

Abstract
Classical swine fever is the most insidious and devastating disease of pigs and wild boars. The virus is closely related to the other members of the genus Pestivirus. The outbreak recorded in Mizoram, India was strategically important as the state shares porous international boundary with East Asian countries. Both immunodiagnostic and molecular techniques were used to confirm the involvement of Classical swine fever virus (CSFV) in this outbreak. Sandwich ELISA and direct FAT could detect CSFV in the tissue samples. RT-nPCR specifically amplified E2 and 5’NTR product of 271 bp. Phylogenetic analysis showed, that the Mizoram isolate (MZ4/69) was very close to the Chinese strain Shimen-HVRI (93.0%) rather than other Indian isolate (CSF-30-03). Present study provides a valuable sequence based molecular data on Indian isolate of CSFV and will be useful in investigation on transmission of such disease from neighbour countries.
Keywords: Classical swine fever virus ELISA, RT-PCR, Sequence analysis
Classical swine fever virus (CSFV) or hog cholera is a highly contagious disease of swine with acute, chronic or persistent infections [14]. The causative virus is a member of the genus Pestivirus of the family Flaviviridae and is closely related both antigenically and structurally to the bovine viral diarrhoea virus (BVDV) and border disease virus (BDV) [9]. Polyclonal antibody based diagnosis fail to distinguish between BVDV and BDV because of poorly defined antigenicity. Type-specific monoclonal antibodies, particularly those directed against E2 could confirm CSFV [7]. Again, PCR using primers directed towards relatively conserved areas of CSFV genes are the best choice to amplify and characterize virus isolates [20]. Genotyping and phylogenetic analysis of Indian isolates of classical swine fever have not been carried out extensively. In this report, outbreaks of CSF were recorded in the Mizoram state of India. The state shares international boundaries with Myanmar and Bangladesh and these porous international boundaries facilitate for migration of pigs from neighbouring countries. Confirmation of the outbreaks and molecular characterization of the virus isolates provide valuable information about epidemiology of CSF in the North Eastern Region of India.
The state of Mizoram, India recorded a high mortality in pigs during the period September 2005 to April 2006. Outbreak of CSF was initially reported at Champai, an Indo-Myanmar boarder district of Mizoram and later it extended to Lunglei and Aizwal district. The disease affected approximately 2,500 pig populations in three districts. Animals of all age groups were affected. Breeds involved in the outbreaks were Large-white Hampshire X local cross and non-descript breeds. Classical clinical symptoms and post-mortem changes of CSFV were recorded in affected pigs and postmortem tissue samples like spleen, tonsil and kidney were collected for confirmation of the CSFV. For detection of viral antigen was done by sandwich ELISA [5] with modification [17] and by direct FAT [1] were conducted.
Classical swine fever viral nucleic acid was amplified following the reverse transcriptase nested polymerase chain reaction (RT-nPCR) technique in spleen, tonsil and kidney samples collected from affected pigs. Total RNA was extracted from tissue samples and standard lapinized virus using RNA-TRI reagent (Ambion) according to manufacturer’s specification. The RT-PCR was standardized using lapinised CSF vaccine virus RNA to amplify parts of E2 and 5′NTR gene segments which served as positive control. The RNA extracted from spleen sample of healthy day old piglet was used as a negative control. Complementary DNA was synthesized using 6 μl of viral RNA and 1 μl of random primer (50 ng/μl) to a total reaction volume of 13 μl. The contents were incubated in a thermal cycler (Perkin Elmer, USA) at 70°C for 5 min and 25°C for 10 min. The reverse transcription was carried out by adding annealed RNAs to the reverse transcription mixture consisting of 4 μl of 5× RT buffer, 1 μl RNAase inhibitor (40 U/μl), 1 μl 10 mM dNTPs mixture, 1 μl Moloney murine leukemia virus reverse transcriptase (200 U/μl). The reverse transcription was carried out at 42°C (1 h) and inactivation of enzyme at 70°C (10 min). Using the PCR primers I & II, V & VI (Table 1); the E2 and 5′NTR genomic regions of CSFV were amplified. The PCR mix in a total volume of 50 μl containing 5.0 μl of 10× PCR buffer, 3.0 μl of 25 mM MgCl2, 1.0 μl of Prim I E2-F (20 pmol/μl), 1.0 μl of Prim II E2-R (20 pmol/μl), 1.0 μl of 10 mM dNTPs, 5.0 μl of cDNA, 0.5 μl (5 U/μl) of Taq DNA polymerase (Qiagen) and 33.5 μl of nuclease free water was subjected to amplification in a thermal cycler at 95°C for 2 min (1 cycle), 95°C for 30 s; 56°C for 45 s; 72°C for 1 min (34 cycles) and 72°C for 1 min. The procedure for the amplification of 5′NTR was similar to that used in the amplification of E2 gene region, except that the annealing temperature was kept at 50°C and primers used were Prim V and VI. For nested-PCR (nPCR) of E2 gene and 5′NTR regions, the procedure was essentially the same except that the template cDNA was replaced by 5 μl of primary PCR amplicons and also the annealing temperature was kept at 58°C for E2 gene and 56°C for 5′NTR. The primers used (Table 1) were internal primers (Prim III & IV) for E2 region gene and (Prim VII & VIII) for 5′NTR gene. The nested-PCR amplified products were analyzed on 1.7% agarose (Amresco) containing ethidium bromide in 0.5× TBE buffer. Electrophoresis was done at 78 V for 1 h 30 min. The ethidium bromide stained DNA bands were visualized using a UV transilluminator (Kodak, biostep, Germany) and compared with 100 bp DNA markers.
Table 1.
Description of the primers used in the present study
| Primer designation | Genomic region | Primer sequence (5′–3′) | Nucleotide position | Reference |
|---|---|---|---|---|
| Prim I | E2-F | AGRCCAGACTGGTGGCCNTAYGA | 2228–2250 | [13] |
| Prim II | E2-R | TTYACCACTTCTGTTCTCA | 2898–2880 | [13] |
| PrimIII (Internal) | E2-F | TCRWCAACCAAYGAGATAGGG | 2477–2497 | [13] |
| Prim IV (Internal) | E2-R | CACAGYCCRAAYCCRAAGTCATC | 2748–2726 | [13] |
| Prim V | 5′NTR-F | CTAGCCATGCCCWYAGTAGG | 94–113 | [10] |
| Prim VI | 5′NTR-R | CAGCTTCARYGTTGATTGT | 514–496 | [10] |
| Prim VII (Internal) | 5′NTR-F | AGCTCCCTGGGTGGTCTA | 146–163 | [10] |
| Prim VIII (Internal) | 5′NTR-R | TGTTTGCTTGTGTTGTATA | 417–399 | [10] |
Nested PCR products generated were subjected to sequencing after purification using QIAquick PCR purification kit (QIAGEN). Sequencing was carried out on automated sequencer (ABI 3100 Genetic Analyzer) using BigDye terminator kit (ABI). Both forward and reverse sequencing was performed on at least two independently generated PCR products. Both E2 and 5′NTR sequences after converting into ‘EditSeq’ files were analysed using ‘MegAlign’ programme of ‘Lasergene’ software (DNASTAR Inc.) by Clustal W method. For comparison various reported CSFV sequences of field isolates, vaccine strain of India and other parts of world were downloaded from GenBank. (Accession numbers: Shimen HVRI-AY775178, CSF30-30-AJ605594, IVRI vac-AJ605598, TZ-05-EF683639, NU4/97-dp-AJ312859, NU1/98-wb-AJ312853, Bresciax-CSF0929, Alfort-CSF0902, Weybridge-X71780, IVRI Vac-AJ605591, CSF30-03-AJ605586, CW2002S-DQ522299).
Highest mortality in young animals was observed during the outbreaks. Percent mortality in pre-weaned and weaned piglets was 50% while 10% in pigs above 1 year of age. The disease showed a variable course and clinical symptoms. The affected animals exhibited clinical signs of inappetance, high fever and constipation followed by diarrhea and haemorrhagic patches in skin, characteristic signs of classical swine fever. There were rashes in the belly, medial aspects of thigh and on the base of ears. Necrotic lesions developed on skin in later stages. Affected pigs showed staggering gait. Pregnant sows either aborted or delivered stillborn fetuses. Agalactia was most common sequele of farrowed sows. On post-mortem examination, pathological changes recorded were button ulcer, extensive haemorrhage in intestine, mesenteric lymph nodes, pharyngeal lymph node, and in palatine tonsils. Kidneys and urinary bladder showed haemorrhagic spots.
The variation in clinical symptoms as well as in the rate of mortality could be attributed to the host resistance factors in different age groups of animals. In field outbreaks, various workers have reported about variation of clinical signs, rate of mortality and reproductive failures among affected pigs [4, 11, 12].
A total of ten tissue samples comprising of spleen (4), tonsils (3) and kidney (3), were screened for presence of CSFV by S-ELISA and direct FAT (Fig. 1). All tissue samples were found positive for CSFV both in ELISA as well as in FAT. Representative tissue samples (spleen-2, kidney-2, tonsil-2) subjected for RT-nPCR yielded expected 271 bp products of target E2 and 5′NTR region of the CSFV (Fig. 2a and b).
Fig. 1.
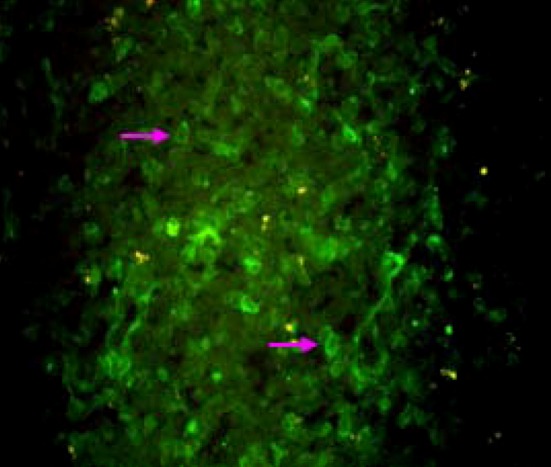
CSFV-infected lymphoid cells of tonsil cryo-section showing bright green fluorescent in FAT (×400)
Fig. 2.
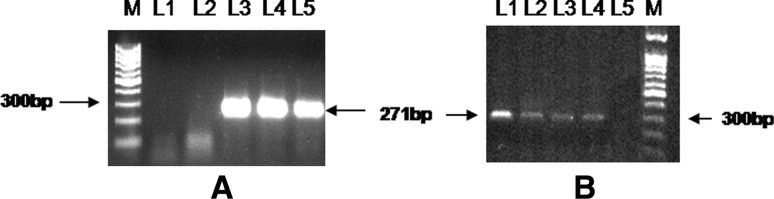
Agarose gel electrophoresis showing amplification of 5’NTR (a) and E2 (b) region of CSFV from infected tissue samples of Pig. In a M-100 bp DNA ladder, L1-NTC, L2-negative sample, L3–L4-positive samples, L5-positive control (LPBP) In b M-100 bp DNA ladder, L1-positive control, L3–L4-positive samples, L5-NTC
Antibody based and RT-nPCR amplification of 5′NTR and E2 region of CSFV confirmed the involvement of the CSF virus in the present outbreaks. Detection of CSFV by RT-PCR in clinical samples was reported by Barkha et al. [3] and in PK-15 cell line by Dhar et al. [6]. Although polyclonal antibody based diagnostics (s-ELISA, FAT) could detect up to Pestivirus group, but the primer sets used for PCR were specific to the nucleotides encoding E2 epitope of CSFV [15]. The Mizoram state experienced repeated outbreaks of CSF since long back but recorded evidence available is early as 1988 by Verma [19]. The state is strategically located near to the international border. It is evident from the annual report of the Govt. of Mizoram [2] that 11,613 pigs either migrated or were imported to the Mizoram from Myanmar, Assam and Bangladesh. The classical swine fever is endemic in Myanmar [8]. There is maximum possibility for spreading of transboundary diseases including CSF to India from these south East Asian countries.
Sequence analysis of E2 region revealed Indian field isolate MZ4/69 having four unique nucleotide changes among 128-nucleotide sequence compared to majority consensus (Fig. 3). These changes were 31C-T, 32A-T, 38T-C and 79C-G. Percent nucleotide similarity of isolate MZ4/69 was maximum (96.1%) with European CSFV strain Weybridge and was minimum (73.4%) with Chinese isolate TZ-05.
Fig. 3.

Alignment of 128 nucleotide sequences of Mizoram CSFV isolate and reference CSF viruses in E2 region. Sequences were aligned using CLUSTAL W method. Residues identical to the consensus are indicated by dots
With lapinised vaccine strain IVRI-Vac, it had percent similarity of 89.1% and with Indian field isolate 30-03 was 86.7%. The percent similarity of MZ4/69 isolate with European isolate Brescia was 85.9%. Phylogenetic analysis (Fig. 4) based on 128 nucleotide E2 gene sequence revealed grouping of MZ4/69 isolate close to Weybridge strain [15] and most distant was Chinese isolate TZ-05. Indian field isolate CSF 30-03 as well as vaccine strain Vac-IVRI were also distantly placed from Indian field isolate MZ4/69. Another Chinese isolate Shimen-HVRI was also closely placed with Indian field isolate MZ4/69 in the phylogenetic tree.
Fig. 4.

Dendrogram showing relationship of Mizoram CSFV isolate with other CSF viruses in E2 region. Sequences are aligned using CLUSTAL W method of DNASTAR and phylogenetic tree was obtained by Mega align software package
Sequence analysis based on 114 nucleotides of 5’NTR showed five unique changes in MZ4/69 isolate as compared to majority consensus (Fig. 5). These changes were 26C–A, 37A–G, 51G–T, 61G–C and 71A–G. Nucleotide similarity of MZ4/69 was maximum (93.8%) with CSFV Alfort strain and was minimum of (88%) with Indian field isolate 30-03.
Fig. 5.

Alignment of 114 nucleotide sequences of Mizoram CSFV isolate and reference CSF viruses in 5′NTR. Sequences were aligned using CLUSTAL W method. Residues identical to the consensus are indicated by dots
The 5′NTR sequence similarity of MZ4/69 isolate with vaccine strain Vac-IVRI was 90.4%. Indian field isolate MZ4/69 was also closer to Chinese isolate Shimen-HVRI and Korean isolate CW2002S with a percent similarity of 93.0 and 92.9%, respectively. Phylogenetically (Fig. 6) also, Indian field isolate MZ4/69 from Mizoram was most distinct due to its separate lineage in the phylogenetic tree, whereas vaccine strain Vac-IVRI and earlier reported Indian field isolate CSFV30-03 were most close to CSFV strain Alfort.
Fig. 6.

Dendrogram showing relationship of Mizoram CSFV isolate with other CSF viruses in 5′NTR. Sequences are aligned using CLUSTAL W method of DNASTAR and phylogenetic tree was obtained by Mega align software package
Analysis of the 5NTR, E2 region and NS5B resulted similar resolution to classify CSFV into three major groups and their sub-groups. Group 1 and its three sub-groups (1.1, 1.2, 1.3) comprise most of the historical strains [13, 15] distributed in most regions of the world. Group 2 containing current viruses, which segregate into sub-groups 2.1, 2.2 and 2.3 cause major epidemics in Europe and in Southeast Asia [15]. Disparate strains of CSFV classified under group 3 are distributed in Taiwan, Korea, Japan, and Thailand and in UK [15, 16, 18]. Our analyses based on E2 and 5NTR nucleotide sequences clearly showed that MZ4/69 strain falls in historic group 1 under the sub-group 1.1 as it showed close relationship with Weybridge (96.1%) and Alfort (93.8%). Interestingly, based on E2 sequence analysis present isolate (MZ4/69) maintains a little distant relationship with Indian isolate CSF 30-03 (86.7%) and with historic Brescia strain (85.9%), which is grouped as 1.2 [13]. It is not appropriate to draw a conclusive evolutionary trend because of paucity of sequence data on Indian isolates of CSFV. Tracing the origin of the Mizoram outbreaks it appears that this isolate (MZ4/69) is very close to the Chinese strain Shimen-HVRI (93.0%) rather than other Indian isolate (CSF 30-03). Geographically, the state Mizoram is close to Myanmar and China. Therefore, along with migration of animals CSFV could spread from these neighbour countries to Mizoram. However, extensive molecular epidemiology on CSFV isolates from different regions of India could give clear picture on evolutionary trend, distribution of various groups and sub-groups of CSFV in this country.
Acknowledgments
The authors are thankful to the Department of Biotechnology, Govt of India for providing financial help under the project entitled “Molecular and immunological characterization and phylogenetic analysis of Indian isolates of classical swine fever virus” to carryout the research.
References
- 1.Anonymous. European Union diagnostic manual on swine fever. Hanover: Germany; 2002.
- 2.Anonymous. Annual report. Animal Husbandry and Veterinary Department, Govt of Mizoram: Mizoram; 2003–2005.
- 3.Barkha R, Tiwari AK, Barman NN, Chaturvedi U, Ravindra PV, Desai G, Subudhi PK, Tiwari S, Kumar S. Detection of classical swine fever virus in various types of clinical samples by RT-PCR. Indian J Virol. 2009;20(1):4–8. [Google Scholar]
- 4.Barman NN, Nath AJ, Hazarika MP, Barman B, Thakuria D. Outbreak of classical swine fever in regularly vaccinated pig herd. Indian J Comp Microbiol Immunol Infect Dis. 2003;24(1):89–90. [Google Scholar]
- 5.Depner KR, Hinrichu U, Bickhardt K, Greiser Wilkie I, Pohlenz J, Moennig LeissB. Evaluation of the enzyme-linked immunosorbent assay for the rapid screening and detection of classical swine fever virus antigens in the blood of pigs. Vet Rec. 1995;14:677–689. doi: 10.20506/rst.14.3.863. [DOI] [PubMed] [Google Scholar]
- 6.Dhar P, Rai A, Verma R. Adaptation of classical swine fever virus (lapinized strain) in PK-15 cells and confirmed by reverse transcription-polymerase chain reaction (RT-PCR) and fluorescent antibody technique (FAT) Indian J Animal Sci. 2008;78(2):135–137. [Google Scholar]
- 7.Edwards S, Moennig V, Wensvoort G. The development of international reference panel of monoclonal antibodies for the differentiation of hog cholera virus from other pestiviruses. Vet Microbiol. 1991;29:101–108. doi: 10.1016/0378-1135(91)90118-Y. [DOI] [PubMed] [Google Scholar]
- 8.Edwards S, Fukusho A, Lefevre PC, Lipowski A, Pejsak Z, Roehe P, Westergaard J. Classical swine fever: the global situation. Vet Microbiol. 2000;73:103–119. doi: 10.1016/S0378-1135(00)00138-3. [DOI] [PubMed] [Google Scholar]
- 9.Francki RIB, Faquet CM, Knudson DL, Brown F. Fifth report of the international committee on taxonomy of viruses. Arch Virol. 1991;2:223–233. [Google Scholar]
- 10.Greiser-Wilke I, Depner K, Fritzemeier J, Haas L, Moennig V. Application of a computer program for genetic typing of classical swine fever virus isolates from Germany. J Virol Methods. 1998;75:141–150. doi: 10.1016/S0166-0934(98)00109-8. [DOI] [PubMed] [Google Scholar]
- 11.Harkness JW. Classical swine fever and its diagnosis. A current view. Vet Rec. 1985;116:288–293. doi: 10.1136/vr.116.11.288. [DOI] [PubMed] [Google Scholar]
- 12.Jindal N, Sharma PC, Mittal D, Tiwari AK, Narang G, Shukla CL. Occurrence of swine fever in vaccinated piggery units in Haryana: detection by RT-PCR. Indian J Virol. 2008;19(1):44–46. [Google Scholar]
- 13.Lowings P, Ibata G, Needham J, Paton D. Classical swine fever virus diversity and evolution. J Gen Virol. 1996;77(6):1311–1321. doi: 10.1099/0022-1317-77-6-1311. [DOI] [PubMed] [Google Scholar]
- 14.Moennig V. Introduction to classical swine fever: virus, disease and control policy. Vet Microbiol. 2000;73:93–102. doi: 10.1016/S0378-1135(00)00137-1. [DOI] [PubMed] [Google Scholar]
- 15.Paton DJ, McGoldrick A, Greiser-Wilke I, Parchariyanon S, Song JY, Liou PP, Stadejek T, Lowings JP, Bjorklund H, Belak S. Genetic typing of classical swine fever virus. Vet Microbiol. 2000;73:137–157. doi: 10.1016/S0378-1135(00)00141-3. [DOI] [PubMed] [Google Scholar]
- 16.Sakoda Y, Ozawa S, Damrongwatanopokin S, Sato M, Ishikawa K, Fukusho A. Genetic heterogenicity of porcine and ruminant viruses mainly isolated in Japan. Vet Microbiol. 1999;65:75–86. doi: 10.1016/S0378-1135(98)00284-3. [DOI] [PubMed] [Google Scholar]
- 17.Singh NK. Immuno and histopathological diagnosis of Hog Cholera in field outbreaks. MVSc Thesis, Assam Agricultural University, Guwahati, India; 2005.
- 18.Singh VK, Saikumar G, Bandyopadhyay SK, Paliwal OP. Phylogenetic analysis of classical swine fever virus (CSFV) by cloning and sequencing of partial 5′ non translated genomic region. Indian J Animal Sci. 2004;74(11):1093–1097. [Google Scholar]
- 19.Verma ND. An outbreak of swine fever in Mizoram state. Indian J Animal Sci. 1988;58(7):774–776. [Google Scholar]
- 20.Vilcek S, Herring AJ, Herring JA, Nettleton PF, Lowings JP, Paton DJ. Pestiviruses isolated from pigs, cattle and sheep can be allocated into at least three genogroups using PCR and restriction endonuclease analysis. Arch Virol. 1994;136:309–323. doi: 10.1007/BF01321060. [DOI] [PubMed] [Google Scholar]