

Abstract
A 5-year-old girl was diagnosed with neurofibromatosis type 2 (NF-2) due to multiple neurofibromas, cafe-au-lait spots, and schwannomas of the brain. During ophthalmologic evaluation, a posterior subcapsular cataract and a gray-green colored subretinal lesion were found in right eye. Fluorescein angiography (FA) revealed a combined hamartoma of the retina and retinal pigment epithelium (CHRRPE). At age 9, she underwent cataract surgery. At this time FA and spectral-domain optical coherence tomography (SD-OCT) were taken. The SD-OCT showed an elevated hyperreflective mass in the retina with prominent attenuation of the inner and outer retina, but minimal attenuation in the photoreceptor layers. The underlying retina appeared to be disorganized and thick (791 µm). This is the first case report of SD-OCT imaging of a CHRRPE associated with NF-2 in a pediatric patient. By using SD-OCT in this patient, we could obtain detailed tumor characteristics, and SD-OCT may be helpful in the diagnosis and management of CHRRPE.
Keywords: Combined hamartoma of retina and retinal pigment epithelium, Neurofibromatoses, Spectral-domain optical coherence tomography
Neurofibromatosis is the most common phakomatosis, with an estimated prevalence of approximately one case per 3000 in the general population [1]. Neurofibromatosis type 2 (NF-2) is a rare autosomal dominant disease with a birth incidence estimated to be one in 33,000 to 40,000 [2,3]. Ocular features of NF-2 include juvenile posterior subcapsular cataracts, combined hamartoma of the retinal and retinal pigment epithelium (CHRRPE), epiretinal membrane (ERM), optic nerve meningioma, optic disc glioma, intraocular schwannoma, and neurotrophic keratopathy [4-9].
CHRRPE is an uncommon, benign hamartomatous malformation involving the retinal pigment epithelium (RPE), neurosensory retina, retinal vessels. and adjacent vitreous [10]. Loss of heterozygosity for the NF-2 gene has been demonstrated in dysplastic or hamartomatous lesions of the retina and optic disc [11].
Here, we present spectral-domain optical coherence tomography (SD-OCT) findings of a CHRRPE associated NF-2 in a pediatric patient.
Case Report
A 5-year-old girl visited the ophthalmology clinic for an ophthalmologic evaluation under the diagnosis of NF-2. Multiple neurofibromas, café-au-lait macules, and multiple schwannomas of both trigeminal nerves from brain magnetic resonance imaging (MRI) lead to the diagnosis of NF-2. At the initial presentation, the visual acuity was 0.05 logarithm of minimum angle resolution (logMAR) of right eye and 0.4 logMAR of left eye. Posterior subcapsular cataract and a gray-green colored subretinal lesion were found in the right eye. There were no specific signs in the left eye. Fundus photography and fluorescein angiography (FA) were performed. FA showed tortuosity of the vessels with localized areas of pinpoint hyperfluorescence and late retinal vascular leakage, corresponding to the retinal lesion in the right eye. These findings were suggestive of a CHRRPE. During follow-up, bilateral acoustic schwannomas and multiple neurogenic tumors in both lungs were found. Also, DNA test revealed pArg198term mutation on the NF-2 gene, which is known to cause NF-2.
At age 9, she underwent phacoemulsification and posterior chamber intraocular lens implantation surgery under general anesthesia. B-scan ultrasonography of the right eye revealed no choroidal excavation or extrascleral extension. After operation, visual acuity was 0.1 logMAR OD and 0.6 logMAR OS. FA and SD-OCT were performed for evaluation of the retinal lesion. FA showed no significant change (Fig. 1). SD-OCT revealed a hyperreflective ERM with traction and retinal folds. Perimacular posterior vitreous detachment was also found. The inner layers of the underlying retina were irregularly thickened (up to 791 µm), but the photoreceptor inner segment/outer segment (IS/OS) junction and RPE layers were not significantly attenuated (Fig. 2). By these examinations, we confirmed the diagnosis of CHRRPE, with relatively less involvement of the photoreceptor layer. We recommended surgical intervention, including pars plana vitrectomy with membrane peeling, but the parents of the patient refused to do so. Therefore, we continued to conduct regular checkups.
Fig. 1.
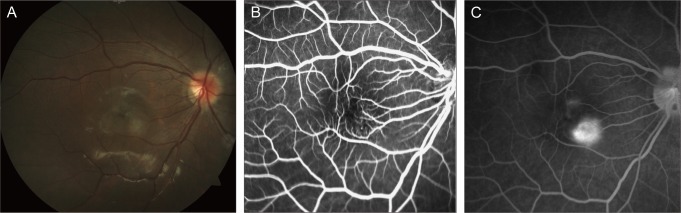
At age 9, (A) fundus photography of the right eye reveals a moderately pigmented and elevated lesion at the inferotemporal vascular arcade, involving the macula. (B) Fundus fluorescein angiography of the right eye demonstrates some areas of pinpoint hyperfluorescence. (C) Late phase of fluorescein angiography demonstrates mild retinal vascular leakage, and shows no significant progression compared with the previous examination.
Fig. 2.

Spectral-domain optical coherence tomography reveals an elevated hyperreflective mass in the retina with mild attenuation of the retinal pigment epithelium and photoreceptor inner segment/outer segment junction. Prominent thickening and attenuation of the inner retina is also noted (A,B). The arrow in (A) represents the hyperreflective epiretinal membrane.
Discussion
NF-2 is a dominantly inherited disorder prone to tumors characterized by the development of multiple schwanommas, meningiomas, and ependymomas, with the great majority of patients developing bilateral schwannoma involvement of the superior vestibular branch of the eighth cranial nerve. The first sign of a more severe multi-tumor disease in early childhood is often a non-8th nerve tumor (including cutaneous tumor) or an ocular presentation. Ophthalmic features are prominent in NF2. Patients often suffer from reduced visual acuity from various causes. Between 60% and 80% of patients have cataracts, which are usually presenile posterior subcapsular cataracts. Optic nerve meningiomas can cause visual loss in the first years of life, and extensive retinal hamartomas can also affect vision.
CHRRPE is a congenital and relatively stable, but it can evolve slowly with age. CHRRPE are usually unilateral. The pigmented elevated masses of CHRRPE are frequently associated with overlying glial tissue that produce retinal distortion, striae, and decreased visual acuity [12]. Histopathologically, CHRRPE lesions demonstrate disorganized retina, hyperplastic glial cells, and vascular tortuosity. The abnormal vitreoretinal interface can cause traction and distortion of the sensory retina. Lesions can also be characterized by predominant tissue subtypes, including melanocytic, vascular, or glial [13]. The glial subtype presents with prominent ERM proliferation and is most amenable to surgical intervention. The clinical presentation can differ based on varying degrees of retinal elevation, vascular tortuosity, and retinal distortion. Diagnosis is most commonly made in childhood by clinical examination and fundus photography. Complications can include retinoschisis, retinal holes, vitreous hemorrhage, choroidal neovascularization, retinal hemorrhages, and exudative retinal detachment [14-16].
One report presented the vitreoretinal interface and retinal microarchitecture findings of CHRRPE using the conventional time domain OCT. Of the 11 patients, 10 had a preretinal membrane, with retinal striae in 9 patients. Retinal anatomic disorganization with loss of identifiable retinal layers at the site of the mass was noted in all patients [17]. After widespread use of SD-OCT, one report described SD-OCT findings of CHRRPE [18]. Compared with the previous report, our patient showed less attenuation of the RPE and photoreceptor IS/OS junction by SD-OCT. Also, prominent thickening and attenuation of the inner retina was noted. We could find out that hamartomatous change affects the inner retina more by SD-OCT. ERM, one of the frequent findings associated with CHRRPE, was another SD-OCT finding in this patient. Partial posterior vitreous detachment was noted at the parafoveal area.
Several studies have concluded that pars plana vitrectomy with membrane peeling has a limited role in the treatment for CHRRPE [10,19-21]. The literature regarding the surgical repair of CHRRPE in children is limited, but recent reports of vitrectomy with membrane peeling for vitreomacular traction and macular distortion associated with CHRRPE have shown promising results [22,23]. Some reports demonstrated improved or stabilized vision after early surgical intervention of ERM in both adults and pediatric patients by vitrectomy and membrane stripping [24,25]. Because there was less attenuation of the photoreceptor IS/OS junction integrity, removal of the ERM could be helpful in this patient, which may help to release the tractional effect of the ERM.
In conclusion, this is unique case report which describes the SD-OCT findings in a Korean child with CHRRPE associated with NF-2. By using the non-invasive SD-OCT that has better resolution, detailed structural images of CHRRPE can be obtained. One unique feature is that the inner retina was more prominently involved than the RPE and photoreceptor IS/OS junction. If the photoreceptor layers are less attenuated in CHRRPE patients, it may be associated with better visual prognosis. Therefore, additional studies using SD OCT would be necessary to fully answer these questions.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.De Laey JJ, Hanssens M. Vascular tumors and malformations of the ocular fundus. Dordrecht: Kluwer Academic Publishers; 1990. pp. 101–120. [PubMed] [Google Scholar]
- 2.Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84:603–618. [PubMed] [Google Scholar]
- 3.Evans DG, Huson SM, Donnai D, et al. A genetic study of type 2 neurofibromatosis in the United Kingdom. I. Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet. 1992;29:841–846. doi: 10.1136/jmg.29.12.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pearson-Webb MA, Kaiser-Kupfer MI, Eldridge R. Eye findings in bilateral acoustic (central) neurofibromatosis: association with presenile lens opacities and cataracts but absence of Lisch nodules. N Engl J Med. 1986;315:1553–1554. doi: 10.1056/NEJM198612113152419. [DOI] [PubMed] [Google Scholar]
- 5.Bouzas EA, Freidlin V, Parry DM, et al. Lens opacities in neurofibromatosis 2: further significant correlations. Br J Ophthalmol. 1993;77:354–357. doi: 10.1136/bjo.77.6.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Landau K, Dossetor FM, Hoyt WF, Muci-Mendoza R. Retinal hamartoma in neurofibromatosis 2. Arch Ophthalmol. 1990;108:328–329. doi: 10.1001/archopht.1990.01070050026011. [DOI] [PubMed] [Google Scholar]
- 7.Kaye LD, Rothner AD, Beauchamp GR, et al. Ocular findings associated with neurofibromatosis type II. Ophthalmology. 1992;99:1424–1429. doi: 10.1016/s0161-6420(92)31789-0. [DOI] [PubMed] [Google Scholar]
- 8.Dossetor FM, Landau K, Hoyt WF. Optic disk glioma in neurofibromatosis type 2. Am J Ophthalmol. 1989;108:602–603. doi: 10.1016/0002-9394(89)90445-5. [DOI] [PubMed] [Google Scholar]
- 9.Freedman SF, Elner VM, Donev I, et al. Intraocular neurilemmoma arising from the posterior ciliary nerve in neurofibromatosis: pathologic findings. Ophthalmology. 1988;95:1559–1564. doi: 10.1016/s0161-6420(88)32983-0. [DOI] [PubMed] [Google Scholar]
- 10.Schachat AP, Shields JA, Fine SL, et al. Combined hamartomas of the retina and retinal pigment epithelium. Ophthalmology. 1984;91:1609–1615. doi: 10.1016/s0161-6420(84)34094-5. [DOI] [PubMed] [Google Scholar]
- 11.Chan CC, Koch CA, Kaiser-Kupfer MI, et al. Loss of heterozygosity for the NF2 gene in retinal and optic nerve lesions of patients with neurofibromatosis 2. J Pathol. 2002;198:14–20. doi: 10.1002/path.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hartnett ME, Trese M, Capone A, et al. Pediatric retina: medical and surgical approaches. Philadelphia: Lippincott Williams and Wilkins; 2005. pp. 231–233. [Google Scholar]
- 13.Schachat AP, Shields JA, Fine SL, et al. Combined hamartomas of the retina and retinal pigment epithelium. Ophthalmology. 1984;91:1609–1615. doi: 10.1016/s0161-6420(84)34094-5. [DOI] [PubMed] [Google Scholar]
- 14.Ryan SJ, Ogden J, Hinton D, Schachat AP. Retina: basic science and inherited retinal disease. 4th ed. Baltimore: Mosby; 2006. pp. 673–678. [Google Scholar]
- 15.Schachat AP, Glaser BM. Retinal hamartoma, acquired retinoschisis, and retinal hole. Am J Ophthalmol. 1985;99:604–605. doi: 10.1016/s0002-9394(14)77975-9. [DOI] [PubMed] [Google Scholar]
- 16.Kahn D, Goldberg MF, Jednock N. Combined retinal-retina pigment epithelial hamartoma presenting as a vitreous hemorrhage. Retina. 1984;4:40–43. doi: 10.1097/00006982-198400410-00006. [DOI] [PubMed] [Google Scholar]
- 17.Shields CL, Mashayekhi A, Dai VV, et al. Optical coherence tomographic findings of combined hamartoma of the retina and retinal pigment epithelium in 11 patients. Arch Ophthalmol. 2005;123:1746–1750. doi: 10.1001/archopht.123.12.1746. [DOI] [PubMed] [Google Scholar]
- 18.Huot CS, Desai KB, Shah VA. Spectral domain optical coherence tomography of combined hamartoma of the retina and retinal pigment epithelium. Ophthalmic Surg Lasers Imaging. 2009;40:322–324. doi: 10.3928/15428877-20090430-19. [DOI] [PubMed] [Google Scholar]
- 19.McDonald HR, Abrams GW, Burke JM, Neuwirth J. Clinicopathologic results of vitreous surgery for epiretinal membranes in patients with combined retinal and retinal pigment epithelial hamartomas. Am J Ophthalmol. 1985;100:806–813. doi: 10.1016/s0002-9394(14)73372-0. [DOI] [PubMed] [Google Scholar]
- 20.Sappenfield DL, Gitter KA. Surgical intervention for combined retinal-retinal pigment epithelial hamartoma. Retina. 1990;10:119–124. doi: 10.1097/00006982-199004000-00006. [DOI] [PubMed] [Google Scholar]
- 21.Mason JO., 3rd Visual improvement after pars plana vitrectomy and membrane peeling for vitreoretinal traction associated with combined hamartoma of the retina and retinal pigment epithelium. Retina. 2002;22:824–825. doi: 10.1097/00006982-200212000-00028. [DOI] [PubMed] [Google Scholar]
- 22.Stallman JB. Visual improvement after pars plana vitrectomy and membrane peeling for vitreoretinal traction associated with combined hamartoma of the retina and retinal pigment epithelium. Retina. 2002;22:101–104. doi: 10.1097/00006982-200202000-00017. [DOI] [PubMed] [Google Scholar]
- 23.Konstantinidis L, Chamot L, Zografos L, Wolfensberger TJ. Pars Plana vitrectomy and epiretinal membrane peeling for vitreoretinal traction associated with combined hamartoma of the retina and retinal pigment epithelium (CHRRPE) Klin Monbl Augenheilkd. 2007;224:356–359. doi: 10.1055/s-2007-962840. [DOI] [PubMed] [Google Scholar]
- 24.Cohn AD, Quiram PA, Drenser KA, et al. Surgical outcomes of epiretinal membranes associated with combined hamartoma of the retina and retinal pigment epithelium. Retina. 2009;29:825–830. doi: 10.1097/IAE.0b013e31819b1788. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Dong F, Dai R, Yu W. Surgical management of epiretinal membrane in combined hamartomas of the retina and retinal pigment epithelium. Retina. 2010;30:305–309. [PubMed] [Google Scholar]