

Abstract
Error correction of sequenced reads remains a difficult task, especially in single-cell sequencing projects with extremely non-uniform coverage. While existing error correction tools designed for standard (multi-cell) sequencing data usually come up short in single-cell sequencing projects, algorithms actually used for single-cell error correction have been so far very simplistic.
We introduce several novel algorithms based on Hamming graphs and Bayesian subclustering in our new error correction tool BAYESHAMMER. While BAYESHAMMER was designed for single-cell sequencing, we demonstrate that it also improves on existing error correction tools for multi-cell sequencing data while working much faster on real-life datasets. We benchmark BAYESHAMMER on both k-mer counts and actual assembly results with the SPADES genome assembler.
Background
Single-cell sequencing [1,2] based on the Multiple Displacement Amplification (MDA) technology [1,3] allows one to sequence genomes of important uncultivated bacteria that until recently had been viewed as unamenable to genome sequencing. Existing metagenomic approaches (aimed at genes rather than genomes) are clearly limited for studies of such bacteria despite the fact that they represent the majority of species in such important studies as the Human Microbiome Project [4,5] or discovery of new antibiotics-producing bacteria [6].
Single-cell sequencing datasets have extremely non-uniform coverage that may vary from ones to thousands along a single genome (Figure 1). For many existing error correction tools, most notably QUAKE [7], uniform coverage is a prerequisite: in the case of non-uniform coverage they either do not work or produce poor results.
Figure 1.
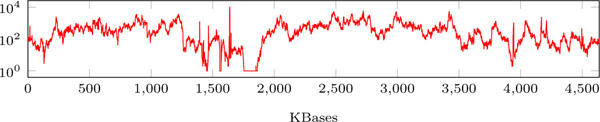
Logarithmic coverage plot for the single-cell E. coli dataset. Logarithmic coverage plot for the single-cell E. coli dataset (similar plot is also given in [2]).
Error correction tools usually attempt to correct the set of k-character substrings of reads called k-mers and then propagate corrections to whole reads which are important to have for many assemblers. Error correction tools often employ a simple idea of discarding rare k-mers, which obviously does not work in the case of non-uniform coverage.
Medvedev et al. [8] recently presented a new approach to error correction for datasets with non-uniform coverage. Their algorithm HAMMER makes use of the Hamming graph (hence the name) on k-mers (vertices of the graph correspond to k-mers and edges connect pairs of k-mers with Hamming distance not exceeding a certain threshold). HAMMER employs a simple and fast clustering technique based on selecting a central k-mer in each connected component of the Hamming graph. Such central k-mers are assumed to be error-free (i.e., they are assumed to actually appear in the genome), while the other k-mers from connected components are assumed to be erroneous instances of the corresponding central k-mers. However, HAMMER may be overly simplistic: in connected components of large diameter or connected components with several k-mers of large multiplicities, it is more reasonable to assume that there are two or more central k-mers (rather than one as in HAMMER). Biologically, such connected components may correspond to either (1) repeated regions with similar but not identical genomic sequences (repeats) which would be bundled together by existing error correction tools (including HAMMER); or (2) artificially united k-mers from distinct parts of the genome that just happen to be connected by a path in the Hamming graph (characteristic to HAMMER).
In this paper, we introduce the BAYESHAMMER error correction tool that does not rely on uniform coverage. BAYESHAMMER uses the clustering algorithm of HAMMER as a first step and then refines the constructed clusters by further subclustering them with a procedure that takes into account reads quality values (e.g., provided by Illumina sequencing machines) and introduces Bayesian (BIC) penalties for extra subclustering parameters. BAYESHAMMER subclustering aims to capture the complex structure of repeats (possibly of varying coverage) in the genome by separating even very similar k-mers that come from different instances of a repeat. BAYESHAMMER also uses a new approach for propagating corrections in k-mers to corrections in the reads. All algorithms in BAYESHAMMER are heavily parallelized whenever possible; as a result, BAYESHAMMER gains a significant speedup with more processing cores available. These features make BAYESHAMMER a perfect error correction tool for single-cell sequencing.
We remark that HAMMER produces only a set of central k-mers but does not correct reads, making it incompatible with most genome assemblers. QUAKE does correct reads but has severe memory limitations for large k and assumes uniform coverage. In contrast, EULER-SR [9] and CAMEL [2] correct reads and do not make strong assumptions on coverage (both tools have been used for single-cell assembly projects [2]) which makes these tools suitable for comparison to BAYESHAMMER. Our benchmarks show that BAYESHAMMER outperforms these tools in both single-cell and standard (multi-cell) modes. We further couple BAYESHAMMER with a recently developed genome assembler SPADES [10] and demonstrate that assembly of BAYESHAMMER-corrected reads significantly improves upon assembly with reads corrected by other tools for the same datasets, while the total running time also improves significantly.
BAYESHAMMER is freely available for download as part of the SPADES genome assembler at http://bioinf.spbau.ru/spades/.
Methods
Notation and outline
Let ∑ = {A, C, G, T} be the alphabet of nucleotides (BAYESHAMMER discards k-mers with uncertain bases denoted N). A k-mer is an element of ∑k, i.e., a string of k nucleotides. We denote the ith letter (nucleotide) of a k-mer x by x[i], indexing them from zero: 0 ≤ i ≤ k - 1. A subsequence of x corresponding to a set of indices I is denoted by x[I]. We use interval notation [i, j] for intervals of integers {i, i + 1,..., j} and further abbreviate x[i, j] = x [{i, i + 1,..., j}]; thus, x = x[0, k - 1]. Input reads are represented as a set of strings R ⊂ Σ* along with their quality values for each r ∈ R. We assume that qr[i] estimates the probability that there has been an error in position i of read r. Notice that in practice, the fastq file format [11] contains characters that encode probabilities on a logarithmic scale (in particular, products of probabilities used below correspond to sums of actual quality values).
Below we give an overview of BAYESHAMMER workflow (Figure 2) and refer to subsequent sections for further details. On Step (1), k-mers in the reads are counted, producing a triple statistics(x) = (countx, qualityx, errorx) for each k-mer x. Here, countx is the number of times x appears as a substring in the reads, qualityx is its total quality expressed as a probability of sequencing error in x, and errorx is a k-dimensional vector that contains products of error probabilities (sums of quality values) for individual nucleotides of x across all its occurrences in the reads. On Step (2), we find connected components of the Hamming graph constructed from this set of k-mers. On Step (3), the connected components become subject to Bayesian subclustering; as a result, for each k-mer we know the center of its subcluster. On Step (4), we filter subcluster centers according to their total quality and form a set of solid k-mers which is then iteratively expanded on Step (5) by mapping them back to the reads. Step (6) deals with reads correction by counting the majority vote of solid k-mers in each read. In the iterative version, if there has been a substantial amount of changes in the reads, we run the next iteration of error correction; otherwise, output the corrected reads. Below we describe specific algorithms employed in the BAYESHAMMER pipeline.
Figure 2.

BAYESHAMMER workflow.
Algorithms
Step (1): computing k-mer statistics
To collect k-mer statistics, we use a straightforward hash map approach [12] that does not require storing instances of all k-mers in memory (as excessive amount of RAM might be needed otherwise). For a certain positive integer N (the number of auxiliary files), we use a hash function h: ∑k →ℤN that maps k-mers over the alphabet Σ to integers from 0 to N - 1.
Algorithm 1 Count k-mers
for each k-mer x from the reads R: do
compute h(x) and write x to Fileh(x).
for i ∈ [0, N - 1]: do
sort Filei with respect to the lexicographic order;
reading Filei sequentially, compute statistics(s) for each k-mer s from Filei.
Step (2): constructing connected components of Hamming graph
Step (2) is the essence of the HAMMER approach [8]. The Hamming distance between k-mers x, y ∈ ∑k is the number of nucleotides in which they differ:
For a set of k-mers X, the Hamming graph HGτ(X) is an undirected graph with the set of vertices X and edges corresponding to pairs of k-mers from X with Hamming distance at most τ, i.e., x, y ∈ X are connected by an edge in HGτ(X) iff d(x, y) ≤ τ (Figure 3). To construct HGτ(X) efficiently, we notice that if two k-mers are at Hamming distance at most τ, and we partition the set of indices [0,k - 1] into τ + 1 parts, then at least one part corresponds to the same subsequence in both k-mers. Below we assume with little loss of generality that τ + 1 divides k, i.e., k = σ (τ + 1) for some integer σ.
Figure 3.
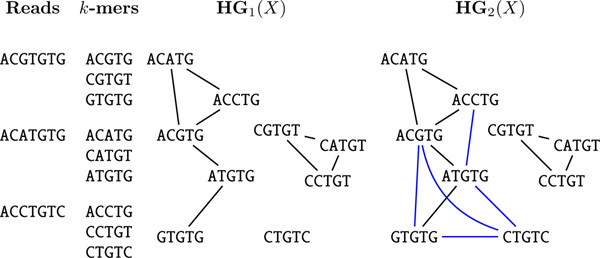
Hamming graphs HG1(X) and HG2(X). Hamming graphs HG1(X) and HG2(X) for X being the set of 4-mers {ACGTG, CGTGT, GTGTG, ACATG, CATGT, ATGTG, ACCTG, CCTGT, CTGTC} of the reads ACGTGTG, ACATGTG, ACCTGTC. Blue edges denote Hamming distance 2.
For a subset of indices I ⊆ [0, k - 1], we define a partial lexicographic ordering ≺I as follows: x ≺I y iff x[I] ≺ y[I], where ≺ is the lexicographic ordering on Σ*. Similarly, we define a partial equality =I such that x =I y iff x[I] = y[I]. We partition the set of indices [0, k - 1] into τ + 1 parts of size σ and for each part I, sort a separate copy of X with respect to ≺I. As noticed above, for every two k-mers x, y ∈ X with d(x, y) ≤ τ, there exists a part I such that x =I y. It therefore suffices to separately consider blocks of equivalent k-mers with respect to =I for each part I. If a block is small (i.e., of size smaller than a certain threshold), we go over the pairs of k-mers in this block to find those with Hamming distance at most τ. If a block is large, we recursively apply to it the same procedure with a different partition of the indices. In practice, we use two different partitions of [0, k - 1]: the first corresponds to contigious subsets of indices (recall that ):
Algorithm 2 Hamming graph processing
procedure HGPROCESS(X, max_quadratic)
Init components with singletons .
for all ϒ ∈ FindBlocksdo
if |ϒ| > max_quadratic then
for all Z ∈ FindBlocks do
ProcessExhaustively
else
ProcessExhaustively.
function FindBlocks
for s = 0,...,τ do
sort a copy of X with respect to , getting Xs.
for s = 0,...,τ do
output the set of equiv. blocks .
procedure PROCESSEXHAUSTIVELY
for each pair x, y ∈ ϒ do
if d(x, y) ≤ τ then join their sets in :
for all do
.
while the second corresponds to strided subsets of indices:
BAYESHAMMER uses a two-step procedure, first splitting with respect to (Figure 4) and then, if an equivalence block is large, with respect to . On the block processing step, we use the disjoint set data structure [12] to maintain the set of connected components. Step (2) is summarized in Algorithm 2.
Figure 4.

Partial lexicographic orderings. Partial lexicographic orderings of a set X of 9-mers with respect to the index sets , , and . Red dotted lines indicate equivalence blocks.
Step (3): Bayesian subclustering
In HAMMER's generative model [8], it is assumed that errors in each position of a k-mer are independent and occur with the same probability ε, which is a fixed global parameter (HAMMER used ε = 0.01). Thus, the likelihood that a k-mer x was generated from a k-mer y under HAMMER's model equals
Under this model, the maximum likelihood center of a cluster is simply its consensus string [8].
In BAYESHAMMER, we further elaborate upon HAMMER's model. Instead of a fixed ε, we use reads quality values that approximate probabilities qx[i] of a nucleotide at position i in the k-mer x being erroneous. We combine quality values from identical k-mers in the reads: for a multiset of k-mers X that agree on the jth nucleotide, it is erroneous with probability Πx∈X qx[j].
The likelihood that a k-mer x has been generated from another k-mer c (under the independent errors assumption) is given by
and the likelihood of a specific subclustering C = C1 ∪... ∪ Cm is
where ci is the center (consensus string) of the subcluster Ci.
In the subclustering procedure (see Algorithm 3), we sequentially subcluster each connected component of the Hamming graph into more and more clusters with the classical k-means clustering algorithm (denoted m-means since k has different meaning). For the objective function, we use the likelihood as above penalized for overfitting with the Bayesian information criterion (BIC) [13]. In this case, there are |C| observations in the dataset, and the total number of parameters is 3 km + m - 1:
• m - 1 for probabilities of subclusters,
• km for cluster centers, and
• 2 km for error probabilities in each letter: there are 3 possible errors for each letter, and the probabilities should sum up to one. Here error probabilities are conditioned on the fact that an error has occurred (alternatively, we could consider the entire distribution, including the correct letter, and get 3 km parameters for probabilities but then there would be no need to specify cluster centers, so the total number is the same).
Algorithm 3 Bayesian subclustering
for all connected components C of the Hamming graph do
m := 1
ℓ1 := 2 log L1(C) (likelihood of the cluster generated by the consensus)
repeat
m := m + 1
do m-means clustering of C = C1 ∪...∪ Cm w.r.t. the Hamming distance; the initial approximation to the centers is given by k-mers that have the least error probability
ℓm := 2 · log Lm(C1,...,Cm) (3 km + m - 1) · log |C|
until ℓm ≤ ℓm-1
output the best found clustering C = C1 ∪...∪ Cm-1
Therefore, the resulting objective function is
for subclustering into m clusters; we stop as soon as ℓm ceases to increase.
Steps (4) and (5): selecting solid k-mers and expanding the set of solid k-mers
We define the quality of a k-mer x as the probability that it is error-free: . The k-mer qualities are computed on Step (1) along with computing k-mer statistics. Next, we (generously) define the quality of a cluster C as the probability that at least one k-mer in C is correct:
In contrast to HAMMER, we do not distinguish whether the cluster is a singleton (i.e., |C| = 1); there may be plenty of superfluous clusters with several k-mers obtained by chance (actually, it is more likely to obtain a cluster of several k-mers by chance than a singleton of the same total multiplicity).
Initially we mark as solid the centers of the clusters whose total quality exceeds a predefined threshold (a global parameter for BAYESHAMMER, set to be rather strict). Then we expand the set of solid k-mers iteratively: if a read is completely covered by solid k-mers we conclude that it actually comes from the genome and mark all other k-mers in this read as solid, too (Algorithm 4).
Step (6): reads correction
After Steps (1)-(5), we have constructed the set of solid k-mers that are presumably error-free. To construct corrected reads from the set of solid k-mers, for each base of every read, we compute the consensus of all solid k-mers and solid centers of clusters of all non-solid k-mers covering this base (Figure 5). This step is formally described as Algorithm 5.
Figure 5.
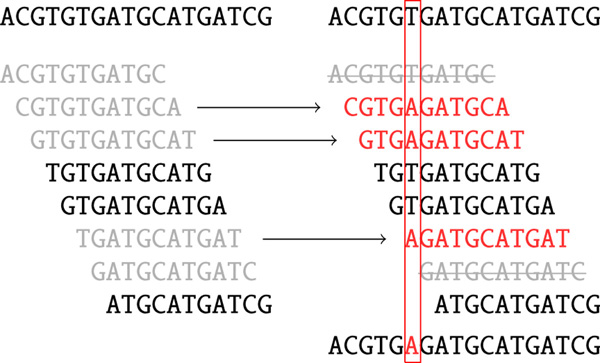
Read correction. Reads correction. Grey k-mers indicate non-solid k-mers. Red k-mers are the centers of the corresponding clusters (two grey k-mers striked through on the right are non-solid singletons). As a result, one nucleotide is changed.
Algorithm 4 Solid k-mers expansion
procedure ITERATIVEEXPANSION(R, X)
while ExpansionStep(R, X) do
function EXPANSIONSTEP(R, X)
for all reads r ∈ R do
if r is completely covered by solid k-mers then
mark all k-mers in r as solid
Return TRUE if X has increased and FALSE otherwise.
Algorithm 5 Reads correction
Input: reads R, solid k-mers X, clusters .
for all reads r ∈ R do
init consensus array υ: [0, |r| - 1] × {A, C, G, T} → ℕ with zeros: υ(j, x[i]):= 0 for all i = 0,...,|r| - 1 and j = 0,...,k - 1
for i = 0,...,|r| - k do
if r[i, i + k - 1] ∈ X (it is solid) then
for j ∈ [i, i + k - 1] do
υ(j, r[i]):= υ(j, r[i]) + 1
if r[i, i + k - 1] ∈ C for some C ∈ then
let x be the center of C
if x ∈ X (r belongs to a cluster with solid center) then
for j ∈ [i, i + k - 1] do
υ(j, x[i]):= υ(j, x[i]) + 1
for i ∈ [0, |r| - 1] do
r[i]:= arg maxa∈Σ υ(i, a).
Results and discussion
Datasets
In our experiments, we used three datasets from [2]: a single-cell E. coli, a single-cell S. aureus, and a standard (multicell) E. coli dataset. Paired-end libraries were generated by an Illumina Genome Analyzer IIx from MDA-amplified single-cell DNA and from multicell genomic DNA prepared from cultured E. coli, respectively These datasets consist of 100 bp paired-end reads with insert size 220; both E. coli datasets have average coverage ≈ 600×, although the coverage is highly non-uniform in the single-cell case.
In all experiments, BAYESHAMMER used k = 21 (we observed no improvements for higher values of k).
k-mer counts
Table 1 shows error correction statistics produced by di erent tools on all three datasets. For a comparison with HAMMER, we have emulated HAMMER with read correction by turning off Bayesian subclustering (HammerExpanded in the table) and both Bayesian subclustering and read expansion, another new idea of BAYESHAMMER (HammerNoExpansion in the table). Note that despite its more complex processing, BAYESHAMMER is significantly faster than other error correction tools (except, of course, for HAMMER which is a strict subset of BAYESHAMMER processing in our experiments and is run on BAYESHAMMER code). BAYESHAMMER also produces, in the single-cell case, a much smaller set of k-mers in the resulting reads which leads to smaller de Bruijn graphs and thus reduces the total assembly running time. Since BAYESHAMMER trims only bad quality bases and does not, like QUAKE, trim bases that it has not been able to correct (it has been proven detrimental for single-cell assembly in our experiments), it does produce a much larger set of k-mers than Quake on a multi-cell dataset.
Table 1.
k-mer statistics.
| Correction tool | Running time | k-mers | Reads | ||||
|---|---|---|---|---|---|---|---|
| Total | Genomic | Non-genomic | % of all genomic k-mers found in reads | % genomic among all k-mers in reads | % reads aligned to genome | ||
| Multi-cell E. coli, total 4,543,849 genomic k-mers | |||||||
| Uncorrected | 187,580,875 | 4,543,684 | 183,037,191 | 99.99 | 2.4 | 99.05 | |
| Quake | 4,565,237 | 4,543,461 | 21,776 | 99.99 | 99.5 | 99.97 | |
| HammerNoExpansion | 30 m | 58,305,738 | 4,543,674 | 53,762,064 | 99.99 | 8.4 | 95.59 |
| HammerExpanded | 36 m | 28,290,788 | 4,543,673 | 23,747,115 | 99.99 | 19.1 | 99.49 |
| BayesHammer | 37 m | 27,100,305 | 4,543,674 | 22,556,631 | 99.99 | 20.1 | 99.62 |
| Single-cell E. coli, total 4,543,849 genomic k-mers | |||||||
| Uncorrected | 165,355,467 | 4,450,489 | 160,904,978 | 97.9 | 2.7 | 79.05 | |
| Camel | 2 h 29 m | 147,297,070 | 4,450,311 | 142,846,759 | 97.9 | 3.0 | 81.25 |
| Euler-SR | 2 h 15 m | 138,677,818 | 4,450,431 | 134,227,387 | 97.9 | 3.2 | 81.95 |
| Coral | 2 h 47 m | 156,907,496 | 4,449,560 | 152,457,936 | 97.9 | 2.8 | 80.28 |
| HammerNoExpansion | 37 m | 53,001,778 | 4,443,538 | 48,558,240 | 97.8 | 8.3 | 81.36 |
| HammerExpanded | 43 m | 36,471,268 | 4,443,545 | 32,027,723 | 97.8 | 12.1 | 86.91 |
| BayesHammer | 57 m | 35,862,329 | 4,443,736 | 31,418,593 | 97.8 | 12.4 | 87.12 |
| Single-cell S. aureus, total 2,821,095 genomic k-mers | |||||||
| Uncorrected | 88,331,311 | 2,820,394 | 85,510,917 | 99.98 | 3.2 | 75.07 | |
| Camel | 5 h 13 m | 69,365,311 | 2,820,350 | 66,544,961 | 99.97 | 4.1 | 75.27 |
| Euler-SR | 2 h 33 m | 58,886,372 | 2,820,349 | 56,066,023 | 99.97 | 4.8 | 75.24 |
| Coral | 7 h 12 m | 83,249,146 | 2,820,011 | 80,429,135 | 99.96 | 3.4 | 75.22 |
| HammerNoExpansion | 58 m | 37,465,296 | 2,820,341 | 34,644,955 | 99.97 | 7.5 | 71.63 |
| HammerExpanded | 1 h 03 m | 23,197,521 | 2,820,316 | 20,377,205 | 99.97 | 12.1 | 76.54 |
| BayesHammer | 1 h 09 m | 22,457,509 | 2,820,311 | 19,637,198 | 99.97 | 12.6 | 76.60 |
For a comparison of BAYESHAMMER with other tools in terms of error rate reduction across an average read, see the logarithmic error rate graphs on Figure 6. Note that we are able to count errors only for the reads that actually aligned to the genome, so the graphs are biased in this way. Note how the first 21 bases are corrected better than others in BAYESHAMMER and both versions of HAMMER since we have run it with k = 21; still, other values of k did not show a significant improvement in either k-mer statistics or, more importantly, assembly results.
Figure 6.
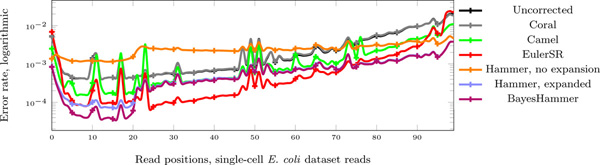
Error reduction. Error reduction by read position on logarithmic scale for the single-cell E. coli, single-cell S. aureus, and multi-cell E. coli datasets.
Assembly results
Tables 2 and 3 shows assembly results by the recently developed SPAdes assembler [10]; SPAdes was designed specifically for single-cell assembly, but has by now demonstrated state-of-the-art results on multi-cell datasets as well.
Table 2.
Assembly results, single-cell E.coli and S. aureus datasets (contigs of length ≥ 200 are used).
| Statistics | BayesHammer | BayesHammer(scaff old) | Coral | Coral (scaff old) | EulerSR | EulerSR (scaff old) | Hammer, expanded | Hammer, no expansion | Hammer, no expansion(scaff old) | Hammer(scaff old) |
|---|---|---|---|---|---|---|---|---|---|---|
| Single-cell E. coli, reference length 4639675, reference GC content 50.79% | ||||||||||
| # contigs (1000 bp) | 191 | 158 | 276 | 224 | 231 | 150 | 195 | 282 | 242 | 173 |
| # contigs | 521 | 462 | 675 | 592 | 578 | 375 | 529 | 655 | 592 | 477 |
| Largest contig | 269177 | 284968 | 179022 | 179022 | 267676 | 267676 | 268464 | 210850 | 210850 | 268464 |
| Total length | 4952297 | 4989404 | 5064570 | 4817757 | 4817757 | 4902434 | 4977294 | 5097148 | 5340871 | 5005022 |
| N50 | 110539 | 113056 | 45672 | 67849 | 74139 | 95704 | 97639 | 65415 | 84893 | 109826 |
| NG50 | 112065 | 118432 | 55073 | 87317 | 77762 | 108976 | 101871 | 68595 | 96600 | 112161 |
| NA50 | 110539 | 113056 | 45672 | 67765 | 74139 | 95704 | 97639 | 65415 | 84841 | 109826 |
| NGA50 | 112064 | 118432 | 55073 | 87317 | 77762 | 108976 | 101871 | 68594 | 96361 | 112161 |
| # misassemblies | 4 | 6 | 9 | 12 | 6 | 8 | 4 | 4 | 7 | 7 |
| # misassembled contigs | 4 | 6 | 9 | 10 | 6 | 8 | 4 | 4 | 7 | 7 |
| Misass. contigs length | 42496 | 94172 | 62114 | 150232 | 47372 | 149639 | 43304 | 26872 | 147140 | 130706 |
| Genome covered (%) | 96.320 | 96.315 | 96.623 | 96.646 | 95.337 | 95.231 | 96.287 | 96.247 | 96.228 | 96.281 |
| GC (%) | 49.70 | 49.69 | 49.61 | 49.56 | 49.90 | 49.74 | 49.68 | 49.64 | 49.60 | 49.68 |
| # mismatches/100 kbp | 11.22 | 11.70 | 8.36 | 9.10 | 5.55 | 5.82 | 12.77 | 54.11 | 52.48 | 13.08 |
| # indels/100 kbp | 1.07 | 8.26 | 9.17 | 12.76 | 0.52 | 47.80 | 0.91 | 1.17 | 7.96 | 8.69 |
| # genes | 4065 + | 4079 + | 3998 + | 4040 + | 3992 + | 4020 + | 4068 + | 4034 + | 4048 + | 4078 + |
| 124 part | 110 part | 180 part | 143 part | 140 part | 107 part | 123 part | 152 part | 136 part | 111 part | |
| Single-cell S. aureus, reference length 2872769, reference GC content 32.75% | ||||||||||
| # contigs (1000 bp) | 95 | 85 | 132 | 113 | 82 | 70 | 114 | 272 | 258 | 101 |
| Total length (1000 bp) | 3019597 | 3309342 | 3055585 | 3066662 | 2972925 | 2993100 | 3033912 | 3389846 | 3405223 | 3509555 |
| # contigs | 260 | 241 | 455 | 423 | 166 | 134 | 312 | 721 | 711 | 292 |
| Largest contig | 282558 | 328686 | 208166 | 208166 | 254085 | 535477 | 282558 | 148002 | 166053 | 328679 |
| Total length | 3081173 | 3368034 | 3160497 | 3166169 | 3008746 | 3020256 | 3111423 | 3575679 | 3594468 | 3584266 |
| N50 | 87684 | 145466 | 62429 | 90701 | 101836 | 145466 | 74715 | 30788 | 34943 | 131272 |
| NG50 | 112566 | 194902 | 87636 | 99341 | 108151 | 159555 | 88292 | 39768 | 45889 | 180022 |
| NA50 | 87684 | 145466 | 62429 | 89365 | 100509 | 145466 | 68711 | 30788 | 34552 | 112801 |
| NGA50 | 88246 | 148064 | 74452 | 90101 | 101836 | 145466 | 88289 | 35998 | 42642 | 148023 |
| # misassemblies | 15 | 17 | 11 | 14 | 4 | 5 | 11 | 14 | 18 | 14 |
| # misassembled contigs | 12 | 14 | 9 | 10 | 4 | 5 | 9 | 14 | 16 | 12 |
| Misass. contigs length | 340603 | 779785 | 478009 | 523596 | 377133 | 918380 | 402997 | 272677 | 324361 | 940356 |
| Genome covered (%) | 99.522 | 99.483 | 99.449 | 99.447 | 99.213 | 99.254 | 99.204 | 98.820 | 98.888 | 99.221 |
| GC (%) | 32.67 | 32.63 | 32.64 | 32.63 | 32.66 | 32.67 | 32.67 | 32.39 | 32.38 | 32.57 |
| # mismatches per 100 kbp | 3.18 | 8.01 | 12.44 | 12.65 | 9.72 | 10.28 | 17.38 | 54.92 | 55.50 | 15.36 |
| # indels per 100 kbp | 2.17 | 2.30 | 15.50 | 15.67 | 3.80 | 4.08 | 3.57 | 2.64 | 2.72 | 3.04 |
| # genes | 2540 + | 2547 + | 2532 + | 2540 + | 2547 + | 2550 + | 2535 + | 2477 + | 2485 + | 2539 + |
| 36 part | 30 part | 45 part | 37 part | 30 part | 27 part | 41 part | 91 part | 85 part | 38 part | |
Table 3.
Assembly results, multi-cell E.coli dataset (contigs of length ≥ 200 are used).
| Statistics | BayesHammer | BayesHammer (sca_old) | Hammer, expanded | Hammer, no expansion | Hammer, no expansion (sca_old) | Hammer (sca_old) | Quake |
|---|---|---|---|---|---|---|---|
| Multi-cell E. coli, 600 coverage, reference length 4639675, reference GC content 50.79% | |||||||
| # contigs (≥ 500 bp) | 103 | 102 | 119 | 238 | 213 | 115 | 165 |
| # contigs (≥ 1000 bp) | 91 | 90 | 99 | 192 | 171 | 96 | 156 |
| Total length (≥ 500 bp) | 4641845 | 4641790 | 4626515 | 4730338 | 4817457 | 4627067 | 4543682 |
| Total length (≥ 1000 bp) | 4633361 | 4633306 | 4611745 | 4696966 | 4787210 | 4612838 | 4537565 |
| # contigs | 122 | 121 | 146 | 325 | 303 | 141 | 204 |
| Largest contig | 285113 | 285113 | 218217 | 210240 | 210240 | 218217 | 165487 |
| Total length | 4647325 | 4647270 | 4635156 | 4756088 | 4844208 | 4635349 | 4555015 |
| N50 | 132645 | 132645 | 113608 | 59167 | 73113 | 113608 | 58777 |
| NG50 | 132645 | 132645 | 113608 | 59669 | 80085 | 113608 | 57174 |
| NA50 | 132645 | 132645 | 113608 | 59167 | 73113 | 113608 | 58777 |
| NGA50 | 132645 | 132645 | 113608 | 59669 | 80085 | 113608 | 57174 |
| # misassemblies | 3 | 3 | 4 | 4 | 7 | 5 | 0 |
| # misassembled contigs | 3 | 3 | 4 | 4 | 7 | 5 | 0 |
| Misassembled contigs length | 44466 | 44466 | 57908 | 15259 | 30901 | 60418 | 0 |
| Genome covered (%) | 99.440 | 99.440 | 99.383 | 98.891 | 98.925 | 99.385 | 98.747 |
| GC (%) | 50.78 | 50.77 | 50.77 | 50.73 | 50.71 | 50.77 | 50.75 |
| N's (%) | 0.00000 | 0.00000 | 0.00000 | 0.00000 | 0.00000 | 0.00000 | 0.00000 |
| # mismatches per 100 kbp | 8.55 | 8.55 | 13.76 | 44.46 | 44.33 | 13.76 | 1.21 |
| # indels per 100 kbp | 0.99 | 0.99 | 1.14 | 0.76 | 0.97 | 1.14 | 0.20 |
| # genes | 4254+45 part | 4254+45 part | 4245+56 part | 4196+72 part | 4204+68 part | 4245+56 part | 4174+62 part |
In the tables, N50 is such length that contigs of that length or longer comprise of the assembly; NG50 is a metric similar to N50 but only taking into account contigs comprising (and aligning to) the reference genome; NA50 is a metric similar to N50 after breaking up misassembled contigs by their misassemblies. NGx and NAx metrics have a more direct relevance to assembly quality than regular Nx metrics; our result tables have been produced by the recently developed tool QUAST [14].
All assemblies have been done with SPADES. The results show that after BAYESHAMMER correction, assembly results improve significantly, especially in the single-cell E. coli case; it is especially interesting to note that even in the multi-cell case, where BAYESHAMMER loses to QUAKE by k-mer statistics, assembly results actually improve over assemblies produced from QUAKE-corrected reads (including genome coverage and the number of genes).
Conclusions
Single-cell sequencing presents novel challenges to error correction tools. In contrast to multi-cell datasets, for single-cell datasets, there is no pretty distribution of k-mer multiplicities; one therefore has to work with k-mers on a one-by-one basis, considering each cluster of k-mers separately. In this work, we further developed the ideas of HAMMER from a Bayesian clustering perspective and presented a new tool BAYESHAMMER that makes them practical and yields significant improvements over existing error correction tools.
There is further work to be done to make our underlying models closer to real life; for instance, one could learn a non-uniform distribution of single nucleotide errors and plug it in our likelihood formulas. Another natural improvement would be to try and rid the results of contamination by either human or some other DNA material; we observed significant human DNA contamination in our single-cell dataset, so weeding it out might yield a significant improvement. Finally, a new general approach that we are going to try in our further work deals with the technique of minimizers introduced by Roberts et al. [15]. It may provide significant reduction in memory requirements and a possible approach to dealing with paired information.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
All authors contributed extensively to the work presented in this paper.
Declarations
The publication costs for this article were funded by the Government of the Russian Federation, grant 11.G34.31.0018.
This article has been published as part of BMC Genomics Volume 14 Supplement 1, 2013: Selected articles from the Eleventh Asia Pacific Bioinformatics Conference (APBC 2013): Genomics. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcgenomics/supplements/14/S1.
Acknowledgements
We thank Pavel Pevzner for many fruitful discussions on all stages of the project. We are also grateful to Andrei Prjibelski and Alexei Gurevich for help with the experiments and to the anonymous referees whose comments have benefited the paper greatly. This work was supported the Government of the Russian Federation, grant 11.G34.31.0018. Work of the first author was also supported by the Russian Fund for Basic Research grant 12-01-00450-a and the Russian Presidential Grant MK-6628.2012.1. Work of the second author was additionally supported by the Russian Fund for Basic Research grant 12-01-00747-a.
References
- Grindberg R, Ishoey T, Brinza D, Esquenazi E, Coates R, Liu W, Gerwick L, Dorrestein P, Pevzner P, Lasken R, Gerwick W. Single cell genome amplification accelerates identification of the apratoxin biosynthetic pathway from a complex microbial assemblage. PLOS One. 2011;6(4):e18565. doi: 10.1371/journal.pone.0018565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitsaz H, Yee-Greenbaum JL, Tesler G, Lombardo MJ, Dupont CL, Badger JH, Novotny M, Rusch DB, Fraser LJ, Gormley NA, Schulz-Trieglaff O, Smith GP, Evers DJ, Pevzner PA, Lasken RS. Efficient de novo assembly of single-cell bacterial genomes from short-read data sets. Nat Biotechnol. 2011;29:915–921. doi: 10.1038/nbt.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishoey T, Woyke T, Stepanauskas R, Novotny M, Lasken R. Genomic sequencing of single microbial cells from environmental samples. Current Opinion in Microbiology. 2008;11(3):198–204. doi: 10.1016/j.mib.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill S, Pop M, Deboy R, Eckburg P, Turnbaugh P, Samuel B, Gordon J, Relman D, Fraser-Liggett C, Nelson K. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312(5778):1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamady M, Knight R. Microbial community profiling for human microbiome projects: tools, techniques, and challenges. Genome Res. 2009;19(7):1141–1152. doi: 10.1101/gr.085464.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Vederas J. Drug discovery and natural products: end of an era or an endless frontier? Science. 2009;325(5937):161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- Kelley DR, Schatz MC, Salzberg SL. Quake: quality-aware detection and correction of sequencing errors. Genome Biology. 2010;11(11):R116. doi: 10.1186/gb-2010-11-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev P, Scott E, Kakaradov B, Pevzner P. Error correction of high-throughput sequencing datasets with non-uniform coverage. Bioinformatics. 2011;27(13):i137–41. doi: 10.1093/bioinformatics/btr208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, Pevzner P. Short read fragment assembly of bacterial genomes. Genome Research. 2008;18:324–330. doi: 10.1101/gr.7088808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich A, Dvorkin M, Kulikov A, Lesin V, Nikolenko S, Pham S, Prjibelski A, Pyshkin A, Sirotkin A, Vyahhi N, Tesler G, Alekseyev M, Pevzner P. SPAdes: a new genome assembler and its applications to single cell sequencing. Journal of Computational Biology. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock P, Fields C, Goto N, Heuer M, Rice P. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010;38(6):1767–1771. doi: 10.1093/nar/gkp1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormen TH, Leiserson CE, Rivest R. Introduction to Algorithms. MIT Press; 2009. [Google Scholar]
- Schwarz G. Estimating the dimension of a model. Annals of Statistics. 1978;6:461–464. doi: 10.1214/aos/1176344136. [DOI] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: Quality Assessment for Genome Assemblies. 2012. [Submitted] [DOI] [PMC free article] [PubMed]
- Roberts M, Hayes W, Hunt BR, Mount SM, Yorke JA. Reducing storage requirements for biological sequence comparison. Bioinformatics. 2004;20(18):3363–3369. doi: 10.1093/bioinformatics/bth408. [DOI] [PubMed] [Google Scholar]