

Abstract
Phosphorylation plays important roles in several processes including synaptic plasticity and memory. The critical role of extracellular signal-regulated kinase (ERK) in these processes is well established. ERK is activated in a sustained manner by different stimuli. However, the mechanisms of sustained ERK activation are not completely understood. Here we show that KCl depolarization-induced sustained ERK activation in the hippocampal slices is critically dependent on protein synthesis and transcription. In addition, the sustained ERK activation requires receptor tyrosine kinase(s) activity. In support of a role for a growth factor in sustained ERK activation, KCl depolarization enhances the level of brain-derived neurotrophic factor (BDNF). Furthermore, BDNF antibody blocks KCl-induced sustained ERK activation. These results suggest a positive feed-back loop in which depolarization-induced BDNF maintains ERK activation in the sustained phase.
Activity-dependent post-translational protein modifications such as phosphorylation play crucial roles in synaptic plasticity and memory. The extracellular signal-regulated kinase (ERK) plays pivotal roles in these processes in invertebrates as well as vertebrates1,2,3. For example, in Aplysia, ERK is activated by a pattern of serotonin application that induces long-term facilitation (LTF) of sensory motor (SN-MN) synapses4,5. In addition, ERK activity is required for LTF of SN-MN synapses6. Furthermore, ERK activity is required for long-lasting forms of memory for sensitization5.
In the vertebrate system, long-term potentiation (LTP) is widely studied as a candidate cellular mechanism of memory7,8. ERK is activated by LTP-inducing stimuli, and ERK activity is required for LTP9,10. Furthermore, ERK is activated by memory training and different kinds of memories such as fear conditioning, spatial memory and taste memory are critically dependent upon ERK activity11,12,13,14,15. The late phases of LTP and memory require translational and transcriptional events. ERK can regulate protein synthesis16 as well as transcription17,18. Thus, ERK has diverse functions that contribute to synaptic plasticity and memory formation.
Several studies have shown that ERK is activated in a sustained manner by different stimuli. For example, Wu et al.19 showed that repeated depolarization events with KCl activate ERK that lasts for at least 1 h (sustained activation). Sharma and colleagues5 found that serotonin induces sustained ERK activation in Aplysia neurons. In addition, sustained ERK activation has been observed with brain-derived neurotrophic factor (BDNF)19,20. Ahmed and Frey21 and Schmitt et al.22 showed that LTP-inducing stimuli cause sustained ERK activation in the hippocampal slices. Swank and Sweatt23 observed a sustained activation profile of ERK after taste memory training. Recently, Michel et al.24 found that associative memory training induces sustained ERK activation in the buccal ganglia of Aplysia. Considerable information is available regarding the processes involved in initial ERK activation3,25. In contrast, little is known about the mechanisms involved in sustained ERK activation. In this study, using KCl depolarization of hippocampal slices, we have investigated the processes that are involved in sustained ERK activation. Part of this study has been published in Society for Neuroscience meeting abstracts26.
Results
KCl Induces sustained ERK activation
We first examined ERK activation in the hippocampal slices immediately and 1 h after treatment with KCl, which leads to Ca++ influx in the cells19,27 and induces long-lasting neuronal plasticity28,29. ERK activation was analyzed using the antibodies that recognize phosphorylated ERK1/2 and the antibodies that recognize total ERK1/2. In agreement with our previous study30 and studies by others31, we found that KCl treatment enhanced ERK activation immediately after the depolarizing stimulus (Fig. 1A). ERK remained activated until 1 h after the stimulus (Fig. 1B), which is referred to as the sustained ERK activation19. Thus, KCl depolarization induces a sustained ERK activation in the hippocampal slices.
Figure 1. KCl depolarization induces sustained ERK activation.

The hippocampal slices were treated with KCl and harvested either immediately (A, Immediate) or 1 h (B, Sustained) after the treatment. The sample blots (A1, B1) and quantified summary data (A2, B2; n = 4 in both sets) show that ERK was activated immediately after KCl treatment and ERK activation was sustained for 1 h after the treatment. Asterisks denote significant difference (p<0.05). In this and others figures, the immediate and sustained ERK activation was examined simultaneously from the slices of the same animal and both samples were resolved on the same gel, but have been shown separately for clarity.
Sustained ERK activation requires protein synthesis
We next examined the processes that may be involved in maintaining ERK activation after depolarizing stimulus. We first tested whether protein synthesis is required for sustained ERK activation. For these experiments, we used emetine, which has previously been used to block protein synthesis32. We examined the sensitivity of ERK activation to protein synthesis inhibition immediately as well as 1 h after the depolarizing stimulus. Consistent with results in the previous section, KCl treatment induced significant ERK activation 1 h after the stimulus. However, when protein synthesis was inhibited using emetine, the sustained ERK activation was completely blocked (Fig. 2A). In contrast, the immediate ERK activation was not affected by protein synthesis inhibition (Fig. 2B). Emetine did not significantly affect basal level of ERK phosphorylation.
Figure 2. KCl-induced sustained ERK activation requires protein synthesis and transcription.
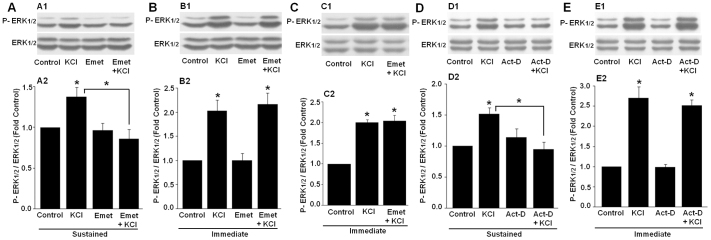
ERK activation in the hippocampal slices was examined 1 h (Sustained) or immediately (Immediate) after KCl treatment. (A) Whereas ERK activation was observed in the control slices 1 h after KCl stimulation, emetine (Emet) completely blocked KCl-induced ERK activation at this time point. (B) The KCl-induced immediate ERK activation was not affected by emetine treatment (KCl and Emet + KCl, p>0.30). Compared to control, emetine alone had no significant effects on ERK activation (Sustained, p>0.90, Immediate, p>0.75). Representative blots are shown in A1 and B1 and summary data are shown in A2 and B2 (n = 7 in both sets). (C) The hippocampal slices were treated with emetine for 1.5 h (same total time of treatment as in the case of sustained ERK activation analysis) before stimulation with KCl. Slices were harvested after KCl treatment and processed for ERK activation analysis. Representative blots (C1) and quantified summary data (C2; n = 3) show that emetine had no effect on immediate ERK activation even with prolonged treatment with emetine. There was no significant difference between KCl and KCl + emetine groups (p>0.93). (D, E) Representative blots (D1 and E1) and summary data (D2 and E2; n = 6 in both sets) show that ERK activation 1 h after KCl stimulation was blocked by the RNA synthesis inhibitor, actinomycin D (Act D; D), but the immediate ERK activation after KCl treatment was unaffected (KCl and Act D + KCl, p>0.42) by actinomycin D (E). Compared to control, actinomycin D alone treatment did not significantly affect basal level of ERK phosphorylation (Sustained, p>0.45, Immediate, p>0.67). Asterisks denote significant difference (p<0.05).
As detailed in the Materials and Methods section, for the effect of emetine on sustained ERK activation, the slices were incubated with emetine for 30 min before KCl stimulation, and they were further incubated for 60 min in the presence of emetine before harvesting them for analysis. However, in case of its effect on immediate ERK activation, the drug incubation was for 30 min before the KCl stimulus. In both cases, emetine was present during the KCl treatment. Hence, we next treated the slices with emetine for 1.5 h before and during KCl stimulation. There was no difference in the extent of ERK activation between the KCl and KCl + emetine treated samples under these conditions (Fig. 2C). These results show that inhibition of sustained ERK activation was not due to longer drug treatment. Thus, whereas ERK activation immediately after the stimulus is independent of protein synthesis, the sustained ERK activation critically requires protein synthesis.
Sustained ERK activation requires transcription
Having established that the KCl-induced sustained ERK activation requires protein synthesis, we next asked whether RNA synthesis is also required for sustained ERK activation. We used a well known transcriptional blocker, actinomycin D33, to inhibit transcription. Similar to the results with protein synthesis inhibition, we found that KCl-induced sustained ERK activation was blocked when actinomycin D was present (Fig. 2D). The immediate ERK activation was not affected by actinomycin D treatment (Fig. 2E). Also, actinomycin D alone did not have significant effect on the basal level of ERK phosphorylation. Thus, KCl-induced sustained ERK activation requires transcription.
Receptor tyrosine kinase activity is required for KCl-induced sustained ERK activation
The results of the previous sections showing that sustained ERK activation requires transcription and protein synthesis raise the possibility that some protein may be synthesized in response to depolarization, which then maintains ERK in an activated state long after the stimulus. We considered the possibility of a growth factor that may be involved in keeping ERK activated after the stimulus. We used a receptor tyrosine kinase (RTK) blocker, K252a19, to block growth factor signalling. The results of these experiments showed that whereas K252a blocked KCl-induced sustained ERK activation (Fig. 3A), the immediate ERK activation was not affected by K252a treatment (Fig. 3B). K252a alone had no significant effects on the basal level of ERK phosphorylation.
Figure 3. KCl-induced sustained ERK activation requires receptor tyrosine kinase activity.

ERK activation in the hippocampal slices was examined 1 h (A, sustained) or immediately (B, immediate) after KCl treatment. Representative blots are shown in A1 and B1, and summary data are shown in A2 and B2 (n = 7 in both sets). ERK was activated in the control slices 1 h after KCl treatment. But, the receptor tyrosine kinase inhibitor (K252a) completely blocked ERK activation at this time point. The ERK activation immediately after the KCl stimulation was unaffected by K252a treatment (KCl and K252a + KCl, p>0.63). Compared to control, K252a did not affect the basal level of ERK phosphorylation (Sustained, p>0.08, Immediate, p>0.41). Asterisks denote significant difference (p<0.05).
KCl treatment enhances the level of brain-derived neurotrophic factor
We next examined whether a growth factor was indeed synthesized after KCl treatment under our experimental conditions. We focused on BDNF that is known to regulate ERK activation. Western blot analysis was performed on total cell extract of control and KCl treated hippocampal slices using BDNF antibody. This analysis showed that the level of mature BDNF protein was significantly increased 1 h after KCl treatment (Fig. 4A).
Figure 4. KCl depolarization increases BDNF levels and BDNF antibody blocks KCl-induced sustained ERK activation.

(A) The hippocampal slices were treated with KCl and the level of mature BDNF protein was examined 1 h after the treatment. Sample Western blots of BDNF and GAPDH proteins are shown in (A1) and quantified summary data are shown in (A2; n = 4). KCl treatment increased BDNF protein level. (B) BDNF antibody blocks KCl-induced sustained ERK activation. The activation of ERK was examined in the hippocampal slices 1 h after KCl treatment. The sample blots (B1) and quantified summary data (B2, n = 6) show that the KCl-induced sustained ERK activation was blocked by BDNF antibody. Compared to control, the BDNF antibody alone did not affect basal ERK activation (p>0.91). Asterisks denotes significant difference (p<0.05).
Anti-BDNF antibody blocks depolarization-induced sustained ERK activation
The experiments in the earlier section raised the possibility that BDNF may be involved in keeping ERK activation sustained. Thus, we next examined whether neutralizing BDNF using antibodies blocks KCl-induced sustained ERK activation. We found that when the hippocampal slices were treated with BDNF antibody, KCl-induced sustained ERK activation was blocked (Fig. 4B). The BDNF antibody alone did not have any significant effect on baseline ERK activation. Collectively, these results suggest that KCl depolarization enhances the level of BDNF that then acts through the receptor tyrosine kinase to keep ERK in an activated state.
Discussion
Posttranslational modifications are known to play crucial roles in the establishment of synaptic plasticity and memory in diverse systems. With respect to phosphorylation, several studies have established a critical role of ERK in these processes. ERK is activated in a sustained manner by different stimuli. However, the mechanisms of sustained ERK activation are not clearly understood. This study shows that protein synthesis and transcriptional events are required for depolarization-induced sustained ERK activation, although ERK activation immediately after the depolarizing stimulus is independent of these processes.
How transcription and protein synthesis processes might be involved in keeping ERK activation sustained after depolarization event? We considered the possibility of involvement of a growth factor in sustained ERK activation. In support of this possibility, we found that whereas the immediate ERK activation does not require RTK activity, the sustained ERK activation is critically dependent upon RTK activity. Previous studies have shown that BDNF synthesis is increased by KCl depolarization of neuronal cells34,35. We found that the level of BDNF is increased 1 h after KCl treatment. Furthermore, BDNF antibody blocks sustained ERK activation. Previous studies have used antibodies against BDNF to study its role in different processes including synaptic plasticity and memory36,37,38. The BDNF antibody used in this study has successfully been used previously to examine BDNF function39,40,41. Alonso and colleagues42 found that injection of BDNF antibody in the hippocampus blocks memory formation. Our results suggest that BDNF is synthesized in response to KCl depolarization and it then acts on the tyrosine kinase receptor to keep ERK in activated state. These results point to a positive feedback loop in the maintenance of ERK activation.
The role of BDNF signalling in synaptic plasticity and memory has been studied extensively. A TrkB ligand is required for LTF and memory for sensitization in Aplysia43. BDNF induces long-lasting enhancement of synaptic strength in the hippocampus44. Chen et al.37 showed that TrkB-IgG fusion protein or BDNF antibody impair LTP in the hippocampal slices. BDNF knock-out mice show deficient LTP45,46. BDNF expression is increased after memory training in different tasks such as water maze, inhibitory avoidance, fear conditioning and radial arm maze47,48,49,50,51. In addition, blocking BDNF function impairs memory. For example, Ma and colleagues showed that BDNF antisense RNA impairs LTP and memory49. Furthermore, BDNF knockout mice show impaired memory52. Other studies also have shown important role of BDNF in LTP and memory. In addition, a role for BDNF and ERK has been shown in memory persistence53. The alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor trafficking plays important roles in synaptic plasticity. KCl enhances surface expression of AMPA receptors in hippocampal neurons54. In addition, BDNF enhances AMPA receptor surface expression in neurons55,56,57,58. Importantly, BDNF-induced AMPA receptor membrane insertion requires ERK activity56,57 Collectively, several studies suggest that BDNF and ERK play important roles in synaptic plasticity and memory. It would be interesting to examine whether the sustained ERK activation observed after LTP induction22 and memory training11,23,59 also depends on protein synthesis, transcription and BDNF signalling.
Methods
Hippocampal slicing and treatment
Six-to-eight week old male Sprague-dawley rats were used for the experiments. The animals were housed under light/dark (12 h/12 h) cycle with rodent chow and water available ad libitum. The animals were sacrificed following the procedure approved by the Institutional Animal Ethics Committee. Halothane (Sigma) was used to anaesthetize the animals before sacrificing. Hippocampal slicing and recovery was performed as described previously30. Slices from both the hippocampi of an animal were pooled. The slices were divided into different treatment groups with each group containing two-to-four slices. The slices were treated with 90 mM KCl for 3 min19,30 in artificial cerebrospinal fluid (aCSF;125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 2 mM CaCl2, 1 mM MgCl2, 25 mM glucose). The concentration of NaCl in KCl-aCSF was reduced making the composition of KCl-aCSF as: 37.5 mM NaCl, 90 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 2 mM CaCl2, 1 mM MgCl2, 25 mM glucose. ERK activation was examined immediately (immediate, slices were put in SDS lysis buffer within 1–2 min of KCl treatment) or 1 h (sustained) after KCl treatment. For immediate ERK activation analysis, slices were rinsed with aCSF after KCl treatment and harvested. For the analysis of ERK activation 1 h after KCl treatment, the slices were treated with KCl for 3 min, rinsed and incubated in aCSF for 1 h before harvesting for protein analysis. When pharmacological inhibitors were used, slices were treated with the inhibitor during the last recovery period for 30 min. The treatment with BDNF antibody was done for 2 h before treatment with KCl. Pharmacological inhibitors or BDNF antibody were included during the KCl treatment also. For the sustained ERK activation analysis, the inhibitors or BDNF antibody were present during the 1 h incubation after KCl treatment in addition to pre-treatment and treatment during the KCl stimulation. To examine the effect of prolonged protein synthesis inhibitor treatment on immediate ERK activation, the slices were treated with emetine for 1.5 h (same total duration of emetine treatment as in the case of sustained ERK activation analysis) before and during the KCl stimulation, rinsed with aCSF and harvested. The protein synthesis inhibitor, emetine (Sigma) was dissolved at 20 mM concentration in aCSF and used at 20 μM final concentration. The stock solution of the transcriptional inhibitor, actinomycin-D (Sigma) was prepared at 25 mM in DMSO, and was used at 25 μM final concentration. The stock solution of the receptor tyrosine kinase inhibitor, K252a, was prepared at 200 μM in DMSO, and was used at 200 nM final concentration. Anti-BDNF antibody (Cat no. AB1513P, Millipore), which was raised against recombinant human BDNF and neutralizes BDNF, but not other neurotrophins (Millipore) was reconstituted in sterile H2O as recommended by the manufacturer at 1 mg/ml concentration and used at 5 μg/ml final concentration. Before use, all the inhibitors were diluted in aCSF to their final concentration. After treatment, entire slice was used to make protein lysate21. The slices were lysed in the lysis buffer (50 mM Tris-Cl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM sodium butyrate, 2% sodium dodecyl sulfate, protease inhibitor cocktail). The samples were boiled for 5 min, centrifuged and the supernatant containing solubilized proteins was stored frozen until use. Protein concentration in the samples was determined using the BCA reagent (Sigma) with bovine serum albumin as standard.
Immunoblotting
Phosphorylation of ERK was examined using immunoblotting method which was carried out essentially as described previously30. Proteins from different samples were resolved by discontinuous polyacrylamide gel electrophoresis. After electrophoresis, the proteins were transferred to nitrocellulose membrane (MDI, India). The blots were first probed with ERK1/2 antibody, stripped and then re-probed with phospho-ERK1/2 antibody. Antibodies against ERK, phospho-ERK and appropriate secondary antibodies were obtained from Cell Signaling Technology. The signal was detected on Hyperfilm (Amersham Biosciences) using enhanced chemiluminescence reagent. NIH Image (NIH, Bethesda, USA) software was used to quantify the band signal. Both, ERK1 and ERK2 or phospho-ERK1 and phospho-ERK2 bands were quantified together. To estimate ERK activation, phospho-ERK1/2 signal was divided by ERK1/2 signal in each sample and then normalized with the control sample5,30. To examine the level of BDNF, hippocampal slices were treated with KCl, incubated in aCSF for 1 h and harvested. Total cell extract from hippocampal slices was used to examine the level of mature BDNF (~14 kDa). The lower portion of the blot was probed with the BDNF antibody with epitope mapping within an internal region of human BDNF (Santa Cruz Biotechnology, Cat no. sc-546). The upper portion was probed with glyceraldyehyde-3-phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnology) antibody, which was used as a loading control. The BDNF signal in each sample was divided by the GAPDH signal and then normalized with the control sample.
Statistical analysis
Data were analyzed by paired student's t-test on phospho-ERK1/2/total-ERK1/2 ratios or BDNF/GAPDH ratios. Differences between groups were considered statistically significant if p-value was <0.05. Data are expressed as mean ± SEM (fold control).
Author Contributions
SKS and CM conceived the project and designed experiments. CM performed experiments. KPS participated in the experiments and helped in the manuscript revision. CM and SKS analysed data and wrote the manuscript. All the authors read and approved the final manuscript.
Acknowledgments
This work was supported by a core grant from Department of Biotechnology, India to National Brain Research Centre. Chinmoyee Maharana was a recipient of Senior Research Fellowship from Council of Scientific and Industrial research, India. We thank Jeet Bahadur Singh and Neha Sharma for their help in some of the experiments.
References
- Adams J. P. & Sweatt J. D. Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu. Rev. Pharmacol. Toxicol. 42, 135–163 (2002). [DOI] [PubMed] [Google Scholar]
- Sharma S. K. & Carew T. J. The roles of MAPK cascades in synaptic plasticity and memory in Aplysia: facilitatory effects and inhibitory constraints. Learn. Mem. 11, 373–378 (2004). [DOI] [PubMed] [Google Scholar]
- Thomas G. M. & Huganir R. L. MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183 (2004). [DOI] [PubMed] [Google Scholar]
- Michael D. et al. Repeated pulses of serotonin required for long-term facilitation activate mitogen-activated protein kinase in sensory neurons of Aplysia. Proc. Natl. Acad. Sci. U. S. A. 95, 1864–1869 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S. K. et al. Differential role of mitogen-activated protein kinase in three distinct phases of memory for sensitization in Aplysia. J. Neurosci. 23, 3899–3907 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K. C. et al. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron 18, 899–912 (1997). [DOI] [PubMed] [Google Scholar]
- Bliss T. V. & Collingridge G., L. . A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39 (1993). [DOI] [PubMed] [Google Scholar]
- Raymond C. R. LTP forms 1, 2 and 3: different mechanisms for the "long" in long-term potentiation. Trends Neurosci. 30, 167–175 (2007). [DOI] [PubMed] [Google Scholar]
- English J. D. & Sweatt J. D. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J. Biol. Chem. 271, 24329–24332 (1996). [DOI] [PubMed] [Google Scholar]
- English J. D. & Sweatt J. D. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J. Biol. Chem. 272, 19103–19106 (1997). [DOI] [PubMed] [Google Scholar]
- Atkins C. M., Selcher J. C., Petraitis J. J., Trzaskos J. M. & Sweatt J. D. The MAPK cascade is required for mammalian associative learning. Nat. Neurosci. 1, 602–609 (1998). [DOI] [PubMed] [Google Scholar]
- Berman D. E., Hazvi S., Rosenblum K., Seger R. & Dudai Y. Specific and differential activation of mitogen-activated protein kinase cascades by unfamiliar taste in the insular cortex of the behaving rat. J. Neurosci. 18, 10037–10044 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum S., Moore A. N., Adams F. & Dash P. K. A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J. Neurosci. 19, 3535–3544 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe G. E., Nadel N. V., Sullivan G. M., Harris A. & LeDoux J. E. Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn. Mem. 6, 97–110 (1999). [PMC free article] [PubMed] [Google Scholar]
- Selcher J. C., Atkins C. M., Trzaskos J. M., Paylor R. & Sweatt J. D. A necessity for MAP kinase activation in mammalian spatial learning. Learn. Mem. 6, 478–490 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher R. J. 3rd., Govindarajan, A. & Tonegawa, S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron 44, 59–73 (2004). [DOI] [PubMed] [Google Scholar]
- Sweatt J. D. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J. Neurochem. 76, 1–10 (2001). [DOI] [PubMed] [Google Scholar]
- Valjent E., Caboche J. & Vanhoutte P. Mitogen-activated protein kinase/extracellular signal-regulated kinase induced gene regulation in brain: a molecular substrate for learning and memory? Mol. Neurobiol. 23, 83–99 (2001). [DOI] [PubMed] [Google Scholar]
- Wu G. Y., Deisseroth K. & Tsien R. W. Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat. Neurosci. 4, 151–158 (2001). [DOI] [PubMed] [Google Scholar]
- Ying S. W. et al. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J. Neurosci. 22, 1532–1540 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed T. & Frey J. U. Plasticity-specific phosphorylation of CaMKII, MAP-kinases and CREB during late-LTP in rat hippocampal slices in vitro. Neuropharmacology 49, 477–492 (2005). [DOI] [PubMed] [Google Scholar]
- Schmitt J. M., Guire E. S., Saneyoshi T. & Soderling T. R. Calmodulin-dependent kinase kinase/calmodulin kinase I activity gates extracellular-regulated kinase-dependent long-term potentiation. J. Neurosci. 25, 1281–1290 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swank M. W. & Sweatt J. D. Increased histone acetyltransferase and lysine acetyltransferase activity and biphasic activation of the ERK/RSK cascade in insular cortex during novel taste learning. J. Neurosci. 21, 3383–3391 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel M., Green C. L., Eskin A. & Lyons L. C. PKG-mediated MAPK signaling is necessary for long-term operant memory in Aplysia. Learn. Mem. 18, 108–117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal S. S., York R. D. & Stork P. J. Extracellular-signal-regulated kinase signalling in neurons. Curr. Opin. Neurobiol. 9, 544–553 (1999). [DOI] [PubMed] [Google Scholar]
- Maharana C. & Sharma S. Mechanisms underlying sustained activation of extracellular signal regulated kinase in the hippocampus. Soc. Neurosci. Abstr. (238.16) (2011). [Google Scholar]
- Nashat A. H. & Langer R. Temporal characteristics of activation, deactivation, and restimulation of signal transduction following depolarization in the pheochromocytoma cell line PC12. Mol. Cell. Biol. 23, 4788–4795 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleck M. W., Palmer A. M. & Barrionuevo G. Potassium-induced long-term potentiation in rat hippocampal slices. Brain Res. 580, 100–105 (1992). [DOI] [PubMed] [Google Scholar]
- Fitzjohn S. M. et al. An electrophysiological characterisation of long-term potentiation in cultured dissociated hippocampal neurones. Neuropharmacology 41, 693–699 (2001). [DOI] [PubMed] [Google Scholar]
- Maharana C., Sharma K. P. & Sharma S. K. Depolarization induces acetylation of histone H2B in the hippocampus. Neuroscience 167, 354–360 (2010). [DOI] [PubMed] [Google Scholar]
- Corvol J. C. et al. Depolarization activates ERK and proline-rich tyrosine kinase 2 (PYK2) independently in different cellular compartments in hippocampal slices. J. Biol. Chem. 280, 660–668 (2005). [DOI] [PubMed] [Google Scholar]
- Sajikumar S., Navakkode S. & Frey J. U. Distinct single but not necessarily repeated tetanization is required to induce hippocampal late-LTP in the rat CA1. Learn. Mem. 15, 46–49 (2008). [DOI] [PubMed] [Google Scholar]
- Frey U., Frey S., Schollmeier F. & Krug M. Influence of actinomycin D, a RNA synthesis inhibitor, on long-term potentiation in rat hippocampal neurons in vivo and in vitro. J. Physiol. 490, 703–711 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F., Hengerer B., Leibrock J., Thoenen H. & Lindholm D. Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J. 9, 3545–3550 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X., Finkbeiner S., Tao X., Finkbeiner S., Arnold D. B., Shaywitz A. J. & Greenberg M. E. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20, 709–726 (1998). [DOI] [PubMed] [Google Scholar]
- Mu J. S., Li W. P., Yao Z. B. & Zhou X. F. Deprivation of endogenous brain-derived neurotrophic factor results in impairment of spatial learning and memory in adult rats. Brain. Res. 835, 259–265 (1999). [DOI] [PubMed] [Google Scholar]
- Chen G., Kolbeck R., Barde Y. A., Bonhoeffer T. & Kossel A. Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J. Neurosci. 19, 7983–7990 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki T. et al. Brain-derived neurotrophic factor-mediated retrograde signaling required for the induction of long-term potentiation at inhibitory synapses of visual cortical pyramidal neurons. Neurosci. Res. 61, 192–200 (2008). [DOI] [PubMed] [Google Scholar]
- Rasika S., Alvarez-Buylla A. & Nottebohm F. BDNF mediates the effects of testosterone on the survival of new neurons in an adult brain. Neuron 22, 53–62 (1999). [DOI] [PubMed] [Google Scholar]
- Freeman A. Y. & Pierce R. C. Neutralization of neutrophin-3 in the ventral tegmental area or nucleus accumbens differentially modulates cocaine-induced behavioral plasticity in rats. Synapse 46, 57–65 (2002). [DOI] [PubMed] [Google Scholar]
- Taylor A. R. et al. Motoneuron programmed cell death in response to proBDNF. Dev. Neurobiol 72, 699–712 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso M. et al. BDNF-triggered events in the rat hippocampus are required for both short- and long-term memory formation. Hippocampus 12, 551–560 (2002). [DOI] [PubMed] [Google Scholar]
- Sharma S. K., Sherff C. M., Stough S., Hsuan V. & Carew T. J. A tropomyosin-related kinase B ligand is required for ERK activation, long-term synaptic facilitation, and long-term memory in aplysia. Proc. Natl. Acad. Sci. U. S. A. 103, 14206–14210 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H. & Schuman E. M. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 267, 1658–1662 (1995). [DOI] [PubMed] [Google Scholar]
- Korte M. et al. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. U. S. A. 92, 8856–8860 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S. L. et al. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16, 1137–1145 (1996). [DOI] [PubMed] [Google Scholar]
- Hall J., Thomas K. L. & Everitt B. J. Rapid and selective induction of BDNF expression in the hippocampus during contextual learning. Nat. Neurosci. 3, 533–535 (2000). [DOI] [PubMed] [Google Scholar]
- Kesslak J. P., So V., Choi J., Cotman C. W. & Gomez-Pinilla F. Learning upregulates brain-derived neurotrophic factor messenger ribonucleic acid: a mechanism to facilitate encoding and circuit maintenance? Behav. Neurosci. 112, 1012–1019 (1998). [DOI] [PubMed] [Google Scholar]
- Ma Y. L., Wang H. L., Wu H. C., Wei C. L. & Lee E. H. Brain-derived neurotrophic factor antisense oligonucleotide impairs memory retention and inhibits long-term potentiation in rats. Neuroscience 82, 957–967 (1998). [DOI] [PubMed] [Google Scholar]
- Mizuno M., Yamada K., Olariu A., Nawa H. & Nabeshima T. Involvement of brain-derived neurotrophic factor in spatial memory formation and maintenance in a radial arm maze test in rats. J. Neurosci. 20, 7116–7121 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattiner L. M., Davis M., French C. T. & Ressler K. J. Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J. Neurosci. 24, 4796–4806 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldt S. A., Stanek L., Chhatwal J. P. & Ressler K. J. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol. Psychiatry 12, 656–670 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekinschtein P. et al. BDNF is essential to promote persistence of long-term memory storage. Proc. Natl. Acad. Sci. U. S. A. 105, 2711–2716 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard L. et al. Transient synaptic activation of NMDA receptors leads to the insertion of native AMPA receptors at hippocampal neuronal plasma membranes. Neuropharmacology 41,700–713 (2001). [DOI] [PubMed] [Google Scholar]
- Caldeira M. V. et al. Brain-derived neurotrophic factor regulates the expression and synaptic delivery of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J. Biol. Chem. 282, 12619–12628 (2007). [DOI] [PubMed] [Google Scholar]
- Li W. & Keifer J. BDNF-induced synaptic delivery of AMPAR subunits is differentially dependent on NMDA receptors and requires ERK. Neurobiol. Learn. Mem. 91, 243–249 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., & Wolf M. E. Brain-derived neurotrophic factor rapidly increases AMPA receptor surface expression in rat nucleus accumbens. Eur. J. Neurosci. 34, 190–198 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin D. A. et al. Brain-derived neurotrophic factor activation of CaM-kinase kinase via transient receptor potential canonical channels induces the translation and synaptic incorporation of GluA1-containing calcium-permeable AMPA receptors. J. Neurosci 32, 8127–8137 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. J., Okutani F., Inoue S. & Kaba H. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase signaling pathway leading to cyclic AMP response element-binding protein phosphorylation is required for the long-term facilitation process of aversive olfactory learning in young rats. Neuroscience 121, 9–16 (2003). [DOI] [PubMed] [Google Scholar]