

Abstract
Background
The recently approved anti-AIDS drug rilpivirine (TMC278, Edurant) is a nonnucleoside inhibitor (NNRTI) that binds to reverse transcriptase (RT) and allosterically blocks the chemical step of DNA synthesis. In contrast to earlier NNRTIs, rilpivirine retains potency against well-characterized, clinically relevant RT mutants. Many structural analogues of rilpivirine are described in the patent literature, but detailed analyses of their antiviral activities have not been published. This work addresses the ability of several of these analogues to inhibit the replication of wild-type (WT) and drug-resistant HIV-1.
Results
We used a combination of structure activity relationships and X-ray crystallography to examine NNRTIs that are structurally related to rilpivirine to determine their ability to inhibit WT RT and several clinically relevant RT mutants. Several analogues showed broad activity with only modest losses of potency when challenged with drug-resistant viruses. Structural analyses (crystallography or modeling) of several analogues whose potencies were reduced by RT mutations provide insight into why these compounds were less effective.
Conclusions
Subtle variations between compounds can lead to profound differences in their activities and resistance profiles. Compounds with larger substitutions replacing the pyrimidine and benzonitrile groups of rilpivirine, which reorient pocket residues, tend to lose more activity against the mutants we tested. These results provide a deeper understanding of how rilpivirine and related compounds interact with the NNRTI binding pocket and should facilitate development of novel inhibitors.
Keywords: HIV, Reverse transcriptase, Rilpivirine
Background
Human immunodeficiency virus type 1 (HIV-1), which causes acquired immunodeficiency syndrome (AIDS), poses important problems for world health with an estimated 2.7 million newly infected individuals in 2010 and an estimated 34 million individuals living with HIV/AIDS worldwide [1]. However, progress is being made in treating HIV/AIDS. Highly active antiretroviral therapy (HAART) is reaching a large number of infected individuals that, until recently, either had no access to treatment, or relied upon ineffective monotherapies. Modern HAART regimens consist of a combination of drugs from several classes that can include nucleoside reverse transcriptase inhibitors (NRTIs), integrase inhibitors, entry inhibitors, CCR5 antagonists, protease inhibitors and non-nucleoside reverse transcriptase inhibitors (NNRTIs) [2]. NNRTIs are used as components of HAART regimens because of their high specificity and modest toxicity [3].
At present, five NNRTIs have been approved for use in humans by the U.S. Food and Drug Administration: rilpivirine (TMC278) (1), etravirine (2), nevirapine (3), efavirenz (4) and delavirdine (5) (Figure 1). Multiple other chemotypes that have NNRTI activity have been reported, including numerous structurally unique small molecules. These compounds bind in the same hydrophobic binding pocket, and the literature shows a distinct progression in NNRTI design [4-7]. Notably, the (E)-3-(4-amino-3,5-dimethylphenyl)acrylonitrile ring system of rilpivirine (Figure 1, top left ring system of 1) led to improved potency against both WT and mutant RTs.
Figure 1.

Chemical structures of approved NNRTIs rilpivirine (1), etravirine (2), nevirapine (3), efavirenz (4) and delaviridine (5).
Although HIV-1 can develop resistance to all of the approved drugs, the first generation NNRTIs were particularly susceptible to the rapid emergence of resistant virus [8]. Rilpivirine and etravirine belong to the chemical class of NNRTIs known as diarylpyrimidines (DAPYs) that was developed through the collaborative efforts of pharma and academia [3,9,10]. The DAPY class of NNRTIs was selected for further study because these compounds are able to bind and inhibit WT RT and a number of clinically relevant NNRTI-resistant mutants. This ability derives from the strategic flexibility of the compounds within the NNRTI binding pocket [11]. Further investigation of the DAPY compounds remains important because they retain activity against a wide range of drug-resistant mutants [12].
Clinical trials of rilpivirine, in combination with two NRTIs, showed that distinct patterns of mutants were selected in patients experiencing virologic failure (VF) compared to treatment regimens with efavirenz [13,14]. Patients receiving rilpivirine were more likely to harbor viruses with NRTI resistance mutations, particularly M184I, than patients receiving efavirenz. Among the NNRTI resistance mutations, E138K was selected most frequently with rilpivirine, while K103N was selected most frequently with efavirenz. The association between M184I and E138K selection in patients receiving rilpivirine is noteworthy, because E138K has been shown to restore replication capacity to viruses harboring M184 mutations [15]. E138K and M184I were detected in 45% and 47%, respectively, of patients receiving rilpivirine and experiencing VF [13]. These mutations were found in 0% and 7%, respectively, of patients experiencing VF in the efavirenz group. By comparison, clinical trials revealed that resistance to etravirine is achieved by a combination of at least three mutations, including V90I, A98G, L100I, K101E/P, V106I, V179D/F, Y181C/I/V and G190A/S [16]. There has been considerable work showing how the DAPY class of NNRTIs bind and inhibit RT [3,10,17-20]. Several groups have designed derivatives of these compounds [20,21]. However, much of the SAR surrounding this class of agents is found primarily in the patent literature.
The activities of a number of key structural analogues of both rilpivirine (1) and etravirine (2) against NNRTI-resistant mutants have not been reported, nor have the interactions of these analogs with HIV-1 RT been evaluated in structural studies. Such information would provide a deeper understanding of how these molecules inhibit WT and drug-resistant RTs and how they can be engineered to enhance their activity against viruses that carry resistance mutations. In addition, novel analogues that select for different patterns of mutations than rilpivirine or etravirine could be used as second-line treatment in patients experiencing VF with either of these drugs, or possibly, in combination with existing drugs. The difference in mutation profile selected by these drugs, despite their structural similarities, suggests that subtle changes can significantly affect the mechanism by which resistance arises. To pursue the goal of identifying analogues that will select novel resistance mutations, we focused our efforts on compounds with specific alterations of the pyrimidine moiety and the 4-amino-benzonitrile moiety of rilpivirine (1) (Figure 2). We were motivated to alter only these structural motifs because the (E)-3-(4-amino-3,5-dimethylphenyl)acrylonitrile ring system has been shown to be an important motif for rilpivirine binding within the induced NNRTI-binding pocket [19].
Figure 2.

Chemical structures of rilpivirine analogues. Analogues differed from rilpivirine (1, boxed) by the addition of an exocyclic moiety to the pyrimidine ring in either of its ‘flipped’ conformations (6–9), the replacement of the pyrimidine ring with a 2,6-purine ring system in ‘flipped’ conformations with or without a protecting group (10–13), replacing the 4-benzonitrile moiety in addition to replacing the pyrimidine ring with a 2,6-purine (14–25), or replacing the pyrimidine ring with a 2,9-purine ring system (26, 27). Differences between each analogue and rilpivirine are indicated in red.
Results and discussion
Inhibitory activity of pyrimidine-based analogues
We examined the effects of rilpivirine and the analogues on HIV-1 replication using a previously described assay and report the concentrations at which luciferase reporter activity was reduced by half relative to uninhibited controls (EC50) with standard deviations in parentheses. In the assay, viral vectors that replicate using WT HIV reverse transcriptase or one of several mutants (L100I, K103N, V106A, E138K, Y181C, Y188L, H221Y and the double mutant K103N/Y181C) were used to infect cells in a single round assay. These mutants were chosen either because they are known NNRTI resistance mutations (L100I, K103N, Y181C, and the K103N/Y181C double mutant), they were selected in patients in rilpivirine clinical trials (E138K and H221Y), or they are positioned to make significant Van der Waals contacts with rilpivirine (V106A and Y188L). All compounds were initially screened against a panel that included at least one RT mutant from all three groups (WT, K103N, E138K, Y181C and Y188L). The cytotoxicity of the compounds was also determined to establish the therapeutic index of each of the compounds.
Results from these analyses are shown in Figure 3. Rilpivirine (1) demonstrated potent activity versus a vector that replicates using WT RT (0.4 ± 0.02 nM) and each of the single RT mutants (the highest value was 2.3 ± 0.2 nM versus the Y188L mutant). The CC50 for 1 was 10 ± 0.6 μM. Pyrimidine analogues that retained the substituted aryl amines in the same structural location and had either a nitro or amine substitution added to the 5-position (Figure 2, analogues 6 and 7) maintained similar activity profiles as 1 with only slight drops in activity versus the Y188L mutant (6.8 ± 0.5 nM and 2.3 ± 0.1 nM, respectively). A ‘flipping’ of this structural pattern (Figure 2, analogues 8 and 9) resulted in a modest drop in potency against the WT RT and K103N mutant and showed more substantial loss of potency against E138K, Y181C and Y188L mutants.
Figure 3.

Activities and cytotoxicities of rilpivirine (1) and its analogues. EC50s, as measured by reduction of luciferase reporter activity, are reported as nM values, CC50s are reported as μM values. Standard deviations are indicated in parentheses. Shading of EC50 values indicates a log scale: no shading = EC50 <1.0 nM, light gray = EC50 between 1.0 nM and 9.9 nM, dark gray = EC50 between 10 nM and 99nM, black = EC50 ≥100 nM. Therapeutic Index (T.I.) is the ratio of CC50/EC50.
Inhibitory activity of purine-based analogues
The closest structural homolog to rilpivirine (1) among the purine derivatives (Figure 2, analogue 11) retained good potency versus the WT as well as the K103N and Y181C mutants but showed a modest drop in activity versus the E138K mutant and a significant drop in activity versus the Y188L mutant (13 ± 0.5 nM). The ‘flipped’ analogue (Figure 2, analogue 13) maintained potency similar to 1 for the WT and each of mutants with the exception of E138K (4.2 ± 0.3 nM). Retaining the p-methoxybenzyl protecting group at the 9-position of the purine reduced the potency of both analogues (Figures 2 and 3, compare 10 to 11 and 12 to 13). Based upon these data, we used 13 as a scaffold and examined the effects of different substituents in place of the 4-benzonitrile moiety.
The results obtained with these analogues (Figure 2, analogues 14–25) show that many of the compounds that are highly active against WT RT show considerable differences in their ability to inhibit the various mutants. Attempts to replace the nitrile functionality of the benzonitrile with other moieties resulted in compounds that were active against the WT and most mutants. However, these analogues (14–19) showed a significant loss of activity versus the Y188L mutant (Figure 3). Replacing the benzonitrile with alternate ring systems was generally successful against WT, but Y188L again conferred resistance (analogues 20–25). The benzo[d]thiazole analogue (21) possessed particularly good activity versus WT and the mutants, including an EC50 of 4.4 nM against Y188L. The derivatives incorporating 2,9-purine ring systems (analogues 26 and 27) were found to possess very different activities. Placement of the benzonitrile moiety on the 9-position resulted in an analogue (26) which showed a significant decrease in activity versus WT and all of the mutants. However, the incorporation of the (E)-3-(3,5-dimethylphenyl)acrylonitrile moiety at the 9-position resulted in a highly active analogue (27) with potencies akin to rilpivirine for WT and all of the mutants except Y188L, which caused a modest drop in activity (9.6 ± 2.2 nM).
Selected analogues assayed against additional mutants
Based on the results obtained with the small panel of mutants, eleven compounds were chosen for testing against a broader panel which included all the mutants listed above (K103N, E138K, Y181C and Y188L), and included rilpivirine (1) as a reference compound. The analogues 6–9, 11, 13, 20, 21, 26 and 27 were selected based on their broad potency against the smaller panel of mutants and/or to determine how their differences from 1 affected their activities. The K103N/Y181C double mutant caused a significant loss of potency for 6, 8, 9, 20, 21, 26 and 27 (Figure 4 and Additional file 1: Table S1), while the analogues 7, 11 and 13 retained good potency against this mutant. Of particular interest, the related analogues 7 and 9 differed dramatically in their ability to inhibit K103N/Y181C RT (EC50 values of 2.2 nM and 47 nM, respectively). These compounds both possess a central 5-aminopyrimidine moiety that is ‘flipped’ between the two analogues. Keeping the pyrimidine ring in the same conformation as in 1 allows 7 to retain activity while 9 suffers ~67-fold loss of potency against the double mutant. A structural explanation for this difference will be discussed in a later section.
Figure 4.

Comparison of EC50 values of selected analogs against an expanded set of mutants. Values for mutants not shown in Figure 3 are included in Additional file 1: Table S1.
Cytotoxicity
In general, the eleven analogues tested against the expanded mutant panel showed lower cytotoxicity compared to the other analogues (Figure 3). Only 20 had a lower CC50 than 1 (2.0 μM for 20 versus 10 μM for 1). CC50 values for analogues 6, 9, 13 and 26 were 1- to 3-fold greater than that of 1. The remaining compounds (7, 8, 11, 21 and 27) had CC50 values greater than 38 μM (up to 115 μM for 11). The resulting therapeutic indices (T.I., ratio of CC50 to EC50) ranged from 4.7x103 (26) to 1.2x106 (11). By comparison, rilpivirine (1) has a T.I. of 2.5x104; so, based on the data obtained in cultured cells, many of these analogues compare favourably to this FDA-approved NNRTI.
Crystal structure of 16 bound to WT RT
The crystal structure of the RT/1 complex has been reported [11]. This structure shows that the (E)-3-(3,5-dimethylphenyl)acrylonitrile moiety occupied an induced hydrophobic tunnel formed between residues Y188, F227, W229 and L234. This moiety is retained in all analogues presented here. To examine the effects of modifications to other regions of the compound, a crystal structure of 16 bound to WT RT was solved at 2.3 Å resolution (Table 1).
Table 1.
X-ray data and refinement statistics
| HIV-1 WT RT/16 | K103N/Y181C mutant HIV-1 RT/21 | |
|---|---|---|
| Protein Data Bank (PDB) accession code |
4I2P |
4I2Q |
| Space group |
C2 |
C2 |
| Cell constants (a, b, c in Å; β in º) |
162.65, 72.99, 109.50; 100.59 |
162.60, 72.95, 108.54; 100.70 |
| Resolution range (Å) |
50.00 – 2.30 |
50.00 – 2.70 |
| Completeness (%) |
98.8 |
99.2 |
| Rmerge |
0.085 |
0.084 |
| Average I/σI |
15.0 |
16.5 |
| Sigma cut-off (I) |
|I| < −3.0σ |
|I| < −3.0σ |
|
Refinement Statistics | ||
| Total no. of atoms (solvent atoms) |
8,258 (309) |
8,094 (146) |
| Resolution (Å) |
2.30 |
2.70 |
| No. of reflections |
55,924 |
34,219 |
| Rwork |
0.212 |
0.215 |
| Rfree |
0.260 |
0.286 |
| Ramachandran statistics (% of residues in favored/disallowed regions) |
99.48/0.52 |
99.90/0.10 |
| RMSD bond length (Å) |
0.009 |
0.009 |
| RMSD bond angles (º) | 1.120 | 1.258 |
Overall, the two compounds bind WT RT very similarly (Figure 5). The (E)-3-(3,5-dimethylphenyl)acrylonitrile moiety occupies the hydrophobic tunnel as seen with 1. In addition, the H-bonds between the central group (pyrimidine in 1, purine in 16) and the backbone amide and carbonyl groups of K101 are maintained. The ring of the 4-anisole group of 16 overlays with the ring of the 4-benzonitrile group of 1. The most pronounced difference is between the bent nature of the methoxy functionality of 4-anisole (16) and the linear nature of the cyano functionality of 4-benzonitrile (1). This difference appears to have only a modest impact on the activities of the two compounds against WT RT (Figure 3).
Figure 5.

Overlay of crystal structures and models of 1 and 16 bound to RT. Co-crystal structures of WT RT bound to 1 (gray) and 16 (green) show only minimal differences in the position of the compound and the conformation of the binding pocket. This is consistent with the similar activity of the two compounds against WT RT (0.2nM for 1 and 0.4nM for 16). Models were generated for each compound bound to Y188L RT (1: magenta, 16:cyan). Both models show the compound positioned approximately one angstrom further into the binding pocket than in the respective WT co-crystal structure. This results in a repositioning of the F227 and W229 side chains and a shift in the overall positioning of the β12-β13 hairpin (shown as a ribbon). These differences in the modeled interactions are consistent with the observed difference in antiviral activities against vectors using Y188L RT (2.3nM for 1 and 29nM for 16).
Starting with the crystal structures of these two compounds bound to WT RT, models were generated of the complexes with Y188L RT. In both models, the compounds are shifted approximately one angstrom relative to their positions in the respective WT RT crystal structures. The linear nature of the cyano functionality of the 4-benzonitrile moiety in 1 allows it to extend past the side chain of F227 with only about 30º rotation around the Cβ-Cγ bond. The 4-anisole’s methoxy group (16) would cause a greater steric clash with the F227 side chain as it is positioned in the WT RT than would the 4-benzonitrile (1). As a result, a shift of the backbone of the β12-β13 hairpin is observed in the Y188L/16 model relative to its position in the WT/16 crystal structure (Figure 5). The F227 Cα is displaced by 1.2 Å and both the Cβ and Cγ atoms are displaced by 1.3 Å. This displacement is propagated throughout the loop including W229; the Cα atom of this residue is displaced by 2.1 Å. The repositioning of this hairpin provides a possible explanation for the observed loss of potency for 16 against Y188L RT.
Crystal structure of 21 bound to K103N/Y181C RT
A structural study comparing rilpivirine binding to WT and mutant RTs revealed that the drug is able to retain its inhibitory activity by adapting its binding mode (wiggling and jiggling) to accommodate changes in the landscape of the binding pocket caused by NNRTI resistance mutations [11]. This hypothesis was tested by analyzing the structures of several mutant RTs with rilpivirine (1) bound and by comparing these binding modes to the binding modes of other NNRTIs, such as tert-butyldimethylsilyl-spiroaminooxathioledioxide (TSAO) [22]. To better understand why one of the rilpivirine analogues was not highly effective at inhibiting mutant RTs, we determined the crystal structure of the K103N/Y181C double-mutant RT in complex with 21 at 2.7 Å resolution (Table 1).
The double mutant K103N/Y181C RT confers ~8-fold resistance to riplivirine (1) and >1000-fold resistance to 21. The overall binding mode of 21 (Figure 6) to the mutant RT is similar to and superimposes well with that of rilpivirine (1) [11]. The (E)-3-(3,5-dimethylphenyl)acrylonitrile moiety of 21, common to both agents, occupies the hydrophobic core of the pocket and interacts with the aromatic side chains of Y188 and W229. Analogue 21 has a bulkier benzothiazole moiety substituted for the benzonitrile group of rilpivirine (1). As a result, the benzothiazole of 21 extends nearly 2 Å towards F227 and moved away from P236 compared to the nitrile group of 1. The benzothiazole group of 21 has extended interactions with the side chains of V106, F227, and Y318. The side chain of F227 is reoriented to accommodate the benzothiazole group of 21, has moved towards P225, and undergoes a ~40º rotation about the Cβ-Cγ bond relative to the RT-rilpivirine structure. Consequently, the flexible 222–226 loop is repositioned; however, the cis-peptide link between P225-P226 observed in the rilpivirine co-crystal is also present in the 21 co-crystal. The benzothiazole moiety of 21 does not have a significant impact on positioning of the β12-β13-β14 sheet, unlike that seen when TSAO or delavirdine binds to RT [22,23].
Figure 6.
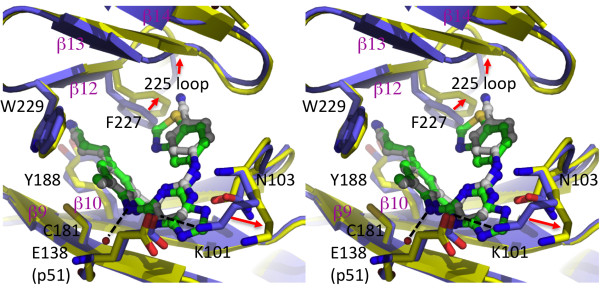
NNRTI-binding pocket superposition of the crystal structures of K103N-Y181C mutant RT/21 (yellow protein and green ligand) and K103N/Y181C mutant RT/rilpivirine (PDB ID. 3BGR; blue protein and gray ligand). The hydrogen bonds are represented as dotted lines, and the most significant structural differences are indicated by red arrows.
In 21, the central pyrimidine ring of rilpivirine (1) is substituted by a purine that extends toward the side chains of K101 (p66) and E138 (p51) at the putative entrance to the inhibitor binding pocket. Both of these side-chains are involved with the entry/exit of NNRTIs into/out of the pocket, and a hydrogen bond exists between the two in most RT/NNRTI structures. In the double mutant K101E/E138K, the H-bond is retained, but these mutations cause a significant loss of potency to several preclinical TSAO-derived NNRTIs [22]. This suggests that the presence of specific amino acids at positions 101 and 138 may affect the binding of other NNRTIs. The positioning of the purine moiety of 21 causes significant rearrangements of the K101 and E138 side chains, which breaks the H-bond between these two residues and forms extensive hydrophobic contacts with methylene groups of both side chains. Interestingly, the N7 atom of the purine ring forms an H-bond with the backbone amide of K101 analogous to that seen with the pyrimidine ring of rilpivirine. The purine moiety also has extensive interactions with L100, the mutated K103N and V106. The mutated K103N side chain has more hydrophobic stacking interactions with the purine moiety of 21 than with the pyrimidine of rilpivirine (1). Our structure indicates that the benzothiazole moiety maintains inhibitor-protein interactions without requiring significant rearrangement of the pocket, apart from reorienting the side chains of K101, E138 (p51), and F227 (Figure 6). Also, the disruption of the K101-E138 H-bond may affect the dissociation of 21 in a manner that is not observable in the co-crystal. The positional constraints on the amino acid residues that are parts of the hydrophobic tunnel and the entrance to the pocket may explain why 21 is less susceptible to pocket mutations.
Modeling other RTs and compounds
Several of the analogues showed significantly different activities against a particular RT mutant despite having only relatively modest structural changes relative to the parent compound. For example, compounds 7 and 9 differ only in the orientation of the central 5-aminopyrimidine moiety. This subtle change results in an EC50 for 9 that is ~21-fold greater than that of 7 against the K103N/Y181C mutant. Models of both of these complexes were generated from the crystal structure of 1 bound to this mutant [11].
The models show no significant changes to either the β12-β13-β14 sheet or the K101-E138 salt bridge. However, the solvent-exposed surface of the pyrimidine is shifted nearly an angstrom when the two models are compared to each other. This positions the exocyclic amine of 9 such that it cannot form the H-bond to the K101 backbone amide seen with 1 (Figure 7). In contrast, this H-bonding is maintained with 7. These models also differ in the orientation of a water molecule forming a bridge to the backbone carbonyl of E138. The exocyclic amine and aryl amine of 7 serve as donors to bind the same oxygen atom of a water molecule. These interactions position the water differently than it appears in the model with 9, where the pyrimidine’s N3 serves as an acceptor and the aryl amine as a donor to bind the same water molecule. The orientation of that water molecule could result in a weaker interaction with the E138 backbone. This subtle change in the compound, and the corresponding loss of activity against the K103N/Y181C mutant, highlights two potential interactions that need to be considered in the design of additional derivatives.
Figure 7.

Models of analogues 7 (cyan) and 9 (magenta) bound to K103N/Y181C RT. (A) Overlay of the two compounds as modeled in the binding pocket of K103N/Y181C HIV-1 RT. (B) The exocyclic amine in 9 cannot H-bond with the backbone NH of K101, as seen in crystal structures of 1. The N3 and linker NH both bind a water molecule that forms a bridge to the backbone carbonyl of E138. (C) The H-bonds with the backbone of K101 are restored in 7. In addition, the exocyclic amine and linker NH both H-bond a water molecule in a different orientation than is seen with 9.
Intramolecular contacts also need to be considered. Analogues 6 and 8 have 5-nitropyrimidine central moieties in the ‘flipped’ orientations. These nitro groups appear capable of forming intramolecular H-bonds with the aryl amines bound to the 6-position of the pyrimidine ring. These interactions may restrict the flexibility of these analogues. This observation is consistent with the similar profiles of 8 and 26 and those of 6 and 27. The potential intramolecular H-bonds may mimic the covalent restriction imposed by the purine ring systems of 26 and 27.
Conclusions
In summary, we have designed and synthesized several small molecules that are structurally related to the recently approved NNRTI rilpivirine (1) and tested them for their ability to inhibit the replication of viral vectors that carry WT RT or several clinically relevant mutant RTs. This data set provides insights into structure-activity relationships of this family of compounds. One agent, a purine analogue with a benzo[d]thiazole appendage at the 6-position (analogue 21), possessed good activity versus all single mutants tested, but showed little activity against the K103N/Y181C double mutant. To understand the molecular basis of this loss of potency, we determined the crystal structure of the double-mutant K103N/Y181C RT in complex with 21. This structure provided insights into how this analogue failed to maintain potency versus specific RT mutants. Molecular modeling also proved useful in assessing how other analogues interact with RT.
Three analogues were identified (7, 11 and 13) that show nearly as broad potency and cytotoxicity comparable to, or more favourable than, that of rilpivirine (1). These three compounds showed some loss of potency (four- to fourteen-fold) against E138K and 11 lost some potency against Y188L (nearly six-fold). It is noted that rilpivirine is comparably active against vectors harbouring either WT or E138K RT in this assay despite the frequent selection of this mutant in clinical trials [13,24]. These trials used treatment regimens that included two NRTIs (TDF/FTC in the ECHO trial and TDF/FTC, zidovudine/lamivudine or abacavir/lamivudine in the THRIVE trial). E138K has been shown to compensate for the loss of viral fitness associated with certain NRTI resistance mutations, particularly M184I or M184V [15]. It is possible that the frequent selection of E138K is driven more by a need to restore fitness to NRTI-resistant RTs than by a significant affect on rilpivirine resistance, consistent with a higher rate of NRTI-resistant mutants selected in the rilpivirine group than in the efavirenz group in the clinical trials [13]. Controlling HIV infection must be done with combinations of drugs that work cooperatively against known resistant mutants. If any of these three analogues select for a different set of mutants than does rilpivirine, they may be useful in patients experiencing virologic failure on a rilpivirine regimen. Such selection experiments, and the synthesis and testing of additional analogues, represent future steps in this continuing effort.
Methods
Synthesis
All compounds shown in Figure 2 were synthesized and assayed for HIV inhibitory activity and cytotoxicity. Synthetic protocols and representative 1H NMR chemical shift data are included as Additional file 1. Briefly, aryl amines were coupled to a PMB-protected 2,6 dichloro-9H-purine starting reagent at the 6-position, followed by addition at the 2-position using a modified Buchwald-Hartwig amination to yield analogues 10-25[25,26]. Analogues 26 and 27 were synthesized by modifying a synthetic protocol described in the patent literature [27].
Virion production
Procedures for virion production and cell-based assay have been reported [28]. Briefly, all cell cultures were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) with 5% (v/v) fetal bovine serum, 5% newborn calf serum, penicillin (50 units/mL) and streptomycin (50 μg/mL) (Quality Biological, Gaithersburg, MD). VSV-g pseudotyped virions were produced by plating 9x105 293 T cells in a 100 mm plate, then transfecting with the pNLNgoMIVR+ΔEnv.LUC (10 μg) and pHCMV-g (3 μg) plasmids by calcium phosphate precipitation on the following day. Transfected cells were incubated at 37°C for 8 hrs, washed twice with PBS, fresh media was added and the cells were returned to the incubator for 48 hrs. The pNLNgoMIVR+ΔEnv.LUC plasmid encodes the HIV-1 genome with an Env deletion and either WT HIV-1 RT, or one of the following mutants: L100I, K103N, V106A, E138K, Y181C, Y188L, H221Y, or K103N/Y181C. Supernatants from each plate were clarified by low-speed centrifugation (3,000xg for 20 min), then treated with 200 U DNase I (Roche Applied Science, Mannheim, Germany) at room temperature for 3 hours. Viral supernatants were stored at −20°C until used.
Infectivity assays
One day prior to infection, 100 μL of human osteosarcoma (HOS) cells were added to each well of a 96-well plate at a density of 4x104 cells/mL. Plates were incubated overnight at 37°C. On the day of infection, 10X drug stocks were prepared by serial dilution in media from an initial 10 mM stock solution in DMSO. Cells in the 96-well plate were pre-treated by the addition of 22 μL of the appropriate drug stock and returned to the incubator for 3 hours. Thawed virus stocks were diluted 1:2 in media, then 100 μL of virus was added to the pre-treated cells, bringing the final drug concentrations to 0.1 nM to 100 nM, with DMSO-treated cells (no drug) serving as a reference. In addition, 12 wells of cells treated with media and no virus served as negative controls. Plates were returned to the incubator for an additional 42 hours, at which time the luciferase activity was determined using Steady-Lite Plus kits (PerkinElmer, Waltham, MA). Background luciferase activity, as measured for the negative controls, was subtracted from the activities measured for each infected well. Drug-free, DMSO-treated wells were normalized to 100% infectivity. Infectivity at each drug concentration was determined as a percentage of these drug-free infections. Drug concentration versus percent infectivity was plotted for each RT-inhibitor combination, and the data were fit to a four-parameter sigmoidal curve using KaleidaGraph 4.0 (Synergy Software, Reading, PA). The inflection point of each curve is reported as the IC50. All values are the mean of triplicate infections.
Cytotoxicity assays
One day prior to treatment, HOS cells were prepared in 96-well plates as described for the infectivity assays, except that 12 wells per plate were treated with cell-free media to serve as negative controls. The following day, serial dilutions of drug stocks were prepared in media from 10 mM stocks in DMSO. Each well received 11 μL of the appropriate drug concentration or DMSO-treated, drug-free media. Final drug concentrations ranged from 0.5 μM to 300 μM. The drug-free wells served as references. Plates were then incubated at 37°C for 48 hours. Cell viability was determined using luciferase reporter ATP-Lite kits (PerkinElmer). As described for the infectivity assays, drug-free reference wells were normalized to 100%, and viabilities at different drug concentrations were determined as a percentage of these references. Data were plotted and analyzed in the same manner as the IC50 data, with the inflection points of the curves reported as CC50.
Crystallography
RT was expressed and purified as described previously [29]. RT (WT and mutant) was co-crystallized with 16 and 21 at 4°C by vapor diffusion in micro-seeded hanging drops containing 1.2 μL each of 20 mg/mL protein (in a solution of 10 mM Tris pH 8.0, 75 mM NaCl, 0.30% (w/v) β-octyl glucopyranoside, 2% (v/v) DMSO, and 0.5 mM 16 or 21 pre-incubated for 10 min at 25°C) and a reservoir solution containing 50 mM imidazole pH 6.6, 100 mM ammonium sulfate, 15 mM manganese sulfate, 10 mM spermine, 5 mM TCEP, 11% (w/w) PEG 8000, and 5% PEG 400. The chosen crystals were soaked for ~20 s in a solution containing 50 mM imidazole pH 6.6, 50 mM ammonium sulfate, 15 mM manganese sulfate, 10 mM spermine, 12% (w/w) PEG 8000, 6% (w/w) PEG 400, and 26% (v/v) ethylene glycol. The crystal was subsequently flash-cooled and stored in liquid N2. Data collection was performed at the Cornell High Energy Synchrotron Source (CHESS) F1 beamline. The diffraction data were indexed, processed, scaled and merged using HKL2000I[30]. The structure was refined using Phenix [31] and model building was done using COOT [32]. The crystallographic data and refinement statistics are listed in Table 1.
Computer modeling
All computer modeling was done using MOE 2009.10 or MOE2011.10 (Chemical Computing Group, Montreal, Quebec, Canada). The model of 1 bound to Y188L RT was based on the previously reported crystal structure of the WT RT/1 complex (PDB ID: 2ZD1 [11]). Models of complexes with 16 and 21 were based on the crystal structures presented in the current work (PDB IDs 4I2P and 4I2Q, respectively). Models of 7 and 9 bound to K103N/Y181C RT were based on the K103N/Y181C RT/1 crystal structure (PDB ID: 3BGR [11]).
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BJ performed the infectivity and cytotoxicity assays and carried out the molecular modeling. GP, GR, HB, JS, DM and CT contributed to the synthesis of compounds. DP, JB, KD and EA performed the crystallographic studies. EA, CT and SH designed the experiments. BJ, KD, DM, EA, CT and SH drafted the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Synthetic schemes used to synthesize purine analogues of rilpivirine. Scheme 1 was used to synthesize analogues 10-25 while Scheme 2 was used to synthesize analogues 26 and 27.
Contributor Information
Barry C Johnson, Email: barryj2@mail.nih.gov.
Gary T Pauly, Email: paulyg@mail.nih.gov.
Ganesha Rai, Email: bantukallug@mail.nih.gov.
Disha Patel, Email: pateld06@eden.rutgers.edu.
Joseph D Bauman, Email: bauman@cabm.rutgers.edu.
Heather L Baker, Email: bheather@mail.nih.gov.
Kalyan Das, Email: kalan@cabm.rutgers.edu.
Joel P Schneider, Email: schneiderjp@mail.nih.gov.
David J Maloney, Email: maloneyd@mail.nih.gov.
Eddy Arnold, Email: arnold@cabm.rutgers.edu.
Craig J Thomas, Email: craigt@mail.nih.gov.
Stephen H Hughes, Email: hughesst@mail.nih.gov.
Acknowledgements
This research was supported by the Intramural Research Programs of the National Cancer Institute, the National Human Genome Research Institute, the National Center for Advancing Translational Sciences, the Intramural AIDS Targeted Antiviral Program (IATAP), and an R37 Merit Award AI 27690 to E.A. We acknowledge the Cornell High Energy Synchrotron Source (CHESS) for X-ray diffraction data collection.
References
- HIV/AIDS. http://www.who.int/topics/hiv_aids/en/
- Hartman TL, Buckheit RW Jr. The continuing evolution of HIV-1 therapy: identification and development of novel antiretroviral agents targeting viral and cellular targets. Mol Biol Int. 2012;2012:401965. doi: 10.1155/2012/401965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Zhan P, De Clercq E, Liu X. Strategies for the design of HIV-1 non-nucleoside reverse transcriptase inhibitors: lessons from the development of seven representative paradigms. J Med Chem. 2012;55:3595–3613. doi: 10.1021/jm200990c. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Bollini M, Thakur VV, Domaoal RA, Spasov KA, Anderson KS. Efficient discovery of potent anti-HIV agents targeting the Tyr181Cys variant of HIV reverse transcriptase. J Am Chem Soc. 2011;133:15686–15696. doi: 10.1021/ja2058583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Chamberlain PP, Stamp A, Short SA, Weaver KL, Romines KR, Hazen R, Freeman A, Ferris RG, Andrews CW. et al. Structural basis for the improved drug resistance profile of new generation benzophenone non-nucleoside HIV-1 reverse transcriptase inhibitors. J Med Chem. 2008;51:5000–5008. doi: 10.1021/jm8004493. [DOI] [PubMed] [Google Scholar]
- Su DS, Lim JJ, Tinney E, Wan BL, Young MB, Anderson KD, Rudd D, Munshi V, Bahnck C, Felock PJ. et al. Biaryl ethers as novel non-nucleoside reverse transcriptase inhibitors with improved potency against key mutant viruses. J Med Chem. 2009;52:7163–7169. doi: 10.1021/jm901230r. [DOI] [PubMed] [Google Scholar]
- Sweeney ZK, Harris SF, Arora SF, Javanbakht H, Li Y, Fretland J, Davidson JP, Billedeau JR, Gleason SK, Hirschfeld D. et al. Design of annulated pyrazoles as inhibitors of HIV-1 reverse transcriptase. J Med Chem. 2008;51:7449–7458. doi: 10.1021/jm800527x. [DOI] [PubMed] [Google Scholar]
- Adams J, Patel N, Mankaryous N, Tadros M, Miller CD. Nonnucleoside reverse transcriptase inhibitor resistance and the role of the second-generation agents. Ann Pharmacother. 2010;44:157–165. doi: 10.1345/aph.1M359. [DOI] [PubMed] [Google Scholar]
- De Corte BL. From 4,5,6,7-tetrahydro-5-methylimidazo[4,5,1-jk](1,4)benzodiazepin-2(1H)-one (TIBO) to etravirine (TMC125): fifteen years of research on non-nucleoside inhibitors of HIV-1 reverse transcriptase. J Med Chem. 2005;48:1689–1696. doi: 10.1021/jm040127p. [DOI] [PubMed] [Google Scholar]
- Das K, Clark AD Jr, Lewi PJ, Heeres J, De Jonge MR, Koymans LM, Vinkers HM, Daeyaert F, Ludovici DW, Kukla MJ. et al. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J Med Chem. 2004;47:2550–2560. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- Das K, Bauman JD, Clark AD Jr, Frenkel YV, Lewi PJ, Shatkin AJ, Hughes SH, Arnold E. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistance mutations. Proc Natl Acad Sci USA. 2008;105:1466–1471. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azijn H, Tirry I, Vingerhoets J, de Bethune MP, Kraus G, Boven K, Jochmans D, Van Craenenbroeck E, Picchio G, Rimsky LT. TMC278, a next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI), active against wild-type and NNRTI-resistant HIV-1. Antimicrob Agents Chemother. 2010;54:718–727. doi: 10.1128/AAC.00986-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimsky L, Vingerhoets J, Van Eygen V, Eron J, Clotet B, Hoogstoel A, Boven K, Picchio G. Genotypic and phenotypic characterization of HIV-1 isolates obtained from patients on rilpivirine therapy experiencing virologic failure in the phase 3 ECHO and THRIVE studies: 48-week analysis. J Acquir Immune Defic Syndr. 2012;59:39–46. doi: 10.1097/QAI.0b013e31823df4da. [DOI] [PubMed] [Google Scholar]
- Cohen CJ, Andrade-Villanueva J, Clotet B, Fourie J, Johnson MA, Ruxrungtham K, Wu H, Zorrilla C, Crauwels H, Rimsky LT. et al. Rilpivirine versus efavirenz with two background nucleoside or nucleotide reverse transcriptase inhibitors in treatment-naive adults infected with HIV-1 (THRIVE): a phase 3, randomised, non-inferiority trial. Lancet. 2011;378:229–237. doi: 10.1016/S0140-6736(11)60983-5. [DOI] [PubMed] [Google Scholar]
- Xu HT, Asahchop EL, Oliveira M, Quashie PK, Quan Y, Brenner BG, Wainberg MA. Compensation by the E138K mutation in HIV-1 reverse transcriptase for deficits in viral replication capacity and enzyme processivity associated with the M184I/V mutations. J Virol. 2011;85:11300–11308. doi: 10.1128/JVI.05584-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzarin A, Campbell T, Clotet B, Johnson M, Katlama C, Moll A, Towner W, Trottier B, Peeters M, Vingerhoets J. et al. Efficacy and safety of TMC125 (etravirine) in treatment-experienced HIV-1-infected patients in DUET-2: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet. 2007;370:39–48. doi: 10.1016/S0140-6736(07)61048-4. [DOI] [PubMed] [Google Scholar]
- Chen X, Zhan P, Li D, De Clercq E, Liu X. Recent advances in DAPYs and related analogues as HIV-1 NNRTIs. Curr Med Chem. 2011;18:359–376. doi: 10.2174/092986711794839142. [DOI] [PubMed] [Google Scholar]
- Guillemont J, Pasquier E, Palandjian P, Vernier D, Gaurrand S, Lewi PJ, Heeres J, de Jonge MR, Koymans LM, Daeyaert FF. et al. Synthesis of novel diarylpyrimidine analogues and their antiviral activity against human immunodeficiency virus type 1. J Med Chem. 2005;48:2072–2079. doi: 10.1021/jm040838n. [DOI] [PubMed] [Google Scholar]
- Janssen PA, Lewi PJ, Arnold E, Daeyaert F, de Jonge M, Heeres J, Koymans L, Vinkers M, Guillemont J, Pasquier E. et al. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2- pyrimidinyl]amino]benzonitrile (R278474, rilpivirine) J Med Chem. 2005;48:1901–1909. doi: 10.1021/jm040840e. [DOI] [PubMed] [Google Scholar]
- Liang YH, Feng XQ, Zeng ZS, Chen FE, Balzarini J, Pannecouque C, De Clercq E. Design, synthesis, and SAR of naphthyl-substituted Diarylpyrimidines as non-nucleoside inhibitors of HIV-1 reverse transcriptase. ChemMedChem. 2009;4:1537–1545. doi: 10.1002/cmdc.200900212. [DOI] [PubMed] [Google Scholar]
- Sun LQ, Zhu L, Qian K, Qin B, Huang L, Chen CH, Lee KH, Xie L. Design, synthesis, and preclinical evaluations of novel 4-substituted 1,5-diarylanilines as potent HIV-1 Non-nucleoside reverse transcriptase inhibitor (NNRTI) drug candidates. J Med Chem. 2012;55:7219–7229. doi: 10.1021/jm3007678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K, Bauman JD, Rim AS, Dharia C, Clark AD Jr, Camarasa MJ, Balzarini J, Arnold E. Crystal structure of tert-butyldimethylsilyl-spiroaminooxathioledioxide-thymine (TSAO-T) in complex with HIV-1 reverse transcriptase (RT) redefines the elastic limits of the non-nucleoside inhibitor-binding pocket. J Med Chem. 2011;54:2727–2737. doi: 10.1021/jm101536x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnouf RM, Ren J, Hopkins AL, Ross CK, Jones EY, Stammers DK, Stuart DI. Unique features in the structure of the complex between HIV-1 reverse transcriptase and the bis(heteroaryl)piperazine (BHAP) U-90152 explain resistance mutations for this nonnucleoside inhibitor. Proc Natl Acad Sci USA. 1997;94:3984–3989. doi: 10.1073/pnas.94.8.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen CJ, Molina JM, Cahn P, Clotet B, Fourie J, Grinsztejn B, Wu H, Johnson MA, Saag M, Supparatpinyo K. et al. Efficacy and safety of rilpivirine (TMC278) versus efavirenz at 48 weeks in treatment-naive HIV-1-infected patients: pooled results from the phase 3 double-blind randomized ECHO and THRIVE Trials. J Acquir Immune Defic Syndr. 2012;60:33–42. doi: 10.1097/QAI.0b013e31824d006e. [DOI] [PubMed] [Google Scholar]
- Kelley JL, Linn JA, Krochmal MP, Selway JW. 9-Benzyl-6-(dimethylamino)-9H-purines with antirhinovirus activity. J Med Chem. 1988;31:2001–2004. doi: 10.1021/jm00118a025. [DOI] [PubMed] [Google Scholar]
- Wolfe JP, Buchwald SL. Scope and limitations of the Pd/BINAP-catalyzed amination of aryl bromides. J Org Chem. 2000;65:1144–1157. doi: 10.1021/jo9916986. [DOI] [PubMed] [Google Scholar]
- Guillemont JEG, Paugam M, Delest BFM, Heeres J, Lewi PJ, Janssen PAJ, Arts FJJH. HIV inhibiting bicyclic pyrimidine derivatives. USA: Office USPaT ed; 2008. [Google Scholar]
- Vu BC, Boyer PL, Siddiqui MA, Marquez VE, Hughes SH. 4′-C-methyl-2′-deoxyadenosine and 4′-C-ethyl-2′-deoxyadenosine inhibit HIV-1 replication. Antimicrob Agents Chemother. 2011;55:2379–2389. doi: 10.1128/AAC.01290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman JD, Das K, Ho WC, Baweja M, Himmel DM, Clark AD Jr, Oren DA, Boyer PL, Hughes SH, Shatkin AJ, Arnold E. Crystal engineering of HIV-1 reverse transcriptase for structure-based drug design. Nucleic Acids Res. 2008;36:5083–5092. doi: 10.1093/nar/gkn464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Crystallography of biological macromolecules. Boston: Kluwer Academic Publishers; 2001. [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synthetic schemes used to synthesize purine analogues of rilpivirine. Scheme 1 was used to synthesize analogues 10-25 while Scheme 2 was used to synthesize analogues 26 and 27.