

Abstract
Most newborn screening (NBS) laboratories use second-tier molecular tests for cystic fibrosis (CF) using dried blood spots (DBS). The Centers for Disease Control and Prevention’s NBS Quality Assurance Program offers proficiency testing (PT) in DBS for CF transmembrane conductance regulator (CFTR) gene mutation detection. Extensive molecular characterization on 76 CF patients, family members or screen positive newborns was performed for quality assurance. The coding, regulatory regions and portions of all introns were sequenced and large insertions/deletions were characterized as well as two intronic di-nucleotide microsatellites. For CF patient samples, at least two mutations were identified/verified and four specimens contained three likely CF-associated mutations. Thirty-four sequence variations in 152 chromosomes were identified, five of which were not previously reported. Twenty-seven of these variants were used to predict haplotypes from the major haplotype block defined by HapMap data that spans the promoter through intron 19. Chromosomes containing the F508del (p.Phe508del), G542X (p.Gly542X) and N1303K (p.Asn1303Lys) mutations shared a common haplotype subgroup, consistent with a common ancient European founder. Understanding the haplotype background of CF-associated mutations in the U.S. population provides a framework for future phenotype/genotype studies and will assist in determining a likely cis/trans phase of the mutations without need for parent studies.
Keywords: Cystic fibrosis, CFTR, Mutation, Newborn screening, Haplotype
1. Introduction
Cystic fibrosis (CF) is a childhood-onset inherited disorder that significantly shortens the life span of affected individuals. The CF transmembrane conductance regulator (CFTR, OMIM ID: 602421) gene is located on chromosome 7 and codes for a protein that is expressed in the respiratory epithelial cells, gastrointestinal system (pancreatic and liver ductals), the vas deferens in males, mucous-secreting cervical cells in women and sweat gland ductals [1]. Defects in the CFTR gene that alter structure, function or expression of this protein can lead to malfunctions or disease processes in the lungs and upper respiratory tract, gastrointestinal tract, pancreas, liver, sweat glands and genitourinary tract [2]. CF is an autosomal recessive disorder that affects approximately 1:4000 people of Western European, North American and Australasian descent. There are over 1850 mutations in the CFTR gene, of which many have been linked to CF while others have unknown or no known consequences [3,4]. Newborn screening (NBS) for CF first became feasible in 1979 when the dried blood spot (DBS) specimens from newborn infants diagnosed with CF were shown to have elevated levels of immunoreactive trypsinogen (IRT) [5], leading to an IRT/IRT screening method [6]. After the discovery of the CFTR gene in 1989, screening expanded to include an IRT/DNA method [7,8]. Data from early screening programs prompted the Centers for Disease Control and Prevention (CDC) to recommend the inclusion CF in NBS panels [9].
CDC’s Newborn Screening and Molecular Biology Branch houses the NBS Quality Assurance Program (NSQAP), co-sponsored by the Association of Public Health Laboratories (APHL), which offers proficiency testing (PT) programs for both IRT and CFTR mutation detection [10,11]. At the end of 2010, ~84% of newborns were screened for CF using the IRT/DNA screening algorithm, which was the first widespread DNA based screening assay [12]. NSQAP’s CFTR mutation collection contains a wide variety of CF-causing mutations including the 23 recommended by the American College of Medical Genetics (ACMG) [13,14] providing quarterly PT challenges to 60 participating laboratories. The aims of this analysis were to verify and identify all CFTR mutations in a diverse group of CF cases and some carriers using comprehensive molecular detection methods. The results from the molecular analysis allowed the prediction of haplotypes for the identified CFTR mutations, providing a detailed molecular framework for the CFTR gene. This framework may assist in understanding why certain genotype combinations may be expressed phenotypically more than others.
2. Materials and methods
2.1. Samples and specimen processing
NSQAP’s CF Mutation Detection PT program received samples from 72 adult or adolescent CF patients or family members [11]. Four N1303K DBS specimens were provided by the California Department of Public Health NBS Program; two from patients diagnosed with CF and two from patients diagnosed with CF-related metabolic syndrome [15]. CDC’s Human Research Protection Office did not consider this study to be human research as all specimens are de-identified and cannot be traced back to the donor. DNA was extracted either from three 3 mm DBS punches (Whatman Grade filter 903 paper, Kent, United Kingdom) using Qiagen’s QIAamp DNA Micro kit or from 250 μl of blood using QIAamp DNA Mini kit (Valencia, California). DNA concentrations were determined by quantitative PCR of the RNase P gene (Applied Biosystems, Foster City, California) using a standard curve made from pooled human genomic DNA (Roche Applied Science, Indianapolis, Indiana).
2.2. PCR amplification and automated florescent sequencing
The CFTR gene was amplified and sequenced using VariantSEQr CFTR (set RSS000010013, Applied Biosystems) with the following modifications: amplification using HotStarTaq Master Mix (Qiagen), unused primer and nucleotide removal using ExoSAP-IT (USB Corporation, Cleveland, Ohio), sequencing using BigDye Terminator (BDT) Ready Reaction kit ver 1.1 and sequencing reaction purification using Applied Biosystem’s BigDye XTerminator. Since Variant SEQr kit provided incomplete coverage for exons 10, 14, 15, intronic mutation c.3717+12191C>T (3849+10kbC→T) and the 3′ UTR, and exon 1 failed to amplify, published primer sequences were used to detect exon 14 [16,17], exon 15 and the intron 22 mutation [16,17]. Primer sequences for exons 1, 10, 14 and the 3′ UTR were custom designed (Table 1). Amplification for custom designed regions used Qiagen’s HotStarTaq Master Mix with 1 μM of forward and reverse primer. Cycling parameters included a 10 min denaturing step at 95 °C followed by 34–38 cycles at 95 °C for 30 s, specific primer annealing temperature for 30 s, and 72 °C for 1 min; followed by an extension at 72 °C for 8 min and an indefinite hold at 4 °C (Table 1). Unused primer and nucleotides were removed using ExoSAP-IT. Cycle sequencing reactions consisted of 1 μl of PCR product, 0.5 μl of BDT ver1.1, 1.75 μl of 5× sequencing buffer, and 3.2 pmol of primer. Sequencing reactions were purified and electrophoresed with run module BDx_Rapid-Seq36_POP7 and data was analyzed using SeqScape software from Applied Biosystems. DNA sequence was aligned with Genbank CFTR genomic reference sequence NG_016465.
Table 1.
Oligonucleotides used for amplification and sequencing. Primer locations are given according to base position in the cDNA [4].
| Primer | Sequence | Locationa | Target region | Annealing temp. | Cycle no. |
|---|---|---|---|---|---|
| CFTR-ex1F | GGGGCGGCGAGGGAGCGAAGG | c.-327_-307 | Exon 1 | 62 °C | 34 |
| CFTR-ex1R | GTTTGGAGACAACGCTGGCCTT | c.43_22 | |||
| CFTR-ex10F | tgtaaaacgacggccagtCAGTGTAATGGATCATGGGCCATGT | c.1210-131_1210-107 | Exon 10 | 61 °C | 38 |
| CFTR-ex10R | caggaaacagctatgaccGCTCGCCATGTGCAAGATACAGT | c.1392+267_1392+245 | |||
| CFTR-ex14-1F | tgtaaaacgacggccagtGAGAGACCCCGAGGATAAATGATTTGC | c.1767-299_1767-273 | Exon 14 | 64 °C | 38 |
| CFTR-ex14-1R | caggaaacagctatgaccCCATGAGTTTTGAGCTAAAGTCTGGCT | c.1936_1910 | |||
| CFTR-ex14-2F | tgtaaaacgacggccagtGCTGTGTCTGTAAACTGATGGCTAACA | c.1767-1_1792 | Exon 14 | 60 °C | 38 |
| CFTR-ex14-2R | caggaaacagctatgaccCCTCTCCCTGCTCAGAATCTGGTA | c.2239_2216 | |||
| CFTR-ex14-3F | tgtaaaacgacggccagtCCAGACTTTAGCTCAAAACTCATGGGA | c.1912_1938 | Exon 14 | 60 °C | 38 |
| CFTR-ex14-3R | caggaaacagctatgaccAGTGTGTCATCAGGTTCAGGACAGA | c.2326_2302 | |||
| CFTR-3′ UTRF | tgtaaaacgacggccagtATGAATCACCTTTTGGTCTGGAGGGA | c.*1313_*1338 | 3′ UTR | 60 °C | 38 |
| CFTR-3′ UTRR | caggaaacagctatgaccGACAAGTGAACGCATCTGAAAACAACA | c.*1587_*1561 |
Note: Amplicons containing an M13 tail, indicated by lower case, were sequenced using M13F and M13R primers.
Location follows mutation database cDNA sequence [4].
2.3. MLPA analysis
The SALSA MLPA kit P091 CFTR (MRC Holland, Amsterdam, Netherlands) was performed with modifications: 5% glycerol was added to the denaturation step, which was extended from 5 to 10 min. Peak sizes were determined using the internal lane standard, LIZ500, with Applied Biosystems’s GeneMapper software and imported into MRC-Holland’s Coffalyser software to determine deletions and/or insertions.
2.4. Microsatellite analysis
Microsatellites IVS8CA and IVS17bCA were amplified in a multiplex PCR using primer sequences described previously [18] modified for capillary electrophoresis fluorescent detection on the ABI 3730 DNA Analyzer. Microsatellites were amplified with Phusion High-Fidelity Master Mix (Finnzymes, Espoo, Finland). An allelic ladder, constructed with in-house samples, was characterized by fragment analysis and sequencing. Peak sizes were determined using the internal lane standard, LIZ500, and the number of repeats was determined using the GeneMapper software (Applied Biosystems).
Analysis of microsatellite IVS17bTA was performed using previously defined primers [18] with several different polymerase enzymes (AmpliTaqGold DNA Polymerase with Gold Buffer and Buffer II (Applied Biosystems), Phusion HF and Phusion GC Master Mix (Finnzymes), Herculease II Fusion DNA Polymerase (Stratagene), EmeraldAmp Max HS PCR Master Mix (Takara) and HotStarTaq Master Mix (Qiagen)) and PCR additives (3–5% DMSO, 8% glycerol, and 1 M Betaine), however, all conditions gave a cluster of 2–5 fragments for each allele.
2.5. Statistical analysis
Linkage disequilibrium (LD) data from the International HapMap Project was examined using the HapMap Genome Browser (Phase 1, 2 and 3—merged genotypes and frequencies). An LD plot of the logarithm of the odds (LOD score) of the Human Genome Diversity Cell Line Panel (HGDP) CEPH population with HAPMAP preset defaults was used to characterize the LD between a given marker pair in this population (Fig. 1). The diamond color where two SNPs intersect indicates the level of LD; darker shades indicate a higher LD while lighter shades indicate lower LD. Gray regions represent missing data points. Haplotypes were predicted from the 34 sequence variants and two intronic microsatellites with an expectation maximization (EM) algorithm in JMP Genomics (SAS, Cary, North Carolina) using the PROC HAPLOTYPE procedure. The CFTR polymorphisms segregated into two haplotype blocks and compared to the HAPMAP LD plot. The two haplotype blocks assort independently, determined by contingency χ2, and CFTR mutations were assigned to specific predicted haplotypes through association frequency.
Fig. 1.
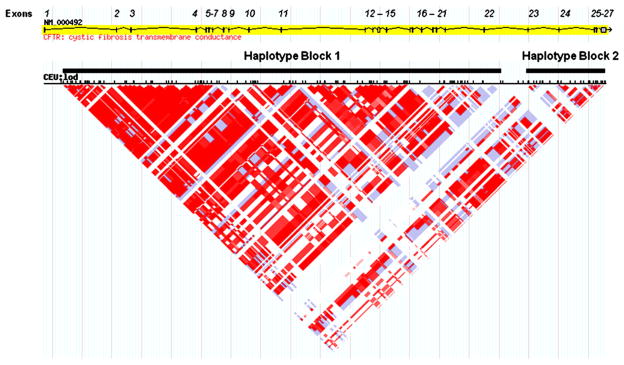
Linkage disequilibrium plot of the CFTR gene from the Hap Map Project Data. Schematic diagram of the CFTR gene on chromosome 7. Exons are represented by vertical lines or boxes (exon number is indicated above each exon). The linkage disequilibrium (LD) plot uses the logarithm of the odds (LOD score) to characterize the LD between a given marker pair in the HapMap population data. White and blue regions indicate no LD (LOD<2); shades of pink/red indicate regions of LD (LOD≥2). The intensity of the red box color is proportional to the strength of the LD property of the marker pair.
3. Results
3.1. Molecular analysis of CFTR chromosomes
DNA sequence data and large deletion analysis of the CFTR gene was performed on 152 unique chromosomes from CF patient or carrier specimens to identify and verify CF-causing mutations and likely benign polymorphisms. In addition to the ACMG 23 recommended mutations, 19 other mutations are represented [11]. Supplemental Table S1 contains a complete list of mutations with HGVS standardized nomenclature. Four specimens contained three mutations (Table 2); however, in all cases, the third mutation was not common, was associated with limited clinical information in the CF mutation database, and was not part of the ACMG recommended 23 mutations. The I1027T mutation, the third mutation in two specimens, has been found in cis with the F508del mutation greater than 5% in the Brittany population (western France) [19]. A number of sequence variants (N=34) with no known consequences also were identified, and were present most often in the non-coding regions of the CFTR gene. Five of these sequence variants have not been described previously in either of the CF Mutation database or NCBI’s dbSNP [4,20]. These new sequence variants are located in non-coding regions: c.1209+ 43T>G (Intron 9, ss432791813), c.1767-231T>C (Intron 13, ss432791824), c.1767-132A>G, (Intron 13, ss432791829), c.*94C>T (3′ UTR, ss432791834) and c.*1823C>T (3′ UTR, ss432791840).
Table 2.
CF specimens with 3 mutations.
| CFTR mutation 1 | Gene location | CFTR mutation 2 | Gene location | CFTR mutation 3 | Gene location |
|---|---|---|---|---|---|
| S549N | Ex12 | 3120+1G→A | Intron 18 | −102T→A | Promoter |
| F508del | Ex11 | G542X | Ex12 | 185+4A→T | Intron1 |
| F508del | Ex11 | F508del | Ex11 | I1027T | Ex19 |
| F508del | Ex11 | W1282X | Ex23 | I1027T | Ex19 |
3.2. Haplotype analysis of CFTR chromosomes
The analysis of the CFTR sequence variants and mutations identified in this study was based on predicted haplotype blocks from the HGDP-CEPH population from the International HapMap project. The HapMap predicts one major haplotype block that spans the promoter through exon 22 (Fig. 1) [21]. Twenty-five sequence variants and 2 microsatellites were characterized in this major block and were used to predict haplotypes with greater than 95% probability. In four samples, 2184delA/394delTT, 3905insT/1248+1G→A, 3120+ 1G→A/−102T→A and 3120+1G→A/L467P, the predicted haplotype could not be unambiguously assigned to one of the two CF-causing mutations. The minor HapMap block spanning intron 23 through the 3′ UTR was randomly associated with the block 1 haplotypes (p=0.850) and was not informative (data not shown).
Among the 144 haplotypes assigned to a CF-causing mutation, there were 34 classes of which four contained the F508del mutation (N=63 chromosomes) (Table 3). Three F508del containing haplotypes were identical except for the number of repeats found at the IVS8CA microsatellite and the fourth was a probable product of a recombination event. The two most common F508del containing haplotypes differed only by the number of repeats of the IVS8CA microsatellite; 32 chromosomes contained 17 repeats and 29 chromosomes contained 23 repeats. The F508del haplotype that contained 23 repeats of the IVS8CA microsatellite was identical to the predicted haplotypes associated with G542X (N=6 of 6), N1303K (N=6 of 6), del Ex17a, b and 18 (N=1 of 1), and 3849+10 kb C→T (N=1 of 2).
Table 3.
Predicted Haplotypes from CF Patient Samples.
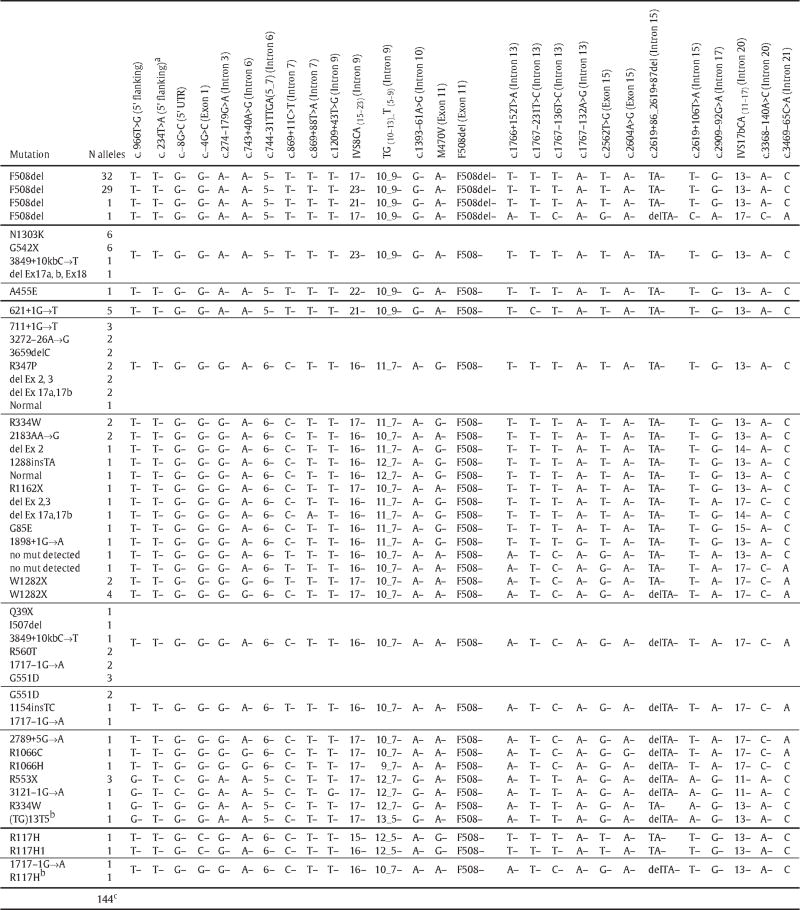
|
Variation found in a sample where the haplotype could not be predicted.
Mutations associated with patients with CF related metabolic syndrome (CRMS) [15].
Haplotypes could not be predicted from 8 of the 152 chromosomes.
3.3. Molecular analysis of the R117H mutation
The R117H mutation, known to be variable in its disease expression, was found in three specimens. Two had an almost identical predicted haplotype including the 5T variant, with the exception of 15 versus 16 IVS8CA repeats. The third R117H predicted haplotype contained a 7T variant and multiple differences from the other two R117H haplotypes. The haplotype containing the R117H mutation with the 7T variant was identical to the 1717-1G→A containing haplotype; however the significance is unknown since each is only found in one chromosome (Table 3). A single CF-related metabolic syndrome chromosome containing the 5T variant in the absence of R117H had a unique haplotype quite different from all other R117H containing haplotypes.
4. Discussion
The goal of this analysis was to characterize the 72 PT specimens in NSQAP’s CFTR Mutation Detection repository, which are used to assist NBS laboratories in ensuring accuracy when they utilize a molecular second tier test for CF. This comprehensive analysis resulted in the identification of 34 classes of predicted haplotypes that span the CFTR gene promoter through intron 18. These haplotypes are consistent with the LD defined by the International HapMap project. Half (N=77) of the 152 chromosomes examined shared a common haplotype subgroup which was associated with three of the most prevalent CF-causing mutations, F508del, G542X, and N1303K.
To understand CFTR mutations, a previous study assessing the origin of 27,177 CF chromosomes from 29 European countries and three North African countries described the five most common CF-causing mutations: F508del (66.8%), G542X (2.6%), N1303K (1.6%), G551D (1.5%) and W1282X (1.0%) [22]. Similarly, Bobadilla et al. described the five most common CF-causing mutations in the U.S., which included F508del (68.6%), G542X (2.4%), G551D (2.1%), W1282X (1.4%) and N1303K (1.3%) [23]. Hence, F508del, G542X, and N1303K are the more common mutations in Caucasians from Europe and the United States. A study on Spanish CF patients suggested an ancient common origin of these three mutations by showing that they all carried a common haplotype subgroup as defined by the IVS8CA, IVS17bCA and IVS17bTA microsatellites [24]. The haplotypes identified in the present study were consistent with the Spanish patient findings, showing that F508del, G542X, and N1303K again share a common haplotype subgroup [24]. Of our 62 F508del containing chromosomes (excluding the one probable recombinant), 29 predicted haplotypes are identical across 27 polymorphisms from the promoter to intron 21 to the G542X containing haplotypes (N=6) and the N1303K containing haplotypes (N=6). The remaining 33 F508del containing haplotypes are identical to this common haplotype except that they contain either 17 or 21 di-nucleotide repeats of the intron 9 IVS8CA microsatellite versus 23. Taken together, these data from a heterogeneous group of CF patients living in the United States support the existence of a common ancient European ancestral haplotype that independently gave rise to these three CF mutations.
A recent study examined the association of common CFTR gene variants and their potential influence on body composition and survival in a non-CF population in rural Ghana [25]. As expected, the LD across the CFTR gene in this Ghana population was not as strong as that seen in a population of European descent; however, they reported specific intron 11 haplotype (comprised of 4 SNPs) associations with young versus old study participants as well as with lower or higher weight in children less than 5 years old. While further studies are necessary to understand if these associations are by chance or if they have any direct influence on health, it provides further motivation to understand the molecular framework of the CFTR gene.
Molecular analysis of CF, as with many autosomal recessive disorders, is complicated by the imperfect correlation between mutations in the CFTR gene and CF phenotype [26,27]. In order to better understand the consequences of the greater than 1850 CFTR mutations, Rowntree and Harris have classified CFTR mutations into five groups, where each classification describes the mechanism by which a group of mutations disrupt CFTR function [27]. To further explain the inability to correlate phenotype and genotype in a supposedly simple “single-gene” disorder, Dipple and McCabe have proposed that there are two functional thresholds relating mutant protein function to phenotype. When protein function is below the first threshold, a severe phenotype will always be observed, whereas if protein function is above the second threshold, the phenotype will be consistently mild. Between these two thresholds, mutations will not necessarily correlate with phenotype and thus should be viewed as “complex traits” [28]. Complex traits are influenced by functional activity thresholds, modifier gene and system dynamics, thereby blurring the lines between genotype phenotype correlations [29–31]. For CF, there are numerous studies that also describe modulatory effects of different genes on the severity of phenotype in CF patients (e.g. mannose-binding lectin 2 and transforming growth factor beta 1) [27,32].
To further define the phenotypic heterogeneity in patients who have the same CF causing mutations, several molecular studies have analyzed the context of disease causing mutations. Researchers found that the phenotypic severity of a CF-causing mutation could be impacted by the genomic context of the CFTR gene as seen when R117H is in cis with the 5T variant and when S1251N is in cis with F508C [33,34]. Thus, the haplotypes defined in this study may assist newborn screeners and clinicians in predicting the cis/trans phase for mutations and poly T variants with variable expression without requiring parent studies. For example, when a newborn specimen is positive for R117H and either F508del, G542X or N1303K, and also carries both an 5T and a 9T variant, a clinician could use haplotype information to proceed with a strong probability that the 9T variant is in cis with F508del, G542X or N1310K and not R117H (Table 3). This study also shows that R117H may reside on the opposite chromosome from nearly all of the listed mutations in Table 3 because it has a different polymorphism background. Additionally, more extensive haplotype studies may allow researchers to determine what if any phenotypic effects results when a particular variant such as 5T are in cis or trans with an identified CF-causing mutation.
This study has defined 144 CFTR haplotypes associated with many CF-causing mutations, laying the groundwork for future research on the molecular structure of the CFTR gene and its potential influence on phenotypic heterogeneity in CF patients. In addition, these extensively characterized specimens will enhance the quality of the PT challenges offered by NSQAP to participating NBS laboratories.
Supplementary Material
Acknowledgments
The authors wish to thank Dr Cédric Le Marechal from Laboratoire de génétique moléculaire et d’histocompatibilité, Génétique moléculaire et génétique épidémiologique, CHU Hôpital Morvan in Brest, France for helpful comments and review. Sean Mochal was funded by the Research Participation Program at the Centers for Disease Control and Prevention (CDC), National Center for Environmental Health’s Division of Laboratory Sciences, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and CDC. Dr. Philip Farrell was supported by NIH grant DK 34108. All work performed was supported by CDC.
Abbreviations
- ACMG
American College of Medical Genetics
- APHL
Association of Public Health Laboratories
- CDC
Centers for Disease Control and Prevention
- CEPH
Centre de’Etude du Polymorphism Humain
- CF
Cystic Fibrosis
- CFTR
Cystic Fibrosis Transmembrane Conductance Regulator gene
- DBS
Dried Blood Spot
- EM
Expectation Maximization
- HGDP
Human Genome Diversity Cell Line Panel
- HGVS
Human Genome Variation Society
- IRT
Immunoreactive Trypsinogen
- LD
Linkage Disequilibrium
- LOD
Logarithm of Odds
- NBS
Newborn Screening
- NCBI
National Center for Biotechnology Information
- NSQAP
Newborn Screening Quality Assurance Program
- PT
Proficiency Testing
- SNPs
Single Nucleotide Polymorphisms
Footnotes
Author disclosure: Dr. Hannon, Emeritus Chief (retired) from the Newborn Screening and Molecular Biology Branch of CDC serves on the NBS Scientific Advisory Council for Advanced Liquid Logic, Inc., Research Triangle Park, North Carolina, and the Georgia Governor’s Public Health Advisory Council. Dr. Hannon also provides consulting services to National Newborn Screening and Genetic Resource Center in Austin, Texas and PerkinElmer, Inc. in Waltham, Massachusetts.
Supplementary materials related to this article can be found online at doi:10.1016/j.ymgme.2011.10.013.
Contributor Information
M. Hendrix, Email: mhendrix@cdc.gov.
C.N. Greene, Email: cgreene@cdc.gov.
S. Mochal, Email: sean.t.mochal@emory.edu.
M.C. Earley, Email: mearley@cdc.gov.
P.M. Farrell, Email: pmfarrell@wisc.edu.
M. Kharrazi, Email: Marty.Kharrazi@cdph.ca.gov.
W.H. Hannon, Email: whannon@bellsouth.net.
P.W. Mueller, Email: pmueller@cdc.gov.
References
- 1.Rock MJ. Newborn screening for cystic fibrosis. Clin Chest Med. 2007;28:297–305. doi: 10.1016/j.ccm.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Welsh MJ, Ramsey BW, Accurso FJ, Cutting GR. Cystic fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 5121–5188. [Google Scholar]
- 3.Pratt VM, Caggana M, Bridges C, Buller AM, DiAntonio L, Highsmith WE, Holtegaard LM, Muralidharan K, Rohlfs EM, Tarleton J, Toji L, Barker SD, Kalman LV. Development of genomic reference materials for cystic fibrosis genetic testing. J Mol Diagn. 2009;11:186–193. doi: 10.2353/jmoldx.2009.080149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsui L. Cystic Fibrosis Centre at the Hospital for Sick Children in Toronto. 2010. Cystic Fibrosis Mutation Database. [Google Scholar]
- 5.Crossley JR, Elliott RB, Smith PA. Dried-blood spot screening for cystic fibrosis in the newborn. Lancet. 1979;1:472–474. doi: 10.1016/s0140-6736(79)90825-0. [DOI] [PubMed] [Google Scholar]
- 6.Hammond KB, Abman SH, Sokol RJ, Accurso FJ. Efficacy of statewide neonatal screening for cystic fibrosis by assay of trypsinogen concentrations. N Engl J Med. 1991;325:769–774. doi: 10.1056/NEJM199109123251104. [DOI] [PubMed] [Google Scholar]
- 7.Comeau AM, Parad RB, Dorkin HL, Dovey M, Gerstle R, Haver K, Lapey A, O’Sullivan BP, Waltz DA, Zwerdling RG, Eaton RB. Population-based newborn screening for genetic disorders when multiple mutation DNA testing is incorporated: a cystic fibrosis newborn screening model demonstrating increased sensitivity but more carrier detections. Pediatrics. 2004;113:1573–1581. doi: 10.1542/peds.113.6.1573. [DOI] [PubMed] [Google Scholar]
- 8.Gregg RG, Wilfond BS, Farrell PM, Laxova A, Hassemer D, Mischler EH. Application of DNA analysis in a population-screening program for neonatal diagnosis of cystic fibrosis (CF): comparison of screening protocols. Am J Hum Genet. 1993;52:616–626. [PMC free article] [PubMed] [Google Scholar]
- 9.Grosse SD, Boyle CA, Botkin JR, Comeau AM, Kharrazi M, Rosenfeld M, Wilfond BS. Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Recomm Rep. 2004;53:1–36. [PubMed] [Google Scholar]
- 10.Li L, Zhou Y, Bell CJ, Earley MC, Hannon WH, Mei JV. Development and characterization of dried blood spot materials for the measurement of immunoreactive trypsinogen. J Med Screen. 2006;13:79–84. doi: 10.1258/096914106777589623. [DOI] [PubMed] [Google Scholar]
- 11.Earley MC, Laxova A, Farrell PM, Driscoll-Dunn R, Cordovado S, Mogayzel PJ, Jr, Konstan MW, Hannon WH. Implementation of the first worldwide quality assurance program for cystic fibrosis multiple mutation detection in population-based screening. Clin Chim Acta. 2011;412:1376–1381. doi: 10.1016/j.cca.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.NNSGRC. National Newborn Screening and Genetics Resource Center. Austin: [Google Scholar]
- 13.Grody WW, Cutting GR, Klinger KW, Richards CS, Watson MS, Desnick RJ. Laboratory standards and guidelines for population-based cystic fibrosis carrier screening. Genet Med. 2001;3:149–154. doi: 10.1097/00125817-200103000-00010. [DOI] [PubMed] [Google Scholar]
- 14.Watson MS, Cutting GR, Desnick RJ, Driscoll DA, Klinger K, Mennuti M, Palomaki GE, Popovich BW, Pratt VM, Rohlfs EM, Strom CM, Richards CS, Witt DR, Grody WW. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med. 2004;6:387–391. doi: 10.1097/01.GIM.0000139506.11694.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borowitz D, Parad RB, Sharp JK, Sabadosa KA, Robinson KA, Rock MJ, Farrell PM, Sontag MK, Rosenfeld M, Davis SD, Marshall BC, Accurso FJ. Cystic Fibrosis Foundation practice guidelines for the management of infants with cystic fibrosis transmembrane conductance regulator-related metabolic syndrome during the first two years of life and beyond. J Pediatr. 2009;155:S106–S116. doi: 10.1016/j.jpeds.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Highsmith WE, Burch LH, Zhou Z, Olsen JC, Boat TE, Spock A, Gorvoy JD, Quittel L, Friedman KJ, Silverman LM, et al. A novel mutation in the cystic fibrosis gene in patients with pulmonary disease but normal sweat chloride concentrations. N Engl J Med. 1994;331:974–980. doi: 10.1056/NEJM199410133311503. [DOI] [PubMed] [Google Scholar]
- 17.Lucarelli M, Narzi L, Piergentili R, Ferraguti G, Grandoni F, Quattrucci S, Strom R. A 96-well formatted method for exon and exon/intron boundary full sequencing of the CFTR gene. Anal Biochem. 2006;353:226–235. doi: 10.1016/j.ab.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 18.Morral N, Estivill X. Multiplex PCR amplification of three microsatellites within the CFTR gene. Genomics. 1992;13:1362–1364. doi: 10.1016/0888-7543(92)90071-y. [DOI] [PubMed] [Google Scholar]
- 19.Fichou Y, Genin E, Le Marechal C, Audrezet MP, Scotet V, Ferec C. Estimating the age of CFTR mutations predominantly found in Brittany (Western France) J Cyst Fibros. 2008;7:168–173. doi: 10.1016/j.jcf.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 20.NCBI. Single Nucleotide Polymophism. [Google Scholar]
- 21.The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 22.Estivill X, Bancells C, Ramos C. Geographic distribution and regional origin of 272 cystic fibrosis mutations in European populations. The Biomed CF Mutation Analysis Consortium. Hum Mutat. 1997;10:135–154. doi: 10.1002/(SICI)1098-1004(1997)10:2<135::AID-HUMU6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 23.Bobadilla JL, Macek M, Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations—correlation with incidence data and application to screening. Hum Mutat. 2002;19:575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- 24.Morral N, Nunes V, Casals T, Chillon M, Gimenez J, Bertranpetit J, Estivill X. Microsatellite haplotypes for cystic fibrosis: mutation frameworks and evolutionary tracers. Hum Mol Genet. 1993;2:1015–1022. doi: 10.1093/hmg/2.7.1015. [DOI] [PubMed] [Google Scholar]
- 25.Kuningas M, van Bodegom D, May L, Meij JJ, Slagboom PE, Westendorp RG. Common CFTR gene variants influence body composition and survival in rural Ghana. Hum Genet. 2010;127:201–206. doi: 10.1007/s00439-009-0762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kerem E, Corey M, Kerem BS, Rommens J, Markiewicz D, Levison H, Tsui LC, Durie P. The relation between genotype and phenotype in cystic fibrosis—analysis of the most common mutation (delta F508) N Engl J Med. 1990;323:1517–1522. doi: 10.1056/NEJM199011293232203. [DOI] [PubMed] [Google Scholar]
- 27.Rowntree RK, Harris A. The phenotypic consequences of CFTR mutations. Ann Hum Genet. 2003;67:471–485. doi: 10.1046/j.1469-1809.2003.00028.x. [DOI] [PubMed] [Google Scholar]
- 28.Dipple KM, McCabe ER. Phenotypes of patients with “simple” Mendelian disorders are complex traits: thresholds, modifiers, and systems dynamics. Am J Hum Genet. 2000;66:1729–1735. doi: 10.1086/302938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dipple KM, McCabe ER. Modifier genes convert “simple” Mendelian disorders to complex traits. Mol Genet Metab. 2000;71:43–50. doi: 10.1006/mgme.2000.3052. [DOI] [PubMed] [Google Scholar]
- 30.Dipple KM, Phelan JK, McCabe ER. Consequences of complexity within biological networks: robustness and health, or vulnerability and disease. Mol Genet Metab. 2001;74:45–50. doi: 10.1006/mgme.2001.3227. [DOI] [PubMed] [Google Scholar]
- 31.Scriver CR, Waters PJ. Monogenic traits are not simple: lessons from phenylketonuria. Trends Genet. 1999;15:267–272. doi: 10.1016/s0168-9525(99)01761-8. [DOI] [PubMed] [Google Scholar]
- 32.Dorfman R, Sandford A, Taylor C, Huang B, Frangolias D, Wang Y, Sang R, Pereira L, Sun L, Berthiaume Y, Tsui LC, Pare PD, Durie P, Corey M, Zielenski J. Complex two-gene modulation of lung disease severity in children with cystic fibrosis. J Clin Invest. 2008;118:1040–1049. doi: 10.1172/JCI33754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalin N, Dork T, Tummler B. A cystic fibrosis allele encoding missense mutations in both nucleotide binding folds of the cystic fibrosis transmembrane conductance regulator. Hum Mutat. 1992;1:204–210. doi: 10.1002/humu.1380010305. [DOI] [PubMed] [Google Scholar]
- 34.Kiesewetter S, Macek M, Jr, Davis C, Curristin SM, Chu CS, Graham C, Shrimpton AE, Cashman SM, Tsui LC, Mickle J, et al. A mutation in CFTR produces different phenotypes depending on chromosomal background. Nat Genet. 1993;5:274–278. doi: 10.1038/ng1193-274. [DOI] [PubMed] [Google Scholar]
- 35.Jambhekar SK, Carroll JL, Keiles S. Report of two patients with associated conditions in addition to cystic fibrosis. J Cyst Fibros. 2010;9:269–271. doi: 10.1016/j.jcf.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 36.Petreska L, Koceva S, Plaseska D, Chernick M, Gordova-Muratovska A, Fustic S, Nestorov R, Efremov GD. Molecular basis of cystic fibrosis in the Republic of Macedonia. Clin Genet. 1998;54:203–209. doi: 10.1111/j.1399-0004.1998.tb04285.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.