

Abstract
Hepatitis C virus (HCV) is a major public health problem and a leading cause of chronic liver disease. An estimated 180 million people are infected worldwide. In this study, we developed a loop-mediated isothermal amplification (LAMP) assay for rapid detection of HCV genomic RNA and compared the sensitivity of LAMP with nested-PCR. A total of 30 blood samples from HCV-infected patients were analyzed with six primers targeting conserved sequences of the HCV 5′UTR within 70 min, under isothermal conditions at 62 °C. Then, visualized by gel electrophoresis with ethidium bromide staining and detected by the naked-eye after adding SYBR Green I. All samples positive for HCV by nested PCR were confirmed by LAMP method. When visualized by gel electrophoresis and ethidium bromide staining, the HCV LAMP assay products appeared in a ladder pattern, with many bands of different sizes. The HCV LAMP product could also be detected by the naked-eye after adding SYBR Green I to the reaction tube and observing a color change from orange to green in positive samples. The HCV LAMP had the same sensitivity as a nested-PCR assay, the detection limit for the both systems were found to be 10 copies/mL of HCV RNA. The LAMP assay reported here is superior for rapid amplification, simple operation, and easy detection and will be useful for rapid and reliable clinical diagnosis of HCV in areas with limited resources, such as developing countries.
Keywords: HCV, Loop-mediated isothermal amplification, Nested-PCR
Introduction
Hepatitis C virus belongs to the Flaviviridae family and is the only member of the Hepacivirus genus [2]. Monitoring by the World Health Organization has suggested that a minimum of 2–3 % of the world’s population is chronically infected with HCV [1, 23]. HCV infection is a major cause of chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma [2]. Therapeutic options are improving, but are still limited, and no vaccine is available to prevent new infections [21, 22]. The commercially-available diagnostic tests are based on enzyme immunosorbent assays (EIA) for the detection of HCV-specific antibodies; recombinant immunoblot assays (RIBA) are often used as a supplemental test for confirmation of a positive EIA result. Both types of single-use kits represent significant monetary costs for hospitals and private clinics [20]. Testing for circulating HCV by genomic sequence amplification (e.g. polymerase chain reaction, and branched DNA assay) has been successfully utilized for confirmation of serological results, as well as to assess the effectiveness of antiviral therapy [4, 21, 24]. However, both the immunoassay and PCR assays require expensive equipment [3, 18, 19, 21]. There is a need for the development of sensitive, simple, cost-effective, and rapid diagnostic techniques for HCV detection. In 2000, Notomi et al. [17] reported a novel nucleic acid amplification method, termed the loop-mediated isothermal amplification (LAMP), that amplifies target sequences with high specificity, efficiency, and speed under isothermal conditions [5, 10]. The only equipment needed for the LAMP reaction is a regular laboratory water bath or a heat block that furnishes a constant temperature of 62–65 °C [16, 17]. Thus, this method could potentially be a valuable tool for the rapid diagnosis of infectious diseases in both commercial and hospital laboratories of developing countries [6, 7, 11, 15]. In this study, we developed the LAMP method for the detection of the Hepatitis C virus genome and compared the sensitivity of LAMP with those of nested-PCR using clinical samples.
Materials and Methods
Clinical Samples
A total of 30 blood samples were obtained from patients who were clinically diagnosed with HCV infection between February 2009 and June 2010. The samples were serologically diagnosed using an HCV immunoglobulin G EIA kit (DiaPro, Milano, Italy) according to the manufacturer’s instructions. In 21 samples HCV genotypes were determined based on the HCV genome sequencing of 5′-UTR regions by the restriction fragment length polymorphism (RFLP) method. All of the samples were transported to the laboratory under a strict cold chain and stored at −70 °C until further investigation.
RNA Extraction and cDNA Synthesis
Genomic viral RNA was extracted from samples by using a RNX® (plus) buffer (CinaGen Co., Tehran, Iran) and following the manufacturer’s protocol. Complementary DNA (cDNA) was synthesized from 5 μL of eluted RNA using random hexamer primers and the Revert Aid First Strand cDNA kit (K1621; Fermentas, Germany) according to the manufacturer’s instructions. The cDNA product was subjected to nested-PCR and LAMP.
Nested-PCR
The sensitivity and specificity of the LAMP method were compared to the nested-PCR technique by using the same templates. Primers used in the nested-PCR assay were constructed according to Kazemi et al. [14]. The location and sequences of nested-PCR primers are given in Table 1. In brief, the PCR reaction was performed in a total volume of 25 μL containing 20 μL PCR reaction buffer, 1 μL out-F HCV primer, 1 μL out-R HCV primer, 1.5 U SmarTaq DNA polymerase (CinaGen Co., Tehran, Iran) and 2 μL cDNA to amplify a 296-bp fragment. In the second round of PCR, 2 μL of undiluted first-round PCR product was added to 20 μL of a PCR mixture similar to the first but using In-F and In-R primers to amplify a 253-bp fragment. The cycle conditions were identical for both rounds, and were: denaturation at 95 °C for 1 min; 30 cycles of amplification at 94 °C for 1 min, 60 °C for 1 min, and 72 °C for 1 min; and a final extension at 72 °C for 5 min. The reactions were carried out in a Mastercycler Gradient Thermocycler (Eppendorf, Hamburg, Germany). PCR products were visualized using 2 % ethidium bromide (EtBr)-stained agarose gel electrophoresis.
Table 1.
Details of nested-PCR and LAMP primers for detection of Hepatitis C virus genomic RNA
| Primer name | Length | Genome position | Sequence | |
|---|---|---|---|---|
| PCR primer | Out-F | 19 | 6–24 | 5′-CTGTGAGGAACTACTGTCT-3′ |
| Out-R | 21 | 282–302 | 5′-GGTGCACGGTCTACGAGACCT-3′ | |
| In-F | 19 | 24–42 | 5′-TTCACGCAGAAAGCGTCTA-3′ | |
| In-R | 21 | 257–277 | 5′-GGGCACTCGCAAGCACCCTAT-3′ | |
| LAMP primer | F3 | 19 | 88–106 | 5′-TCCCGGGAGAGCCATAGTG-3′ |
| B3 | 21 | 254–274 | 5′-CACTCGCAAGCACCCTATCAG-3′ | |
| FIPa | 43 | 148–166 108–127 |
5′-GATCCAAGAAAGGACCCGGTTTTTCTGCGGAACCGGTGAGTAC-3′ | |
| BIPa | 43 | 179–197 232–251 |
5′-CCTGGAGATTTGGGCGTGCTTTTAGTACCACAAGGCCTTTCGC-3′ | |
| Loop F | 17 | 129–145 | 5′-TCATCCTGGCAATTCCG-3′ | |
| Loop B | 18 | 202–222 | 5′-GCGAGACTGCTAGCCGAG-3′ |
aFIP (F1c-TTTT-F2) consists of F2 and F1c sequences as shown on Fig. 1. BIP (B1c-TTTT-B2) consists of B2 and B1c sequences. B2c sequence complementary to B2, F1c sequence complementary to F1
Primer Design for LAMP Reaction
The oligonucleotide primers used for LAMP amplification of HCV were designed from 5′UTR region sequences obtained from GenBank (Accession No: GQ418245). First, the 5′UTR sequences from different genotypes were aligned by the Mega 4 software (http://www.megasoftware.net) to identify conserved regions. From the aligned sequences, the potential target region of the 187-bp corresponding to genome positions 88–274 was selected, and LAMP primers were designed by the GeneRunner software (http://www.generunner.net/). A set of six primers (two outer, two inner, and two loop) were designed. Primer specificity was tested using the Mega 4 software against the collected HCV complete genome genotypes (available in the GenBank-EMBL database, www.EBI.ac.uk) as well as the BLAST program (available at http://www.ncbi.nih.nlm.gov). The detailed sequences of primers used for amplification of HCV are shown in Fig. 1.
Fig. 1.

Location of the LAMP primers in 5′UTR region of the Hepatitis C virus genome RNA. This sequence represents positions 88–274 in the Hepatitis C virus GenBank sequence (Accession No: GQ418245). The primers included two outer primers (F3 and B3), two inner primers (Forward Inner Primer, FIP, which contains a sequence complementary to F1 linked with the F2 sequence; Backward Inner Primer, BIP, which includes a B1 sequence linked with the sequence complementary to B2), as well as two loop primer (loop F and loop B which are located between F1 and F2 and between B1 and B2, respectively)
LAMP Reaction
The LAMP reaction was carried out in a 25 μL reaction mixture composed of 1.6 μM each of FIP and BIP, 0.2 μM each of F3 and B3, 0.4 μM each of loop-F and loop-B, 2.5 μL Thermopol buffer (20 mM Tris-HCl (pH 8.8), 10 mM KCl, 10 mM (NH4)2SO4, 9 mM MgSO4 and 0.1 % Triton X-100), 1.4 mM deoxynucleoside triphosphate mix, 1 M Betain (Sigma-Aldrich, St. Louis, MO, USA), 3 μL of target cDNA, and distilled water. The mixtures were heated to 95 °C for 5 min, and then chilled on ice prior to addition of 8 U of Bst DNA polymerase large fragment (New England Biolabs). Immediately after addition of the polymerase, the mixture was incubated at 62 °C for 70 min in a heating block (SBH 130D; Stuart Scientific, Staffordshire, UK) and then heated at 80 °C for 10 min to terminate the reaction. Samples which the copy numbers of virus RNA had previously been quantified by real-time PCR were used as positive controls. LAMP products were subjected to 2 % agarose gel electrophoresis stained with EtBr and visualized under UV light. In addition, 1 μL of SYBR Green I (Invitrogen lot: 49743A) diluted 1:10 was added directly to the LAMP products. The solution turned green if LAMP reaction products were present, otherwise it remained orange; coloration was evaluated under natural light and UV light (302 nm; via handheld UV torch lamp).
Optimization of HCV LAMP Reaction Conditions
Varying concentrations of the FIP, BIP, F3, B3, loop-F, and loop-B primers were evaluated, as well as use of only four of the primers (excluding loop-F and loop-B). Time of reaction was varied from 45 to 90 min in order to determine the optimal detection time for HCV genomic RNA.
Specificity and Sensitivity of the HCV LAMP Reaction
Specificity of LAMP primers for HCV was tested against positive and negative controls and for nonspecific attachment to other viruses, including CMV, HBV and HSV-1. Sample for which the copy numbers of virus RNA had previously been determined by the quantitative Cobas Amplicor HCV Monitor Test (Roche Diagnostic Systems, Pleasanton, CA, USA), was used for analysis of the sensitivity of LAMP. HCV RNA was extracted from sample containing 104 copies/mL and serially diluted at a ratio of 1:10 (104, 103, 102, 10, 1). The results were compared with those obtained from nested-PCR.
Results
Optimization of the HCV LAMP Reaction
The success of LAMP amplification relies on the specificity of designed primer sets. The primers used in this study were selected to target the highly conserved regions of the 5′UTR of HCV, based upon multiple sequence alignment of all of the known circulating genotypes by using the Mega4 software. Our primers can detect a large part of the HCV genotype (available in the GenBank-EMBL database, www.EBI.ac.uk). The details of the primers, with regard to their positions in the genomic sequences, are shown in Table 1, and a schematic representation of the LAMP primer design is depicted in Fig. 1. The LAMP reaction was performed using nested-PCR positive RNA samples as templates to determine the optimal primer combination and duration of reaction. The amplicon was formed using either four or six primers, as described. The reaction with four primers produced a detectable LAMP product after 90 min at 62 °C, while reaction with six primers generated detectable product after 70 min. When 1.5 U of Taq DNA polymerase was used to supplement the reaction, the amplification product was detected 20 min earlier (at 50 min after reaction initiation).
Detection of LAMP Products by Alternate Methods
As observed by agarose gel electrophoresis, the LAMP assay was able to amplify the 187-bp target sequence of the 5′UTR of HCV at 62 °C after 70 min of reaction time. The amplification pattern presented as ladder-like, due to the formation of a mixture of stem-loop DNAs with various stem lengths and cauliflower-like structures with multiple loops formed by annealing between alternately inverted repeats of the target sequence in the same strand (Fig. 2a). Results obtained with the visual detection methods correlated with the agarose gel electrophoresis results (Fig. 2b).
Fig. 2.
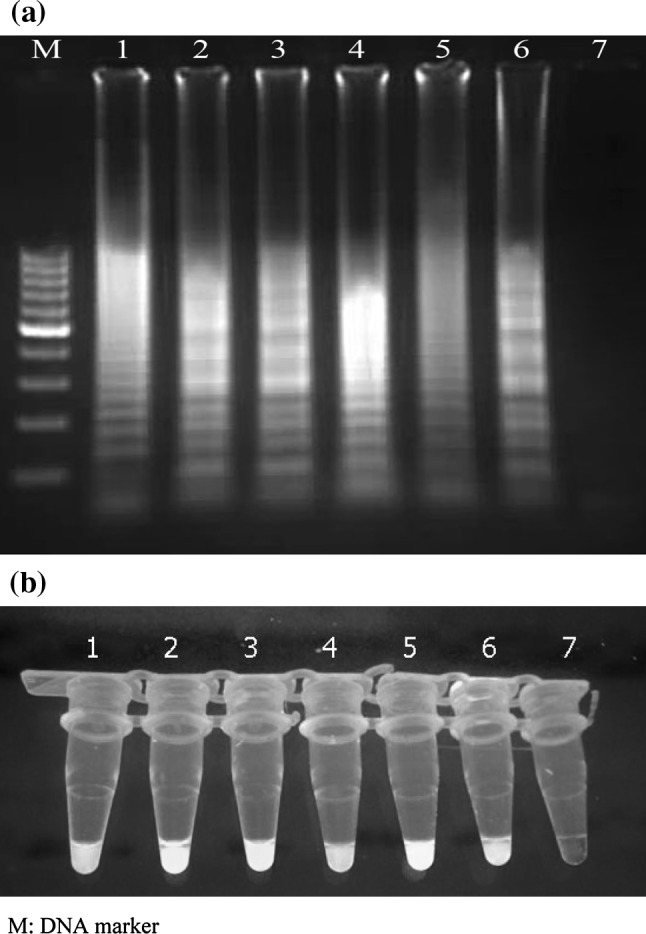
(a) Electrophoretic analysis of LAMP amplified products on a 2 % agarose gel; lane M 100-bp DNA ladder marker (Fermentas, Genruler, Germany), lane 1–5 LAMP product of Hepatitis C virus, lane 6 LAMP product of positive control, lane 7 LAMP without target RNA, negative control. (b) SYBR Green I fluorescent dye-mediated monitoring of HCV LAMP amplification. Visual observation of green fluorescence of DNA binding SYBR Green I under UV light, changed to green in the case of positive amplification, whereas in negative control having no amplification, the original orange color is retained; tubes 1–5 LAMP product of HCV, tube 6 LAMP products of positive control, tube 7 LAMP without target RNA. (Color figure online)
Specificity and Sensitivity of the HCV LAMP Reaction
Our primers demonstrated a high degree of specificity for Hepatitis C virus by amplifying only HCV template. Negative results were obtained with all of the other viruses tested. When the sensitivity of this assay was tested by 10-fold serial dilutions of RNA molecules obtained from clinical samples and compared to the results from nested-PCR, it was found that both test systems were able to detect as few as 10 copies/mL. A 100 % concordance between the two test systems, with regard to sensitivity, was observed. The limits for the HCV LAMP reaction under optimal conditions (using six primers at 62 °C for 70 min) are shown in Fig. 3.
Fig. 3.

Agarose gel demonstrating the sensitivity of LAMP and nested-PCR methods. The LAMP and nested-PCR methods were carried out using serial 10-fold dilutions of Hepatitis C virus RNA prepared from sample containing 104 copies/mL (a) LAMP reaction, (b) Nested PCR reaction; lane M 100-bp ladder size markers (Fermentase, Genruler, Germany), lanes 1–5 104 copies/mL, 103 copies/mL 102 copies/tube, 101 copies/mL and 100 copies/mL
Discussion
The Hepatitis C virus is a major public health problem and a leading cause of chronic liver disease. Predictive mathematical models of HCV epidemiology have suggested that mortality will continue to increase over the next two decades [7]. Globally, an estimated 170 million individuals are chronically infected [1, 23]. Rapid detection and accurate identification of HCV in patients at all stages of infection are crucial for immediate initiation of proper antiviral treatment. Clinical diagnostics are currently based upon antibody detection assays [4, 21]. However, several disadvantages of such serological methods exist, including the potential for problematic cross-reactions with similar, common viruses and the inability to discriminate between previous exposure and current infection [7, 8, 13]. Recently, more sensitive molecular-based diagnostic methods have been developed using reverse transcription-PCR (RT-PCR). In fact, the nested and real-time PCR techniques have been demonstrated as sensitive and specific for detection of HCV [4, 21]. Unfortunately, nested-PCR is particularly susceptible to PCR product contamination from the first-round PCR products, which may be introduced during the sample transfer step, prior to the second round of amplification. The frequency of false–positive results due to such carryover contamination has been reported to range from 4 to 31 % [3, 9]. Moreover, both nested and real-time PCR-based methods require high-precision instrumentation and elaborate methods for the detection of the amplified products [12]. Thus, these methods have been described as cumbersome to adapt to routine clinical use and have been avoided, especially by peripheral health care settings and private clinics [19]. In response to this issue, we developed the HCV LAMP assay to achieve detection of HCV RNA in clinical samples. Compared to PCR and real-time PCR, the LAMP approach is significantly simpler to perform and its detection ability is more sensitive [17, 18]. No thermal cycler instrument is required because no heat denaturation step is used with the template DNAs. In addition, the specificity of the reaction is extremely high because it uses six primers that recognize eight distinct regions on the target sequence. One of the most attractive features of the LAMP assay is the ability for the user to monitor the success of amplification with the naked-eye, as facilitated by active incorporation of the SYBR Green I dye [19]. The alternative product detection system using ethidium bromide has several limitations, such as generation of hazardous waste and sensitivity that is 25–100 times less than that of SYBR Green I. Since the initial development of LAMP, which was for detection of hepatitis B virus DNA, the assay has been successfully modified for clinical diagnosis of emerging pathogens, including bacteria, viruses and parasites. In this study, the LAMP reaction was adapted to be performed at 62 °C for 90 and 70 min using four and six primers, respectively, and with a standard heat block as the only equipment, to detect HCV. In addition, we demonstrated that by adding 1.5 U of Taq DNA polymerase to the reaction mixture allowed for HCV RNA to be detected as early as 50 min. The reason is not clearly understood, but it seems that may be due to activity of Taq DNA polymerase as an alternative starter when the first outer primer attaches to target sequence. Among the 30 EIA-positive serum samples, only 27 cases were positive by nested-PCR and LAMP; three cases were negatives by both procedures (Table 2). Our results demonstrated that LAMP has a high sensitivity, equal to those for the nested-PCR method. The results obtained from serial 10-fold dilutions of RNA from positive clinical samples showed that both systems were able to detect as few as 10 copies/mL. The LAMP primers used in this research have been described for the first time. As expected, our primers and the developed method were able to detect major genotypes of HCV in Iran (1a, 1b, 3a and 2) as well as a large part of the HCV genotype that was registered in NCBI. Our primers also demonstrated a high degree of specificity for the HCV virus, yielding no false-positive results with any of the other viruses tested. This attribute is a result of the choice of sequence-specific primers, which were designed according to highly conserved sequences from six different genotypes of HCV virus. Use of SYBR Green I for visual inspection of LAMP amplification products was a simple and superior technique; by adding 1 μL of diluted SYBR Green I and observing under UV light the result could be obtained, so post-amplification processes such as electrophoresis and EtBr staining were obviated. Although the sensitivity of detecting HCV by the LAMP reaction was equivalent to the nested-PCR test, it is considered superior because it is a simpler, cost- and time-effective procedure. The ability to obtain accurate results within 50–70 min after extraction of the viral genome and cDNA synthesis is particularly amenable to private clinics and hospital-based settings and will contribute to laboratory-based surveillance studies.
Table 2.
Result of virological examination: serology, nested-PCR and LAMP using clinical samples obtained from 30 patients suspected of Hepatitis C virus infection
| Methods | No. of Results | |
|---|---|---|
| Positive | Negative | |
| HCV IgG EIA | 30 | 0 |
| Nested-PCR | 27 | 3 |
| LAMP | 27 | 3 |
In conclusion, the HCV LAMP assay developed in this study is useful for rapid clinical diagnosis of HCV infection in developing countries, as it does not require the use of sophisticated equipment or skilled personnel.
Acknowledgments
The authors are grateful to the Islamic Azad University, Jahrom and Shahrekord branches and Almahdi Clinical Laboratory for their executive support of this project.
References
- 1.Alter MJ. Epidemiology of Hepatitis C virus infection. World J Gastroenterol. 2007;13:2436–2441. doi: 10.3748/wjg.v13.i17.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brass V, Moradpour D, Blum HE. Molecular virology of Hepatitis C virus (HCV): 2006 update. Int J Med Sci. 2006;3:29–34. doi: 10.7150/ijms.3.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caia T, Loub GQ, Yangc J, Xu D, Meng ZH. Development and evaluation of real-time loop-mediated isothermal amplification for hepatitis B virus DNA quantification: a new tool for HBV managemen. J Clin Virol. 2008;41:270–276. doi: 10.1016/j.jcv.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 4.Chevaliez S, Pawlotsky JM. Hepatitis C virus serologic and virologic tests and clinical diagnosis of HCV related liver disease. Int J Med Sci. 2006;3:35–40. doi: 10.7150/ijms.3.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Enomoto Y, Yoshikawa T, Ihira M, Akimoto S, Miyake F, Usui C, Suga S, Suzuki K, Kawana T, Nishiyama Y, Asano Y. Rapid diagnosis of Herpes simplex virus infection by a loop-mediated isothermal amplification method. J Clin Microbiol. 2005;43:951–955. doi: 10.1128/JCM.43.2.951-955.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enosawa M, Kageyama S, Sawai K, Watanabe K, Notomi T, Onoe S, Mori Y, Yokomizo Y. Use of loop-mediated isothermal amplification of the IS900 sequence for rapid detection of cultured Mycobacterium avium subsp. paratuberculosis. J Clin Microbiol. 2003;41:4359–4365. doi: 10.1128/JCM.41.9.4359-4365.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghany MG, Strader DB, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;4:1335–1374. doi: 10.1002/hep.22759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glynn SA, Wright DJ, Kleinman SH, Hirschkorn D, Tu Y, Heldebrant C, Smith R, Giachetti C, Gallarda J, Busch MP. Dynamics of viremia in early Hepatitis C virus infection. Transfusion. 2005;45:994–1002. doi: 10.1111/j.1537-2995.2005.04390.x. [DOI] [PubMed] [Google Scholar]
- 9.Gretch DR, Wilson JJ, Carithers RL, Corazon Dela Rosa JR, Han JH, Corey L. Detection of Hepatitis C virus RNA: comparison of one-stage polymerase chain reaction (PCR) with nested-set PCR. J Clin Microbiol. 1993;31:289–291. doi: 10.1128/jcm.31.2.289-291.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ihira M, Yoshikawa T, Enomoto Y, Akimoto S, Ohashi M, Suga S, Nishimura N, Ozaki T, Nishiyama Y, Notomi T, Ohta Y, Asano Y. Rapid diagnosis of Human herpesvirus 6 infection by a novel DNA amplification method, loop-mediated isothermal amplification. J Clin Microbiol. 2004;42:140–145. doi: 10.1128/JCM.42.1.140-145.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwamoto T, Sonobe T, Hayashi K. Loop-mediated isothermal amplification for direct detection of Mycobacterium tuberculosis complex, M. avium, and M. intracellulare in sputum samples. J Clin Microbiol. 2003;41:2616–2622. doi: 10.1128/JCM.41.6.2616-2622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeffrey J, Germer BS, Nizar N. Advances in the molecular diagnosis of hepatitis C and their clinical implications. Mayo Clin Proc. 2001;76:911–920. doi: 10.4065/76.9.911. [DOI] [PubMed] [Google Scholar]
- 13.Kalantar-Zadeh K, Miller LG, Daar ES. Diagnostic discordance for Hepatitis C virus infection in hemodialysis patients. Am J Kidney Dis. 2005;46:290–300. doi: 10.1053/j.ajkd.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 14.Kazemi B, Tafvizi F, Badehpour M. Determination of HCV genotypes in Iran. Biotechnology. 2005;4:139–143. doi: 10.3923/biotech.2005.139.143. [DOI] [Google Scholar]
- 15.Mori N, Motegi Y, Shimamura Y, Ezaki T, Natsumeda T, Yonekawa T, Ota Y, Notomi T, Nakayama T. Development of a new method for diagnosis of Rubella virus infection by reverse transcription–loop-mediated isothermal amplification. J Clin Microbiol. 2006;44:3268–3273. doi: 10.1128/JCM.00803-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagamine K, Hase T, Notomi T. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol Cell Probes. 2002;16:223–229. doi: 10.1006/mcpr.2002.0415. [DOI] [PubMed] [Google Scholar]
- 17.Notomi TH, Okayama H, Masubuchi T, Yonekawa T, Watanabe K, Amino N, Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28. [DOI] [PMC free article] [PubMed]
- 18.Parida MM, Sannarangaiah S, Dash PK, Rao PVL, Morita K. Loop mediated isothermal amplification (LAMP): a new generation of innovative gene amplification technique; perspectives in clinical diagnosis of infectious diseases. Rev Med Virol. 2008;18:407–421. doi: 10.1002/rmv.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parida MM, Santhosh SR, Dash PK, Tripathi NK, Saxena P, Ambuj S, Sahni AK, Lakshmana Rao PV, Morita K. Development and evaluation of reverse transcription–loop-mediated isothermal amplification assay for rapid and real-time detection of Japanese encephalitis virus. J Clin Microbiol. 2006;44:4172–4178. doi: 10.1128/JCM.01487-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pawlotsky JM. Use and interpretation of virological tests for hepatitis C. Hepatology. 2002;36:65–73. doi: 10.1002/hep.1840360709. [DOI] [PubMed] [Google Scholar]
- 21.Strader DB, Wright T, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C. Hepatology. 2004;39:1147–1171. doi: 10.1002/hep.20119. [DOI] [PubMed] [Google Scholar]
- 22.Strickland GT, El-Kamary SS, Klenerman P, Nicosia A. Hepatitis C vaccine: supply and demand. Lancet Infect Dis. 2008;8:379–386. doi: 10.1016/S1473-3099(08)70126-9. [DOI] [PubMed] [Google Scholar]
- 23.Sy T, Mazen JM. Epidemiology of Hepatitis C virus (HCV) infection. Int J Med Sci. 2006;3:41–46. doi: 10.7150/ijms.3.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong T, Lee SS. Hepatitis C: a review for primary care physicians. Can Med Assoc J. 2006;174:649–659. doi: 10.1503/cmaj.1030034. [DOI] [PMC free article] [PubMed] [Google Scholar]