

Summary
DNA double-strand break (DSB) signaling and repair are critical for cell viability, and rely on highly coordinated pathways whose molecular organization is still incompletely understood. Here, we show that heterogeneous nuclear ribonucleoprotein U-like (hnRNPUL) proteins 1 and 2 play key roles in cellular responses to DSBs. We identify human hnRNPUL1 and 2 as binding partners for the DSB sensor complex MRE11-RAD50-NBS1 (MRN) and demonstrate that hnRNPUL1 and 2 are recruited to DNA damage in an interdependent manner that requires MRN. Moreover, we show that hnRNPUL1 and 2 stimulate DNA-end resection and promote ATR-dependent signaling and DSB repair by homologous recombination, thereby contributing to cell survival upon exposure to DSB-inducing agents. Finally, we establish that hnRNPUL1 and 2 function downstream of MRN and CtBP-interacting protein (CtIP) to promote recruitment of the BLM helicase to DNA breaks. Collectively, these results provide insights into how mammalian cells respond to DSBs.
Keywords: DNA damage, DNA repair, DNA double-strand breaks, DNA-end resection, heterogeneous nuclear ribonucleoproteins
Introduction
DNA double-strand breaks (DSBs) represent the most toxic form of DNA damage, which, if inaccurately repaired, can contribute to various human disorders including cancer, neurodegeneration and immunodeficiency (Friedberg et al., 2006; Jackson and Bartek, 2009). To counteract the deleterious effects of DNA damage and preserve the integrity of their genetic material, cells have evolved elaborate surveillance pathways, collectively termed the DNA damage response (DDR), which detect, signal and repair DNA lesions (Harper and Elledge, 2007; Polo and Jackson, 2011). In particular, DSB detection by the MRE11-RAD50-NBS1 (MRN) sensor complex (Rupnik et al., 2009) in higher eukaryotes recruits and activates the checkpoint kinase ATM (Difilippantonio and Nussenzweig, 2007; Falck et al., 2005; Lee and Paull, 2005; Uziel et al., 2003), which then promotes DSB signaling by phosphorylating multiple targets including the effector kinase Chk2 and the histone variant H2AX (Matsuoka et al., 2007). Concomitantly, DNA repair proteins are recruited to DSBs to resolve the damage by two major pathways (Hartlerode and Scully, 2009; Pardo et al., 2009): non-homologous end-joining (NHEJ; Lieber, 2008) and homologous recombination (HR; San Filippo et al., 2008), the latter being restricted to S and G2 phases of the cell cycle. HR but not NHEJ relies on the presence of a sister chromatid and requires extensive nucleolytic processing (resection) of DNA ends to generate stretches of single-stranded DNA (ssDNA) that will invade the homologous template. In addition to promoting HR, Replication protein A (RPA)-coated ssDNA also recruits the checkpoint kinase ATR together with its interaction partner, ATRIP, resulting in ATR-dependent signaling of DSBs and Chk1 phosphorylation (Cimprich and Cortez, 2008; Cortez et al., 2001; Zou and Elledge, 2003).
Although it is well established that DNA ends are resected in human cells through the concerted actions of the MRN complex and several auxiliary factors including CtBP-interacting protein (CtIP; Clerici et al., 2005; Limbo et al., 2007; Sartori et al., 2007), Bloom syndrome helicase (BLM), and the EXO1 exonuclease (Budd and Campbell, 2009; Gravel et al., 2008; Mimitou and Symington, 2008; Zhu et al., 2008), precisely how these factors are recruited to DNA breaks in a coordinated manner remains elusive. Moreover, it is not known whether additional proteins mediate or control DSB resection. Here, by using a combination of biochemistry and cell biology, we identify human heterogeneous nuclear ribonucleoprotein U-like (hnRNPUL) proteins 1 and 2 as being required for effective DSB resection, contributing to both ATR-dependent signaling and DSB repair by HR. We also establish that hnRNPUL1 and 2 act downstream of MRN and CtIP by facilitating the recruitment of BLM helicase to DNA breaks.
Results
Identification of hnRNPUL1 and 2 as MRN binding partners
Previous work has shown that di-phosphorylated Ser-Asp-Thr-Asp (SDTD) repeat motifs on MDC1 mediate interactions with NBS1, a subunit of the MRN complex (Chapman and Jackson, 2008; Hari et al., 2010; Lloyd et al., 2009; Melander et al., 2008; Spycher et al., 2008; Williams et al., 2009; Wu et al., 2008). This suggested to us that the MDC1 SDTD phosphopeptide could be used to identify additional MRN-binding proteins. Indeed, in addition to binding MRN, the MDC1 SDTD phosphopeptide, but not its non-phosphorylated derivative, specifically retrieved another prominent protein of ~125 kDa from HeLa cell nuclear extracts, which was identified by mass spectrometry as hnRNPUL2 (Figure 1A). As hnRNPUL2 contains no defined phospho-dependent interaction motifs and is negatively charged under physiological conditions (theoretical isoelectric point: 4.85) making electrostatic interactions with the phospho-peptide unlikely, this suggested that hnRNPUL2 bound the SDTD phosphopeptide indirectly via interactions with MRN. Consistent with this idea, while in vitro translated hnRNPUL2 on its own showed very weak binding to the MDC1 SDTD phosphopeptide, this binding was strongly enhanced in the presence of the MRN subunit NBS1 (Figure 1B). Additionally, GFP-tagged hnRNPUL2 co-immunoprecipitated with MRN subunits from cell extracts in a manner that was unaffected by treatment with the DNA-damaging agent camptothecin (CPT; Figures 1C-1E). Notably, the hnRNPUL2-related protein hnRNPUL1 (also known as E1B-55K-associated protein 5; E1B-AP5) that is involved in ATR signaling in response to viral infection (Blackford et al., 2008) also co-precipitated with hnRNPUL2 (Figure 1C), and reciprocally co-immunoprecipitated with MRN subunits and the MRN-binding factor CtIP from crude cell extracts (Figures 1F and 1G). Furthermore, in vitro experiments showed that, of the individual MRN subunits, only NBS1 directly bound hnRNPUL1 and 2 (Figure 1H), and revealed that both hnRNPUL proteins bound NBS1 within its C-terminal region (Figure 1I). By using hnRNPUL1 deletion constructs, we localized the NBS1 binding region to the middle part of hnRNPUL1 that contains its BBS and RGG domains, and established that deleting either of these domains impaired the ability of hnRNPUL1 to interact with the NBS1 C-terminus (Figures 1J-L). Collectively, these results established that hnRNPUL1 and 2 bind the MRN complex, and suggested an involvement for these hnRNPUL proteins in cellular responses to DSBs.
Figure 1. hnRNP U-like proteins bind MRN and CtIP.

(A) Silver-stained gel (originally from Chapman and Jackson, 2008) of proteins retrieved from HeLa nuclear extracts with MDC1 SDTD phosphopeptide (PP) or unphosphorylated peptide (-). Bands were identified by liquid chromatography mass spectrometry.
(B) [35S]-labeled in vitro translated hnRNPUL2 pulled-down with MDC1 SDTD phosphopeptide or beads alone as negative control, in the presence or absence of unlabelled in vitro translated NBS1.
(C) GFP-hnRNPUL2 (GFP-UL2) immunoprecipitates from HEK293 cells contain MRN subunits and hnRNPUL1 (UL1).
(D) MRN immunoprecipitates from HEK293 cells transiently expressing GFP-hnRNPUL2.
(E) RAD50 immunoprecipitates from HEK293 cells transiently expressing GFP-hnRNPUL2 that were treated or not with camptothecin (CPT). RPA phosphorylation (RPA S4/8-P) is a readout of DNA damage.
(F) hnRNPUL1 immunoprecipitates from human LCLs (lymphoblastoid cells) using two different antibodies (Ab-1, Ab-2) contain the MRN complex and CtIP.
(G) MRN and CtIP immunoprecipitates from human LCLs contain hnRNPUL1.
(H-K) GST-pull-downs using the indicated GST-fusions, as revealed by Coomassie staining, and [35S]-labeled proteins. The asterisk on panel I indicates a degradation product. Mutant forms of hnRNPUL1 are as shown on the scheme.
(L) Domain organization of hnRNPUL1 and 2: SAP (SAF-A/B, Acinus and PIAS motif), SPRY (SPIa/Ryanodine receptor domain), NK (putative nucleoside/nucleotide kinase domain), BBS (BRD7-binding site), RGG (arginine and glycine-rich region, RNA and ssDNA binding), PP (proline-rich region). Percentages of identity are indicated (total and for individual colored domains). N, M and C refer to hnRNPUL1 deletion mutants.
Dual behavior of hnRNPUL protein localization after DNA damage
To gain insights into how hnRNPUL proteins might contribute to the DDR, we analyzed their localization upon induction of DNA damage. This revealed that both hnRNPUL1 and 2 were excluded from sites of laser-induced damage (Figure 2A left columns). Moreover, the exclusion was rapid, being detectable within the first few minutes after damage induction, and persisted over time, mirroring H2AX phosphorylation (γH2AX; Figure S1A). Exclusion was also observed from ionizing radiation-induced foci (IRIF) and from 53BP1 OPT domains (Figure 2A middle and right columns) that appear in G1 cells to mark spontaneous DSBs generated as a result of incomplete DNA replication in the preceding cell cycle (Harrigan et al., 2011; Lukas et al., 2011). Exclusion did not appear to result from protein degradation, since global hnRNPUL1 levels were not detectably affected by DNA damage and because hnRNPUL1 exclusion from laser micro-irradiated nuclear regions still occurred in the presence of the proteasome inhibitor MG132 or the autophagy inhibitor 3-methyladenine (Figures S1B and S1C). While exploring the mechanism for this exclusion, we found that exclusion of hnRNPUL1 was impaired by deletion of its RGG-rich RNA binding domain (Gabler et al., 1998) and paralleled the inhibition of RNA synthesis at sites of laser-induced and IR-induced DNA damage (Figures S1D and S1E). Furthermore, while we found that the majority of hnRNPUL1/2 in undamaged cells was resistant to detergent extraction, the proteins became totally extractable after RNase treatment (Figure S1F and data not shown). These results suggested that localization of hnRNPUL proteins in the nucleus of an undamaged cell depends on the presence of RNA, and that their exclusion from DNA damage sites is a readout of local inhibition of transcription at these locations (Shanbhag et al., 2010).
Figure 2. hnRNPUL recruitment to and exclusion from sites of DNA damage.

(A) Exclusion of hnRNPUL1 and GFP-hnRNPUL2 from sites of laser damage (30 min after micro-irradiation), IRIF (9 h post 10 Gy IR) and 53BP1 OPT domains in U2OS cells. Arrowheads indicate major IRIF and OPT domains.
(B) Live imaging of GFP-hnRNPUL1 and 2 recruitment to laser damage in stable U2OS cells pre-treated with DRB. Pre: before micro-irradiation. Note that GFP-hnRNPUL1 is expressed at close to endogenous levels as shown in Figure S2D.
(C) GFP-hnRNPUL1 recruitment to laser damage (γH2AX) revealed by RNase treatment, which solubilizes GFP-hnRNPUL1 except at DNA damage sites. Nucleolin immunodetection is used as positive control for RNase treatment.
(D) GFP-hnRNPUL1 recruitment to laser damage 2 min after micro-irradiating U2OS cells analyzed by immunostaining as indicated.
(E) Recruitment of GFP-hnRNPUL1 and 2 to sites of laser damage (red line: laser path) analyzed by live cell imaging 1 min after irradiation in U2OS cells treated with DRB and the indicated siRNAs (siLuci: control). Efficiency of NBS1 depletion is confirmed by immunofluorescence on cells fixed 1 h after laser damage (right panels, where the top row represents the same cells by live imaging 1 min after laser micro-irradiation). Percentages indicate proportions of damaged cells showing recruitment of hnRNPUL proteins to laser lines (obtained by scoring >100 cells in two independent experiments).
(F) Recruitment of wild-type (WT) or mutant HA-hnRNPUL1 proteins to sites of laser damage analyzed by immunofluorescence 2 min after micro-irradiation in U2OS cells pre-treated with DRB. Expression of the mutant proteins, referring to the scheme in Figure 1, is shown on the western-blot panel. Scale bars, 10 μm. See also Figures S1, S2 and S3.
As an approach to see whether hnRNPUL protein localization might also respond more directly to DNA damage and not solely reflect local changes in transcription, we examined their localization after DNA damage in cells where transcription was globally inhibited (Figure S1G; note that under these conditions, cellular responses to DSBs were not markedly affected, as shown in Figures S2A and S2B). When using either 5,6 dichloro-β-D-ribofuranosylbenzimidazole (DRB) or α-amanitin to inhibit transcription by RNA polymerase II, we detected early and transient recruitment of hnRNPUL proteins to sites of laser-induced damage (Figure 2B and data not shown; see Figures S2E and S2F for endogenous hnRNPUL1). In line with these findings, accumulation of hnRNPUL1 at DNA damage sites was also observed when cells were treated with RNase prior to antibody detection (Figure 2C), indicating that a proportion of hnRNPUL proteins is recruited to damaged chromatin in a RNA-independent manner and becomes readily detectable when the RNA-dependent pool is extracted. Moreover, hnRNPUL recruitment to damage sites was also detected without transcription inhibition or RNase treatment when increased laser damage was used on cells expressing tagged hnRNPUL proteins (Figure S2G), or when laser damage was targeted to heterochromatic regions that are largely devoid of active transcription (Figure S2H). Collectively, these data indicated the existence of two hnRNPUL protein pools: one involved in RNA metabolism while the other engages with sites of DSBs.
The recruitment of hnRNPUL proteins to DNA damage was not restricted to S/G2 cells, as established by co-staining with the S/G2 marker Cyclin A (Figure 2D and data not shown). Moreover, as shown in Figure 2E, the accumulations of hnRNPUL1 and 2 at DNA damage sites were largely inter-dependent, which, together with their co-immunoprecipitation (Figure 1C), suggested that hnRNPUL1 and 2 are recruited to sites of DNA damage as part of the same protein complex. Consistent with the interaction we had identified between hnRNPUL1/2 and the MRN complex being functionally important, DNA-damage recruitment of both hnRNPUL1 and 2 was severely impaired by siRNA-mediated depletion of NBS1 or MRE11 (Figure 2E, Figures S3A and S3B and data not shown). Nevertheless, while recruitment of hnRNPUL proteins to DNA damage sites required the presence of the MRN complex, it did not rely on MRN nuclease activity, as shown by using the MRE11 inhibitor Mirin (Dupre et al., 2008), which in fact consistently enhanced hnRNPUL protein accrual at damage sites (Figure S3C).
The above findings demonstrated that hnRNPUL proteins are recruited to DNA damage sites in an MRN-dependent manner. To gain further insights into this recruitment, we examined hnRNPUL1 deletion mutants for their ability to localize to sites of laser-induced DNA damage. This indicated that the C-terminal half of hnRNPUL1, that is involved in NBS1 binding (Figure 1J), was required for the effective accumulation of hnRNPUL1 at DNA-damage sites (Figure 2F). Notably, although we found the BBS domain in the C-terminal part of hnRNPUL1 to contribute to NBS1 binding in vitro (Figure 1J), it was not needed for hnRNPUL1 recruitment under these experimental conditions (Figure 2F; note, however, that interpretation of the cell-based recruitment experiments is complicated by the presence of endogenous hnRNPUL1 and 2 that could contribute to the recruitment of mutant proteins). By contrast, deleting the hnRNPUL1 RGG domain, which also contributes to NBS1 binding (Figure 1J), strongly impaired hnRNPUL1 recruitment to sites of laser-induced DNA damage (Figure 2F). Note also that the defective behavior of the ΔRGG mutant is not due to protein misfolding, since this mutant is proficient for transcriptional regulation (Kzhyshkowska et al., 2003) and for binding the known hnRNPUL1 partner p53 (Barral et al., 2005; data not shown). Together, these data indicated that hnRNPUL protein recruitment to DNA damage largely correlates with the ability to bind NBS1.
hnRNPUL proteins promote ATR-dependent DSB signaling
To investigate the functional importance of hnRNPUL proteins in the DDR, we examined their requirement for cell viability upon exposure to DSB-inducing agents. Thus, we found that siRNA-mediated depletion of hnRNPUL1 or 2 rendered cells more sensitive to ionizing radiation (IR) or the topoisomerase I poison camptothecin (CPT; Figure 3A) but not to ultra-violet light (data not shown), suggesting that hnRNPUL proteins control cellular responses to DSBs. Nevertheless, depletion of hnRNPUL1 or 2 did not impair IR- or camptothecin-induced focus formation by the checkpoint mediator proteins MDC1, BRCA1 and 53BP1 (Figure 3B). Furthermore, hnRNPUL1 or 2 depletion did not markedly affect phosphorylation of the ATM target Chk2 after IR or camptothecin treatment (Figure 3C and data not shown), thereby demonstrating that hnRNPUL1 and 2 are not crucial for ATM-dependent DNA-damage signaling. Similarly, hnRNPUL protein depletion had no detectable effect on the autophosphorylation activity of DNA-dependent protein kinase (DNA-PK), which promotes DSB repair by NHEJ (data not shown; Meek et al., 2008). By contrast, IR-induced and camptothecin-induced phosphorylation of Chk1, which is targeted by the ATM-related kinase ATR (Cimprich and Cortez, 2008), was significantly impaired in hnRNPUL1-depleted cells (Figure 3C). These results are consistent with previous findings in adenovirus-infected cells, where it was shown that efficient ATR signaling requires hnRNPUL1 (Blackford et al., 2008). Similar defects were observed in cells depleted for hnRNPUL2 (Figure 3C; as shown in Figure S4A, hnRNPUL2 depletion did not impair hnRNPUL1 levels). Together, these data demonstrated that hnRNPUL protein depletion affects ATR- but not ATM-dependent signaling after DSB induction.
Figure 3. hnRNPUL depletion causes hypersensitivity to DSB inducing agents and impairs ATR signaling.

(A) Clonogenic survival of HeLa (top panels) and U2OS cells (lower panel) upon hnRNPUL1 and/or 2 depletion (siUL1, siUL2, siUL1+2) compared to control (siControl, siLuci) after treatment with indicated agents. CtIP depletion is used as positive control. Error bars indicate s.e.m. from 3 independent experiments.
(B) Immunostaining with the indicated antibodies in siRNA treated-HeLa cells exposed to 10 Gy of IR (top) or U2OS cells treated with 1 μM CPT (bottom). Scale bars, 10 μm.
(C) Phosphorylation of proteins upon hnRNPUL1 or 2 depletion analyzed by western-blotting of total extracts prepared from HeLa (top panels) or U2OS cells (bottom panel) at the indicated times after cell exposure to 3 Gy IR or 1 μM CPT. See also Figures S4 and S6.
hnRNPUL proteins stimulate DSB resection and HR
Because ATR-dependent signaling of DSBs involves the production of ssDNA by DNA-end resection, we investigated the potential role of hnRNPUL proteins in this process by monitoring phosphorylation of the ssDNA binding protein RPA. Notably, DNA-damage induced phosphorylation of the RPA2 subunit on Ser-4 and Ser-8 was markedly reduced upon depletion of hnRNPUL1 or 2, as shown by western-blot analysis after camptothecin treatment (Figure 4A) and by immunofluorescence detection of this phosphorylation event at sites of laser-induced damage (Figure 4B; note that DNA-damage induction still took place effectively after hnRNPUL1 or 2 depletion, as evidenced by histone H2AX phosphorylation). Importantly, we found that hnRNPUL protein depletion did not markedly perturb DNA replication as measured by bromo-deoxyuridine (BrdU) incorporation, and did not alter the proportion of cells in S/G2, as revealed by Cyclin A immunostaining (Figure S4B), thus indicating that the impact of hnRNPUL1/2 depletion on RPA phosphorylation was not due to indirect cell cycle effects. Furthermore, the RPA and Chk1 phosphorylation defects and the camptothecin hypersensitivity phenotype of hnRNPUL1-depleted cells were rescued by expression of an siRNA-resistant wild-type hnRNPUL1 construct (WT) but not by mutant constructs that do not bind NBS1 and/or are not effectively mobilized to and from DNA damage sites (Figures 4C and 4D). These results thus underline the functional importance of hnRNPUL-protein interaction with NBS1 and hnRNPUL protein relocalization in response to DNA damage.
Figure 4. hnRNPUL depletion impairs DNA end resection and HR.

(A) Western-Blot analysis of RPA phosphorylation (RPA S4/8-P) 90 min after camptothecin treatment (+CPT) on total extracts from U2OS cells treated with the indicated siRNAs (Luci: control). U2OS cells stably expressing GFP-hnRNPUL2 were used to assess hnRNPUL2 depletion (right panel). Cyclin A levels monitor S/G2 cells and Tubulin is used as loading control.
(B) Immunodetection of phosphorylated RPA (RPA S4/8-P) at laser micro-irradiation sites in U2OS cells treated with the indicated siRNAs.
(C) Western blotting of total extracts from HeLa cells transfected with the indicated siRNAs and plasmids (vector alone or hnRNPUL1 encoding plasmids) after exposure to 1 μmM CPT for 1 h. siCtrl : control ; siUL1#3 targets hnRNPUL1 3’UTR; hnRNPUL1 mutants refer to the scheme below.
(D) Clonogenic survival of HeLa cells transfected as in C in response to CPT. Error bars: s.e.m. from 3 separate experiments.
(E-F) hnRNPUL1 depletion in U2OS (top panels) and HeLa cells (bottom panel) prevents laser-, CPT- and IR-induced accumulation of RPA and ssDNA formation (BrdU) at DNA damage sites (γH2AX), which is normally observed in S/G2 cells (Cyclin A-positive).
(G) Immuno-detection of ATRIP at laser-damage sites (left) and by western-blotting of total extracts (right) from U2OS cells treated with indicated siRNAs. Cells were analyzed 1 h after micro-irradiation in all cases. Scale bars, 10 μm.
(H) HR-mediated gene conversion assay. HR efficiencies are normalized to siLuci. Error bars: s.d. from 3 independent experiments. CtIP depletion is used as a positive control. siRNA efficiencies are shown on the western-blot panel. Proportions of cells in S/G2 on the corresponding FACS profiles were estimated by the Dean/Jett/Fox model with FlowJo software. See also Figures S4 and S5.
Consistent with the observed defect in RPA phosphorylation, accumulation of RPA at sites of laser-, camptothecin- or IR-induced damage was impaired in cells depleted of hnRNPUL1 or 2 (Figure 4E, Figures S4C and S4D and data not shown). Furthermore, by using a method to detect BrdU-substituted cellular DNA under non-denaturing conditions (Raderschall et al., 1999), we found that this direct readout of ssDNA production at DNA damage sites was also inhibited by hnRNPUL1 depletion (Figure 4F). In line with these findings, recruitment of the RPA-binding protein ATRIP to DNA damage sites was also defective in hnRNPUL1-depleted cells (Figure 4G; note that ATRIP levels were unaffected by hnRNPUL1 depletion). Importantly, hnRNPUL protein depletion specifically impaired ssDNA formation and RPA accumulation in response to DNA damage and not in the context of replication stress (Figure S5). Collectively, these results demonstrated that hnRNPUL proteins contribute to ATR-dependent signaling by promoting DSB resection. In accord with this, as shown in Figure 4H, hnRNPUL1 or 2 depletion reduced the efficiency of homologous recombination (HR) by about 50% as measured by a chromosomal I-SceI-mediated gene conversion assay (Pierce et al., 2001). From these data, we conclude that hnRNPUL proteins stimulate DSB resection and thereby promote both ATR signaling and DSB repair by HR.
hnRNPUL1/2 act downstream of CtIP to promote BLM recruitment
To gain insight into how hnRNPUL proteins contribute to DSB resection, we examined whether their depletion affected the DNA-damage recruitment of various factors known to be involved in resection. Consistent with our data indicating that hnRNPUL1/2 recruitment to DNA lesions depends on MRN, depletion of hnRNPUL proteins did not prevent MRN accumulation at sites of laser-induced DNA damage (Figure 5A; note that as shown in Figure S2C, this is also the case in the presence of DRB). Similarly, recruitment and DNA damage-induced phosphorylation of CtIP were unaffected by depletion of hnRNPUL proteins (Figures 5B and 5C). These data, together with our observation that hnRNPUL protein depletion does not further sensitize MRE11- or CtIP-depleted cells to camptothecin (Figure 5D), implied that hnRNPUL proteins act in a resection pathway downstream of MRN and CtIP.
Figure 5. hnRNPUL proteins act downstream of MRN and CtIP and promote BLM recruitment to DSBs.

(A-B) Recruitment of NBS1, MRE11, CtIP (endogenous) or GFP-CtIP to laser-damage sites in cells depleted of hnRNPUL1 or 2 compared to controls (siLuci); cells were fixed 1 h after micro-irradiation.
(C) Western-blot analysis of total extracts from U2OS cells treated with indicated siRNAs. Camptothecin treatment (CPT) was 1 μM for 90 min; Tubulin is used as loading control; P indicates CtIP phosphorylated forms.
(D) Clonogenic survival of HeLa cells treated with the indicated siRNAs (siCtrl: control) in response to CPT. Error bars: s.e.m. from 3 independent experiments. Similar results were obtained upon co-depletion of hnRNPUL2 and CtIP (data not shown).
(E) Recruitment of GFP-EXO1 to laser-damage in U2OS cells treated with indicated siRNAs. Cells were fixed 5-10 min after micro-irradiation. The western-blot panel shows expression of full-length GFP-EXO1 in these cells (detected with anti-GFP antibody).
(F) Clonogenic survival as in D.
(G) Defective recruitment of BLM to laser-damage upon hnRNPUL1 or 2 depletion compared to control (siLuci). U2OS cells were fixed 1 h after micro-irradiation; scale bars, 10 μm.
(H) Clonogenic survival of U2OS cells as in D.
(I) Western-blot analysis of GFP immunoprecipitates from HEK293 cells transiently expressing GFP-hnRNPUL1 or 2 (GFP: mock transfection).
(J) Reciprocal co-immunoprecipitation of endogenous hnRNPUL1 and BLM from HeLa cell extracts. Immunoprecipitation experiments were performed with cell extracts prepared in the presence of benzonase, which digests nucleic acids. Similar results were obtained by immunoprecipitation in the presence of ethidium bromide (data not shown), ruling out an indirect interaction via DNA. See also Figures S3, S4 and S6.
Notably, the EXO1 exonuclease, which functions in DSB resection downstream of CtIP, was also efficiently recruited to DNA damage sites following depletion of hnRNPUL1 or 2 (Figure 5E). However, contrary to what we observed in the context of MRN- or CtIP-depleted cells, the co-depletion of hnRNPUL1 and EXO1 caused cells to be more sensitive to camptothecin than upon single depletion of these factors (Figure 5F; similar results were obtained with hnRNPUL2 depletion, data not shown). These data therefore suggested that EXO1 and hnRNPUL1/2 promote resection through parallel mechanisms. Strikingly, while exploring this possibility, we found that depletion of either hnRNPUL1 or 2 markedly and reproducibly impaired the recruitment of the BLM helicase to sites of laser-induced or camptothecin-induced damage (Figure 5G and Figures S4C and S4E; note that, as shown in Figure 5C, BLM expression levels were unaffected by hnRNPUL1/2 depletion). Because BLM depletion also impairs resection and associated events (Gravel et al., 2008), these data suggested that hnRNPUL proteins promote resection at least in part by mediating BLM recruitment. Consistent with this model, which invokes BLM acting downstream of hnRNPUL proteins at sites of DNA damage, BLM depletion did not affect hnRNPUL1 accumulation at DNA damage sites (Figure S3D), and the effects of BLM and hnRNPUL protein depletion on cellular sensitivity to camptothecin were non-additive (Figure 5H).
Significantly, the resection defect in hnRNPUL1-depleted cells was not overcome by BLM over-expression, as established both by RPA phosphorylation and camptothecin sensitivity assays (Figure S3E and data not shown). BLM recruitment to CPT-induced damage was, however, rescued by expression of an siRNA-resistant wild-type hnRNPUL1 construct but not by the ΔBBS mutant (Figure S4F). This could explain why this mutant does not rescue the camptothecin hypersensitivity or resection phenotypes of hnRNPUL1-depleted cells (Figures 4C and 4D) even though it is recruited to DNA damage sites (Figure 2F). Importantly, the observed resection and BLM recruitment defects induced by hnRNPUL protein depletion were not indirect consequences of inhibited ATR signaling (Blackford et al., 2008) because depletion of ATRIP or ATR to prevent such signaling did not impair RPA or BLM recruitment to DNA damage sites and did not significantly impair the efficiency of HR (Figure S6). Collectively, these results established that hnRNPUL proteins contribute to DNA-end resection downstream of MRN and CtIP, and suggested that this is at least partly mediated by them stimulating BLM recruitment to DSBs. Although it is possible that hnRNPUL proteins act via an intermediary factor or affect the DNA substrate in a way that makes it suitable for BLM engagement, we currently favor a more direct role, because we found that hnRNPUL proteins and BLM co-immunoprecipitate (Figures 5I and 5J).
Discussion
Our work has revealed a function for hnRNPUL proteins in cellular responses to DNA damage that is likely to be distinct from their well-established roles in RNA metabolism (Han et al., 2010). Specifically, our findings indicate that hnRNPUL1 and 2 are recruited to sites of DNA damage in an MRN-dependent and RNA-independent manner, and promote effective DNA resection, a process required for ATR-dependent signaling from DSB sites and for DSB repair by HR. In light of these findings, it will be of interest to examine potential DDR roles for other members of the hnRNP family, several of which have recently been linked to the DDR (Haley et al., 2009). In this regard, we note that the hnRNPUL1/2-related protein hnRNPU (also known as SAF-A; scaffold attachment factor-A) was recently identified as a specific substrate for DNA-PK (Berglund and Clarke, 2009; Britton et al., 2009), which controls DSB repair by NHEJ. The DNA-PK target site is not conserved in hnRNPUL1 or 2 however, suggesting that hnRNPU and hnRNPUL proteins might fulfill distinct and complementary functions in response to DSBs, perhaps by promoting NHEJ and HR, respectively.
Interestingly, the recruitment of hnRNPUL proteins to DNA damage sites was readily detectable in heterochromatin domains that are largely devoid of transcription, and also upon transcription inhibition or RNase treatment, both of which solubilize the RNA-associated pool of hnRNPUL proteins. These data indicate that the function of hnRNPUL proteins in response to DNA DSBs is distinct from their previously described roles in RNA metabolism. We also found that hnRNPUL1 protein mobilization to and from DNA damage sites both involve the RGG domain of the protein, suggesting that these differing responses might be mutually exclusive, depending on whether the RGG domain is bound to RNA or not. Our data also establish that exclusion and recruitment of hnRNPUL proteins are both local responses to DNA damage, albeit with different kinetics: recruitment is transient while exclusion is maintained as long as the damage persists. Notably, hnRNPUL protein recruitment was detectable at sites of laser micro-irradiation but not in IRIF (data not shown), presumably because only a relatively small number of hnRNPUL molecules accumulate at DSBs in a transient manner, which precludes visualization of their recruitment to IRIF. This type of behavior is not without precedent because certain well-characterized DSB-binding factors, such as the Ku heterodimer, also do not form detectable IRIF (Bekker-Jensen et al., 2006; Kim et al., 2005).
Recent studies have characterized DNA-end resection as a two-stage process, with initial limited resection mediated by MRN and CtIP subsequently leading to longer-range processing by mechanisms that involve EXO1 and BLM (Bernstein and Rothstein, 2009; Huertas, 2010; Longhese et al., 2010; Mimitou and Symington, 2009; You and Bailis, 2010). A major outstanding challenge is to define the molecular mechanisms underlying the shift from short- to long-range resection. Our findings suggest that hnRNPUL1 and 2 contribute to this key transition in human cells by promoting BLM, but not EXO1, recruitment to DNA damage sites. This model is further supported by our co-depletion analyses, which have indicated that hnRNPUL proteins operate downstream of MRN and CtIP in a resection pathway likely functioning in parallel to EXO1. Interestingly, although hnRNPUL1 and hnRNPUL2 are related in sequence, our findings indicate that they do not act in a redundant manner, as resection defects are observed when either protein is depleted from cells. In line with this, we have found that the accumulations of hnRNPUL1 and 2 at DNA damage sites are largely inter-dependent and that they are epistatic for their effects on cellular sensitivity to DSB-inducing agents, suggesting that they are recruited as part of the same protein complex and function in the same DSB response pathway.
Although we cannot rule out the possibility that hnRNPUL protein depletion may also affect DSB responses indirectly, as a result of mRNA transcription or splicing misregulation, their association with DSB response factors and their recruitment to DSB sites favor them having a more direct role in controlling cellular responses to DSBs. Indeed, our data on the MRN dependence of hnRNPUL1/2 recruitment to DNA damage sites, which largely correlates with NBS1 binding, suggest that direct MRN binding is a key aspect of hnRNPUL1/2 functioning in the DDR. It is possible that hnRNPUL1/2 control the ability of CtIP and MRN to mediate initial resection events. However, our findings that hnRNPUL1 and 2 interact with BLM and are required for BLM recruitment to DNA damage sites provide support to a model in which hnRNPUL1 and 2 promote DSB resection downstream of CtIP by stimulating BLM accrual (Figure 6). Nevertheless, it is possible that hnRNPUL proteins also promote resection via BLM-independent mechanisms, such as by affecting EXO1-mediated events, although we have not seen an impact of hnRNPUL depletion on EXO1 recruitment to DNA damage sites. Another, not necessarily mutually exclusive, possibility is that hnRNPUL proteins allow DSB resection by removing an inhibitory component(s) from DNA damage sites, potentially by mechanisms related to hnRNPUL protein exclusion from DNA-damage regions. Indeed, such a mechanism would help explain why hnRNPUL proteins are not required in recently developed biochemical assays for DNA resection, which so far consist only of resection activators (Cejka et al., 2010; Nimonkar et al., 2011; Niu et al., 2010).
Figure 6. Model for the involvement of hnRNPUL proteins in DSB responses.
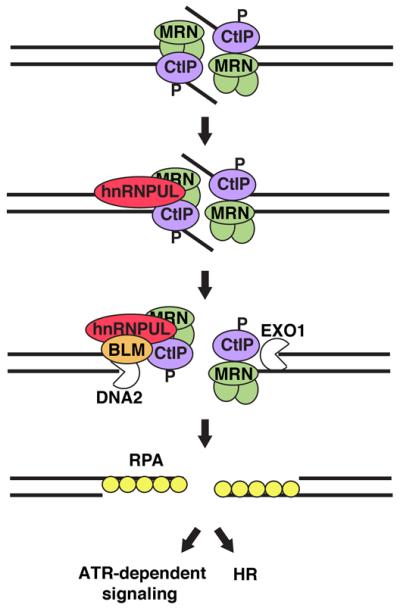
hnRNPUL proteins are recruited to DSBs following MRN and CtIP binding, and contribute to DSB resection by stimulating BLM recruitment. ssDNA resulting from DNA end resection is bound by RPA, promoting ATR-dependent signaling and DSB repair by HR. P: DNA damage-dependent phosphorylation of CtIP; DNA2: DNA replication helicase 2 homolog, nuclease/helicase.
While most known DNA-end resection factors are conserved from yeast to humans, clear counterparts of hnRNPUL proteins are only found in higher eukaryotes. In budding yeast, a physical interaction between the MRN counterpart (MRX) and the BLM ortholog (Sgs1) has been identified, suggesting that MRX may directly recruit Sgs1 to DSBs in this system (Cejka et al., 2010; Chiolo et al., 2005; Niu et al., 2010; Shim et al., 2010), whereas our data demonstrate that additional factors, including hnRNPUL1 and 2, contribute to BLM recruitment in human cells. While the reason for this difference between yeast and human cells is not yet clear, we speculate that it may reflect a need for additional levels of control in mammalian cells, perhaps to better coordinate resection with cell-cycle status, transcription, RNA processing and/or chromatin structure.
Experimental procedures
Cell culture and transfections
Human U2OS, U2OS DR-GFP (M. Jasin), HEK-293, HeLa, A549, LCLs (lymphoblastoid cell line derived from a healthy laboratory donor’s cells transformed with Epstein-Barr virus) and mouse NIH3T3 cells were grown as described in Supplemental material. Cell transfections with plasmid DNA or siRNA duplexes (Table S1) were performed by using Lipofectamine 2000 and Lipofectamine RNAiMax (Invitrogen), respectively, following manufacturer’s instructions. Cells were analyzed 72 h after transfection.
Plasmids
Plasmids are described in Table S2. All constructs were verified by direct sequencing and/or restriction digests. Cloning details and primer sequences (Sigma-Aldrich) are available upon request.
Antibodies
Anti-hnRNPUL1 rabbit polyclonal antibodies were raised against full length GST-hnRNPUL1 as described (Blackford et al., 2008). Other antibodies are commercially available and are listed in Table S3.
DNA damage and drug treatments
IR was delivered by an X-ray generator (Faxitron X-ray Corporation RX-650, 120 kV, 5 mA, dose rate 10 Gy/min). Drug treatments are described in Table S4. For RNase treatment, cells were first permeabilized in 10 mM PIPES pH 7.0, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 0.5% Triton-X-100 for 5 min and then treated with 1 mg/ml RNase A (Roche) in PBS for 10 min at room temperature. Permeabilization and digestion times were both reduced to 2 min to visualize hnRNPUL1 recruitment.
Laser micro-irradiation
Laser micro-irradiation of cells pre-sensitized with BrdU was performed as described (Galanty et al., 2009). When indicated, cell pre-sensitization was omitted and damage was introduced with a multiphoton laser (see Supplemental material for details).
Immunostaining and immunoprecipitation
These procedures are described in Supplemental material.
GST pull-downs
GST pull-downs were carried out as described (Blackford et al., 2008).
Peptide pull-downs and mass spectrometry
Mass spectrometry analyses of proteins pulled-down with biotinylated MDC1 peptides were described in (Chapman and Jackson, 2008). For peptide pull-downs with in vitro translated proteins, 10 μl of in vitro translated products were incubated for 1 h with peptide-bound beads in 0.65 M NaCl, 2 mM EDTA, 25 mM Tris/HCl pH 7.4, 1% NP-40, 10 mg/ml BSA.
Fluorescence-Activated Cell Sorting (FACS)
Cells were fixed in ice-cold 70% ethanol. DNA was stained with 50 μg/ml propidium iodide in PBS containing 0.1% Triton-X-100 and 0.5 mg/ml DNAse-free RNase A (Sigma-Aldrich). Samples were processed on a FACSCalibur flow cytometer equipped with CellQuest software (Becton Dickinson). Results were analyzed using FlowJo software (TreeStar).
Colony forming assays
Survival assays were performed as described (Galanty et al., 2009; see Supplemental material for details).
Homologous recombination assays
I-SceI-mediated gene conversion assays were as described (Galanty et al., 2009; see Supplemental material for details).
Supplementary Material
Figure S1: hnRNPUL1 exclusion from sites of DNA damage is not due to protein degradation and is linked to inhibition of RNA synthesis (related to Figure 2)
Figure S2: Effect of DRB on the cellular response to DSBs and hnRNPUL1 recruitment to DNA damage sites (related to Figure 2)
Figure S3: Role of the MRN complex and BLM in hnRNPUL1 function in response to DNA damage (related to Figures 2 and 5)
Figure S4: Effect of hnRNPUL protein depletion on the S/G2 cell population and on RPA and BLM recruitment to DNA damage sites in S/G2 (related to Figures 3, 4 and 5)
Figure S5: hnRNPUL proteins are not involved in response to replication stress (related to Figure 4)
Figure S6: Inhibition of ATR signaling does not impinge on resection and HR (related to Figures 3 and 5)
Table S1: siRNA sequences
Table S2: Plasmids
Table S3: Primary antibodies
Table S4: Drug treatments
Highlights.
hnRNPUL proteins associate with the DSB sensor complex MRN
hnRNPUL proteins display both exclusion from and MRN-dependent recruitment to DSBs
hnRNPUL proteins stimulate DSB resection, signaling and repair
hnRNPUL proteins promote BLM recruitment to sites of DNA damage
Acknowledgements
We thank L. Bowler (Sussex Proteomics Centre) for mass spectrometry analyses, the imaging facility PICT-IBiSA@BDD of the Institut Curie (Paris, France) for multiphoton laser micro-irradiation, R. Baer, J. Gautier, E. Seto, N. Ellis, J. Falck, M. Lavin and J. Petrini for providing reagents, J. Coates for helping with cell survival experiments, and K. Dry for critical reading of the manuscript. Research in SPJ laboratory is supported by grants from Cancer Research UK (CRUK), the European Union FP7 (GENICA, DDResponse, ERC) and by core infrastructure funding from CRUK and the Wellcome Trust. SPJ receives his salary from the University of Cambridge, supplemented by CRUK. Research in RJG and GSS laboratories is supported by the Medical Research Council and CRUK. The Heinrich-Pette-Institute is supported by the Freie und Hansestadt Hamburg and the Bundesministerium für Gesundheit. This work was supported by a grant from the Deutsche Forschungsgemeinschaft to TD. SEP was funded by the Human Frontier Science Program Organization, Association pour la Recherche sur le Cancer and Marie Curie IEF. ANB and AT were funded by a University of Birmingham Medical School PhD studentship.
References
- Barral PM, Rusch A, Turnell AS, Gallimore PH, Byrd PJ, Dobner T, Grand RJ. The interaction of the hnRNP family member E1B-AP5 with p53. FEBS Lett. 2005;579:2752–2758. doi: 10.1016/j.febslet.2005.03.095. [DOI] [PubMed] [Google Scholar]
- Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, Lukas J. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol. 2006;173:195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund FM, Clarke PR. hnRNP-U is a specific DNA-dependent protein kinase substrate phosphorylated in response to DNA double-strand breaks. Biochem Biophys Res Commun. 2009;381:59–64. doi: 10.1016/j.bbrc.2009.02.019. [DOI] [PubMed] [Google Scholar]
- Bernstein KA, Rothstein R. At loose ends: resecting a double-strand break. Cell. 2009;137:807–810. doi: 10.1016/j.cell.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Bruton RK, Dirlik O, Stewart GS, Taylor AM, Dobner T, Grand RJ, Turnell AS. A role for E1B-AP5 in ATR signaling pathways during adenovirus infection. J Virol. 2008;82:7640–7652. doi: 10.1128/JVI.00170-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton S, Froment C, Frit P, Monsarrat B, Salles B, Calsou P. Cell nonhomologous end joining capacity controls SAF-A phosphorylation by DNA-PK in response to DNA double-strand breaks inducers. Cell Cycle. 2009;8:3717–3722. doi: 10.4161/cc.8.22.10025. [DOI] [PubMed] [Google Scholar]
- Budd ME, Campbell JL. Interplay of Mre11 nuclease with Dna2 plus Sgs1 in Rad51-dependent recombinational repair. PLoS One. 2009;4:e4267. doi: 10.1371/journal.pone.0004267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, Pokharel S, Campbell JL, Kowalczykowski SC. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature. 2010;467:112–116. doi: 10.1038/nature09355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Jackson SP. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008;9:795–801. doi: 10.1038/embor.2008.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiolo I, Carotenuto W, Maffioletti G, Petrini JH, Foiani M, Liberi G. Srs2 and Sgs1 DNA helicases associate with Mre11 in different subcomplexes following checkpoint activation and CDK1-mediated Srs2 phosphorylation. Mol Cell Biol. 2005;25:5738–5751. doi: 10.1128/MCB.25.13.5738-5751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Mantiero D, Lucchini G, Longhese MP. The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J Biol Chem. 2005;280:38631–38638. doi: 10.1074/jbc.M508339200. [DOI] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- Difilippantonio S, Nussenzweig A. The NBS1-ATM connection revisited. Cell Cycle. 2007;6:2366–2370. doi: 10.4161/cc.6.19.4758. [DOI] [PubMed] [Google Scholar]
- Dupre A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, Gautier J. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat Chem Biol. 2008;4:119–125. doi: 10.1038/nchembio.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. Second Edition ASM Press; 2006. [Google Scholar]
- Gabler S, Schutt H, Groitl P, Wolf H, Shenk T, Dobner T. E1B 55-kilodalton-associated protein: a cellular protein with RNA-binding activity implicated in nucleocytoplasmic transport of adenovirus and cellular mRNAs. J Virol. 1998;72:7960–7971. doi: 10.1128/jvi.72.10.7960-7971.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature. 2009;462:935–939. doi: 10.1038/nature08657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley B, Paunesku T, Protic M, Woloschak GE. Response of heterogeneous ribonuclear proteins (hnRNP) to ionising radiation and their involvement in DNA damage repair. Int J Radiat Biol. 2009;85:643–655. doi: 10.1080/09553000903009548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SP, Tang YH, Smith R. Functional diversity of the hnRNPs: past, present and perspectives. Biochem J. 2010;430:379–392. doi: 10.1042/BJ20100396. [DOI] [PubMed] [Google Scholar]
- Hari FJ, Spycher C, Jungmichel S, Pavic L, Stucki M. A divalent FHA/BRCT-binding mechanism couples the MRE11-RAD50-NBS1 complex to damaged chromatin. EMBO Rep. 2010;11:387–392. doi: 10.1038/embor.2010.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol. 2011;193:97–108. doi: 10.1083/jcb.201011083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–168. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol. 2010;17:11–16. doi: 10.1038/nsmb.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Krasieva TB, Kurumizaka H, Chen DJ, Taylor AM, Yokomori K. Independent and sequential recruitment of NHEJ and HR factors to DNA damage sites in mammalian cells. J Cell Biol. 2005;170:341–347. doi: 10.1083/jcb.200411083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kzhyshkowska J, Rusch A, Wolf H, Dobner T. Regulation of transcription by the heterogeneous nuclear ribonucleoprotein E1B-AP5 is mediated by complex formation with the novel bromodomain-containing protein BRD7. Biochem J. 2003;371:385–393. doi: 10.1042/BJ20021281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- Limbo O, Chahwan C, Yamada Y, de Bruin RA, Wittenberg C, Russell P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol Cell. 2007;28:134–146. doi: 10.1016/j.molcel.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd J, Chapman JR, Clapperton JA, Haire LF, Hartsuiker E, Li J, Carr AM, Jackson SP, Smerdon SJ. A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell. 2009;139:100–111. doi: 10.1016/j.cell.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhese MP, Bonetti D, Manfrini N, Clerici M. Mechanisms and regulation of DNA end resection. EMBO J. 2010;29:2864–2874. doi: 10.1038/emboj.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, Lukas J. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol. 2011;13:243–253. doi: 10.1038/ncb2201. [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- Meek K, Dang V, Lees-Miller SP. DNA-PK: the means to justify the ends? Adv Immunol. 2008;99:33–58. doi: 10.1016/S0065-2776(08)00602-0. [DOI] [PubMed] [Google Scholar]
- Melander F, Bekker-Jensen S, Falck J, Bartek J, Mailand N, Lukas J. Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J Cell Biol. 2008;181:213–226. doi: 10.1083/jcb.200708210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. Nucleases and helicases take center stage in homologous recombination. Trends Biochem Sci. 2009;34:264–272. doi: 10.1016/j.tibs.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–362. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu H, Chung WH, Zhu Z, Kwon Y, Zhao W, Chi P, Prakash R, Seong C, Liu D, Lu L, et al. Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature. 2010;467:108–111. doi: 10.1038/nature09318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo B, Gomez-Gonzalez B, Aguilera A. DNA repair in mammalian cells: DNA double-strand break repair: how to fix a broken relationship. Cell Mol Life Sci. 2009;66:1039–1056. doi: 10.1007/s00018-009-8740-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–3242. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raderschall E, Golub EI, Haaf T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc Natl Acad Sci U S A. 1999;96:1921–1926. doi: 10.1073/pnas.96.5.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupnik A, Lowndes NF, Grenon M. MRN and the race to the break. Chromosoma. 2010;119:115–135. doi: 10.1007/s00412-009-0242-4. [DOI] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell. 2010;141:970–981. doi: 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim EY, Chung WH, Nicolette ML, Zhang Y, Davis M, Zhu Z, Paull TT, Ira G, Lee SE. Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 2010;29:3370–3380. doi: 10.1038/emboj.2010.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spycher C, Miller ES, Townsend K, Pavic L, Morrice NA, Janscak P, Stewart GS, Stucki M. Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J Cell Biol. 2008;181:227–240. doi: 10.1083/jcb.200709008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. Embo J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, Classen S, Glover JN, Iwasaki H, Russell P, Tainer JA. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell. 2009;139:87–99. doi: 10.1016/j.cell.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Luo K, Lou Z, Chen J. MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc Natl Acad Sci U S A. 2008;105:11200–11205. doi: 10.1073/pnas.0802885105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z, Bailis JM. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010;20:402–409. doi: 10.1016/j.tcb.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: hnRNPUL1 exclusion from sites of DNA damage is not due to protein degradation and is linked to inhibition of RNA synthesis (related to Figure 2)
Figure S2: Effect of DRB on the cellular response to DSBs and hnRNPUL1 recruitment to DNA damage sites (related to Figure 2)
Figure S3: Role of the MRN complex and BLM in hnRNPUL1 function in response to DNA damage (related to Figures 2 and 5)
Figure S4: Effect of hnRNPUL protein depletion on the S/G2 cell population and on RPA and BLM recruitment to DNA damage sites in S/G2 (related to Figures 3, 4 and 5)
Figure S5: hnRNPUL proteins are not involved in response to replication stress (related to Figure 4)
Figure S6: Inhibition of ATR signaling does not impinge on resection and HR (related to Figures 3 and 5)
Table S1: siRNA sequences
Table S2: Plasmids
Table S3: Primary antibodies
Table S4: Drug treatments