were desalted and concentrated with PerfectPure C18 tips immediately prior to LC-MS/MS analysis. Tandem mass spectra were collected with an LTQ-Orbitrap hybrid mass spectrometer. For the label free samples, we used cells of each population independently and were analyzed with the same methodology. Further details concerning label-free quantification of K-GG modified peptides can be found in supplementary experimental procedures.
ES cell culture, transfection and alkaline phosphatase staining
ES cell lines were maintained on gelatin coated plates in ES media as described previoulsy (Reavie et al., 2010). For the self-renewal screen, Nanog-GFP ES cells (kind gift from Dr. I. Lemischka, Mount Sinai School of Medicine) were transfected with 30nM siRNA (Dharmacon). For differentiation, 12 hours post-transfection, ES media was changed to media −LIF + 5µM RA. GFP fluorescence was measured using an LSRII (BD Biosciences) 48–72 hours post-transfection. C-MycT58AER ES cells were maintained in ES media supplemented with 10nM 4-OHT (kind gift from Dr. Stephen Dalton, University of Georgia). For alkaline phosphatase staining assays cells were fixed and stained with alkaline phosphatase detection kit (Millipore).
Cloning in targeting vectors and engineering of ES cells expressing tagged proteins
cDNAs for Psmd14WT, Psmd14H113Q, Psmd14C120S and eGFP were cloned in frame into the modified and Tet-operated vector pINTA, bearing N-terminal Strep-TagII/Flag (SF) tandem tags (Gloeckner et al., 2009) (kind gift of Dr. R. Bonasio, NYU School of Medicine). The resulting vectors (pINTA-SF-Psmd14 and pINTA-SF-eGFP) were nucleoporatated (Amaxa) into KH2 ES cells engineered to carry an M2rtTA transactivator in the ROSA26 locus, ensuring inducible expression of the tagged cDNA(Beard et al., 2006; Hochedlinger et al., 2005). ES cells were selected with 50µg/mL Zeocin (Invitrogen) for 7 days. Expression of tagged proteins was confirmed by western blot analysis with anti-StrepTagII antibody (StrepMABclassic, IBA).
Flow Cytometry analysis
For intracellular c-Myc staining, cells trypsinized, fixed, and permeabilized using BD cytofix/cytoperm kit following manufactuors protocol. Stainings were performed with rabbit anti-c-Myc (Cell Signaling) followed by goat anti-rabbit Alexa Fluor 647 (Invitrogen).
Immunofluorescence
siRNA transfections were carried out in 96-well black plates (Corning). Cells were fixed with 4% paraformelhyde (BD Biosciences) for 10 minutes, and permeabilized for 10 minutes using 0.1% Triton-X100 (Sigma). Wells were blocked with 3% goat serum (Invitrogen), and stained with primary antibodies overnight for Ubiquitin Lys48-specific (Apu2) (Millipore), and Oct-3/4 (C-10) (Santa Cruz). Secondary antibody staining was performed with Alexa488 or 594 conjugated antibodies (Invitrogen), Alexa594 conjugated Phalloidin (Invitrogen), and DAPI.
Microarray Analysis
Total cellular RNA was isolated using the RNAeasy microkit (Qiagen) using manufacturer's instructions. Following cDNA synthesis and labeling samples were hybridized to Affymetrix mouse 430 chips (Affymetrix Inc) and scanned at the NYU medical center Affymetrix Core Facility. CEL files were loaded into GeneSpring (Agilent) to assess overall quality. Feature intensities for each probe set were condensed into a single intensity value using the MAS 5.0 algorithm.
Cellular Reprogramming
For siRNA silencing during reprogramming, “reprogrammable” MEFs were reverse transfected with 30nM siRNA (Dharmacon) and lipofectamine 2000 (Invitrogen) in a 12 well plate, the following day media was replaced with ES cell media and 2µg/ml Dox (Stadtfeld et al., 2010). 7 days following initial transfection cells were transfected a second time, and day 11 Dox was removed, and cells were cultured in ES media until day 15 post transfection. ES-like colonies were enumerated, and stained for alkaline phosphatase activity (Milipore). For experiments with shRNA silecing, “reprogrammable” MEFs were plated with concentrated pLMP-GFP retrovirus in the presence of 8µg/ml polybrene. The following day GFP+ cells were sorted and plated on mitomycin C treated MEFs, and media was replaced with ES media containing 2µg/ml Dox and replaced every other day for 10 days, where Dox was removed for the last 4 days of cultures. At day 14, ES cell like colonies were enumerated and/or clones isolated.
Statistical analysis
All experiments were preformed in triplicate unless noted and statistical analyses were performed using paired two-tailed Student's t-test assuming experimental samples of equal variance. * p-value <0.05, **p-value< 0.01. siRNA libraries were divided into seven 96-well plates each containing negative and positive controls, as well as, nontarget control. The z-score [z=(x-m)/s] was determined for each plate, where×was the raw score to be standardized, m the mean of the differential expression (% differential expression=% GFP+ cells in target gene- % GFP+ in non-target control) of the plate, and s is the standard deviation of the plate. Each plate was preformed in triplicate and z-score was averaged between triplicate experiments.
Supplementary Material
HIGHLIGHTS.
Shotgun proteomics characterizes the ubiquitin landscape in ES and iPS cells
RNAi screens of UPS members identify novel regulators of pluripotency
Psmd14 deubiquitinase activity is required to maintain pluripotency
c-Myc stabilization by Fbxw7 inhibits differentiation and enhances reprogramming
ACKNOWLEDGEMENTS
We would like to thank Dr. I. Lemischka for the Nanog-GFP reporter ES cell line; Dr. S. Dalton for the c-c-mycT58AER ES cells; Drs. D. Reinberg, R. Bonasio and L-A. Rojas for providing the pINTA-SF vectors; Dr. F. Gonsalves and Dr. Beatrix Ueberheide for helpful discussions; and Dr. H. Li and Dr. T. Liu at the Center for Advanced Proteomics Research, New Jersey School of Medicine, for selected mass spectrometry analysis. Also, the NYU Genome Technology Center (supported in part by NIH/NCI P30 CA016087-30 grant) for expert assistance with microarray experiments. IA is supported by the National Institutes of Health (RO1CA133379, RO1CA105129, RO1CA149655, RO1GM088847), and the NYSTEM Program of the New York State Health Department. E.I.C. is supported by the National Institutes of Health (1U19A1091175-01 and 1S10 RR023680-1). S.M.B. is supported by the NYU Hematology/Oncology NIH training grant (5T32HL007151-33) and the NIH institutional training grant (1T32CA160002-01), B.A-O. is supported by the Alexander von Humboldt Foundation, and A.S. is supported by NYSTEM institutional NYU Stem Cell training grant (C026880). I.A and K.H. are Howard Hughes Medical Institute Early Career Scientists.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession Numbers
All microarray data are deposited in Gene Expression Omnibus (GEO) database with accession number GSE39772.
REFERENCES
- Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (Bortezomib) Cancer investigation. 2004;22:304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- Aghajan M, Jonai N, Flick K, Fu F, Luo M, Cai X, Ouni I, Pierce N, Tang X, Lomenick B, et al. Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat Biotechnol. 2010;28:738–742. doi: 10.1038/nbt.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Yang DY, Xu QZ, Zhang SM, Huo YY, Shang ZF, Wang Y, Wu DC, Zhou PK. DNA-dependent protein kinase catalytic subunit modulates the stability of c-Myc oncoprotein. Molecular cancer. 2008;7:32. doi: 10.1186/1476-4598-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003;17:126–140. doi: 10.1101/gad.224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard C, Hochedlinger K, Plath K, Wutz A, Jaenisch R. Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis. 2006;44:23–28. doi: 10.1002/gene.20180. [DOI] [PubMed] [Google Scholar]
- Busino L, Millman SE, Scotto L, Kyratsous CA, Basrur V, O'Connor O, Hoffmann A, Elenitoba-Johnson KS, Pagano M. Fbxw7alpha- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat Cell Biol. 2012;14:375–385. doi: 10.1038/ncb2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright P, McLean C, Sheppard A, Rivett D, Jones K, Dalton S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development. 2005;132:885–896. doi: 10.1242/dev.01670. [DOI] [PubMed] [Google Scholar]
- Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- Crusio KM, King B, Reavie LB, Aifantis I. The ubiquitous nature of cancer: the role of the SCF(Fbw7) complex in development and transformation. Oncogene. 2010;29:4865–4873. doi: 10.1038/onc.2010.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- Gallery M, Blank JL, Lin Y, Gutierrez JA, Pulido JC, Rappoli D, Badola S, Rolfe M, Macbeth KJ. The JAMM motif of human deubiquitinase Poh1 is essential for cell viability. Mol Cancer Ther. 2007;6:262–268. doi: 10.1158/1535-7163.MCT-06-0542. [DOI] [PubMed] [Google Scholar]
- Gloeckner CJ, Boldt K, Schumacher A, Ueffing M. Tandem affinity purification of protein complexes from mammalian cells by the Strep/FLAG (SF)-TAP tag. Methods Mol Biol. 2009;564:359–372. doi: 10.1007/978-1-60761-157-8_21. [DOI] [PubMed] [Google Scholar]
- Gontan C, Achame EM, Demmers J, Barakat TS, Rentmeester E, van IW, Grootegoed JA, Gribnau J. RNF12 initiates X-chromosome inactivation by targeting REX1 for degradation. Nature. 2012;485:386–390. doi: 10.1038/nature11070. [DOI] [PubMed] [Google Scholar]
- Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. The Journal of biological chemistry. 2003;278:51606–51612. doi: 10.1074/jbc.M310722200. [DOI] [PubMed] [Google Scholar]
- Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121:465–477. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang X, Tian C, Liu M, Wang Y, Tolmachev AV, Sharma S, Yu F, Fu K, Zheng J, Ding SJ. Quantitative proteomic analysis of mouse embryonic fibroblasts and induced pluripotent stem cells using 16O/18O labeling. J Proteome Res. 2012;11:2091–2102. doi: 10.1021/pr300155r. [DOI] [PubMed] [Google Scholar]
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Tanaka K. Regulatory mechanisms involved in the control of ubiquitin homeostasis. Journal of biochemistry. 2010;147:793–798. doi: 10.1093/jb/mvq044. [DOI] [PubMed] [Google Scholar]
- Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E, Martin A. Complete subunit architecture of the proteasome regulatory particle. Nature. 2012;482:186–191. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QR, Xing XB, Chen TT, Li RX, Dai J, Sheng QH, Xin SM, Zhu LL, Jin Y, Pei G, et al. Large scale phosphoproteome profiles comprehensive features of mouse embryonic stem cells. Mol Cell Proteomics. 2011;10:M110 001750. doi: 10.1074/mcp.M110.001750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao B, Jin Y. Wwp2 mediates Oct4 ubiquitination and its own auto-ubiquitination in a dosage-dependent manner. Cell research. 2010;20:332–344. doi: 10.1038/cr.2009.136. [DOI] [PubMed] [Google Scholar]
- Lu R, Markowetz F, Unwin RD, Leek JT, Airoldi EM, MacArthur BD, Lachmann A, Rozov R, Ma'ayan A, Boyer LA, et al. Systems-level dynamic analyses of fate change in murine embryonic stem cells. Nature. 2009;462:358–362. doi: 10.1038/nature08575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NJ, Gregory RI. MicroRNA gene regulatory pathways in the establishment and maintenance of ESC identity. Cell Stem Cell. 2010;7:31–35. doi: 10.1016/j.stem.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A, Onoyama I, Sunabori T, Kageyama R, Okano H, Nakayama KI. Fbxw7-dependent degradation of Notch is required for control of "stemness" and neuronal-glial differentiation in neural stem cells. The Journal of biological chemistry. 2011;286:13754–13764. doi: 10.1074/jbc.M110.194936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Oike Y, Onoyama I, Iwama A, Arai F, Takubo K, Mashimo Y, Oguro H, Nitta E, Ito K, et al. Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes & development. 2008;22:986–991. doi: 10.1101/gad.1621808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A. Epigenetic modifications in pluripotent and differentiated cells. Nat Biotechnol. 2010;28:1079–1088. doi: 10.1038/nbt.1684. [DOI] [PubMed] [Google Scholar]
- Niakan KK, Ji H, Maehr R, Vokes SA, Rodolfa KT, Sherwood RI, Yamaki M, Dimos JT, Chen AE, Melton DA, et al. Sox17 promotes differentiation in mouse embryonic stem cells by directly regulating extraembryonic gene expression and indirectly antagonizing self-renewal. Genes & development. 2010;24:312–326. doi: 10.1101/gad.1833510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, Scholer H, Smith A. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- Orlicky S, Tang X, Neduva V, Elowe N, Brown ED, Sicheri F, Tyers M. An allosteric inhibitor of substrate recognition by the SCF(Cdc4) ubiquitin ligase. Nat Biotechnol. 2010;28:733–737. doi: 10.1038/nbt.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Brumbaugh J, Wenger CD, Tian S, Probasco MD, Bailey DJ, Swaney DL, Tervo MA, Bolin JM, Ruotti V, et al. Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat Methods. 2011;8:821–827. doi: 10.1038/nmeth.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- Ramakrishna S, Suresh B, Lim KH, Cha BH, Lee SH, Kim KS, Baek KH. PEST Motif Sequence Regulating Human NANOG for Proteasomal Degradation. Stem Cells Dev. 2011 doi: 10.1089/scd.2010.0410. [DOI] [PubMed] [Google Scholar]
- Reavie L, Della Gatta G, Crusio K, Aranda-Orgilles B, Buckley SM, Thompson B, Lee E, Gao J, Bredemeyer AL, Helmink BA, et al. Regulation of hematopoietic stem cell differentiation by a single ubiquitin ligase-substrate complex. Nat Immunol. 2010;11:207–215. doi: 10.1038/ni.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaniel C, Lee DF, Lemischka IR. Exploration of Self-Renewal and Pluripotency in ES Cells Using RNAi. Methods Enzymol. 2010;477C:351–365. doi: 10.1016/S0076-6879(10)77018-X. [DOI] [PubMed] [Google Scholar]
- Smith KN, Singh AM, Dalton S. Myc represses primitive endoderm differentiation in pluripotent stem cells. Cell stem cell. 2010;7:343–354. doi: 10.1016/j.stem.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Hochedlinger K. Induced pluripotency: history, mechanisms, and applications. Genes Dev. 2010;24:2239–2263. doi: 10.1101/gad.1963910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Maherali N, Borkent M, Hochedlinger K. A reprogrammable mouse strain from gene-targeted embryonic stem cells. Nat Methods. 2010;7:53–55. doi: 10.1038/nmeth.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szutorisz H, Georgiou A, Tora L, Dillon N. The proteasome restricts permissive transcription at tissue-specific gene loci in embryonic stem cells. Cell. 2006;127:1375–1388. doi: 10.1016/j.cell.2006.10.045. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Thompson BJ, Jankovic V, Gao J, Buonamici S, Vest A, Lee JM, Zavadil J, Nimer SD, Aifantis I. Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J Exp Med. 2008 doi: 10.1084/jem.20080277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Aravind L, Oania R, McDonald WH, Yates JR, 3rd, Koonin EV, Deshaies RJ. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298:611–615. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- Vilchez D, Boyer L, Morantte I, Lutz M, Merkwirth C, Joyce D, Spencer B, Page L, Masliah E, Berggren WT, et al. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature. 2012;489:304–308. doi: 10.1038/nature11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet. 2007;39:380–385. doi: 10.1038/ng1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr Biol. 2004;14:1852–1857. doi: 10.1016/j.cub.2004.09.083. [DOI] [PubMed] [Google Scholar]
- Xu G, Paige JS, Jaffrey SR. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat Biotechnol. 2010;28:868–873. doi: 10.1038/nbt.1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T, Cohen RE. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature. 2002;419:403–407. doi: 10.1038/nature01071. [DOI] [PubMed] [Google Scholar]
- Ye X, Nalepa G, Welcker M, Kessler BM, Spooner E, Qin J, Elledge SJ, Clurman BE, Harper JW. Recognition of phosphodegron motifs in human cyclin E by the SCF(Fbw7) ubiquitin ligase. The Journal of biological chemistry. 2004;279:50110–50119. doi: 10.1074/jbc.M409226200. [DOI] [PubMed] [Google Scholar]
- Ying QL, Nichols J, Chambers I, Smith A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell. 2003;115:281–292. doi: 10.1016/s0092-8674(03)00847-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.