

Summary
The RV144 trial demonstrated 31% vaccine efficacy (VE) at preventing HIV-1 infection1. Antibodies against the HIV-1 envelope variable loops 1 and 2 (V1/V2) domain correlated inversely with infection risk2. We hypothesized that vaccine-induced immune responses against V1/V2 would selectively impact, or sieve, HIV-1 breakthrough viruses. 936 HIV-1 genome sequences from 44 vaccine and 66 placebo recipients were examined. We show that vaccine-induced immune responses were associated with two signatures in V1/V2 at amino-acid positions 169 and 181. VE against viruses matching the vaccine at position 169 was 48% (CI: 18 to 66%; p=0.0036), whereas VE against viruses mismatching the vaccine at position 181 was 78% (CI: 35% to 93%; p=0.0028). Residue 169 is in a cationic glycosylated region recognized by broadly neutralizing and RV144-derived antibodies. The predicted distance between the two signatures sites (21±7 Å), and their match/mismatch dichotomy, suggest that multiple factors may be involved in the protection observed in RV144. Genetic signatures of RV144 vaccination in V2 complement the finding of an association between high V1/V2 binding antibodies and reduced risk of HIV-1 acquisition and provide evidence that vaccine-induced V2 responses plausibly played a role in the partial protection conferred by the RV144 regimen.
Main
Vaccination with the RV144 regimen (ALVAC-HIV and AIDSVAX B/E gp120) afforded an estimated 31% protection against HIV-1 acquisition1. Two immune correlates of infection risk were identified: plasma IgA antibodies to Env were associated with increased risk, and IgG binding to Env-V1/V2 with decreased risk2. These analyses compared HIV-1 infected and uninfected vaccine recipients, and established correlates of risk3,4, which are not necessarily predictive of protection as immune responses are not randomized among vaccinees. In contrast, sieve analyses5–7 compare breakthrough viruses in vaccine and placebo recipients, leveraging the randomization to causally attribute observed differences between HIV-1 sequences to the vaccine. Sieve analyses look for evidence that vaccine-induced immune responses selectively block certain viruses and/or drive escape mutations post-infection, and interrogate treatment differences in HIV-1 sequences derived at the time of HIV-1 diagnosis as evidence for this effect.
We hypothesized that RV144 vaccine-induced antibodies to Env-V1/V2 could selectively prevent HIV-1 infections by certain variants, and that this effect would be evident in the V1/V2 region of breakthrough viruses. To test this hypothesis, we examined the relationship between vaccine status and V1/V2 sequence characteristics using 936 HIV-1 sequences from 110 breakthrough infections: 44 vaccine and 66 placebo recipients (focusing on subjects infected with HIV-1 CRF01_AE, i.e. 110 of the 121 characterized infections that occurred after the first immunization). One epidemiologically-known transmission pair was confirmed by phylogenetic reconstructions; thus, sensitivity analyses were performed after removing sequences from the individual who was the second to become HIV-1 infected to preserve independence of infection events.
Our analysis focused on HIV-1 sequences corresponding to the glycoprotein 70 gp70-V1/V2 clade B reagent used to identify the correlate of risk (AA120 to 204 of the reference sequence HXB2). Supplementary Methods S1 summarizes the pre-filtering of sites that was performed as pre-specified to increase statistical power. First, sites that were invariant or where there was little confidence in the alignment were excluded. Then, sites were selected following two approaches. The first approach, termed `contact residues', required that sites were both a) known contact residues for monoclonal antibodies or had been implicated as such in neutralization sensitivity assays8–11, and b) `hotspots' of vaccine-induced binding antibody reactivity from a linear peptide binding microarray analysis of uninfected RV144 vaccine recipients. The `contact residues' approach yielded eight sites for analysis: HXB2-positions 120, 124, 165, 166, 168, 169, 171, 181. The second approach, called EPIMAP (Epitope Prediction by Interrogating conformational ensembles of glycosylated antigens with a Multi-oriented Antibody Probe), was based on structural predictions of antibody epitopes: thousands of potential antibody epitopes (centered on gp120 surface residues for the three Env in the vaccine) were predicted and used to rank V1/V2 sites by their likelihood of being antibody targets. The EPIMAP approach yielded 12 high-ranking sites: HXB2-positions 160, 166, 168–173, 178, 179, 181, 197.
To test if vaccination (through vaccine-induced antibodies to Env-V1/V2) protected against acquisition of certain HIV-1 variants, we adapted the primary analysis method of Rerks-Ngarm and colleagues1 to assess VE against HIV-1 genotypes differing at the 15 selected sites identified by either the `contact residues' or EPIMAP approaches (Supplementary Table S1 and Figure S2; note that VE is estimated based on all RV144 participants, i.e. 8,197 vaccine and 8,198 placebo recipients). We found that VE significantly differed for HIV-1 genotypes defined by whether they presented a residue matching the vaccine insert (or not) at position 169 (p=0.034) and 181 (p=0.024) (Table 1). The estimated cumulative HIV-1 incidence curves in the vaccine and placebo groups illustrate VE for two genotypes (K169 and I181X) and no efficacy for the opposite residues (Supplementary Figure S2) (diagnostic tests did not show significant evidence of violation of the proportional hazards assumption) – and the cumulative HIV-1 incidence curves showed a trend toward waning vaccine effect on genotype over time, analogous to the temporal effect on efficacy seen in RV1441,12. The estimated VE against viruses matching the vaccine at position 169 (K169) was 48% (p=0.0036; 95% CI: 18%, 66%), whereas VE versus 169-mismatched viruses was not significant. VE against viruses differing from the vaccine insert at 181 (181X) was 78% (p=0.0028; 95% CI: 35%, 93%), whereas VE versus vaccine-matched viruses at that site was not significant. Furthermore, VE against viruses that were both 169-matched and 181-mismatched was 80% (p=0.0046; CI: 31%, 94%). These results suggest that vaccine-induced immune responses to the Env-V2 may have blocked infections with viruses matching the vaccine at K169 and differing from the vaccine at I181. We applied the statistical method of Haynes and colleagues2 to test whether gp70-V1/V2 antibodies and V2 `hotspot' antibodies were correlates of risk of infection for specific HIV-1 genotypes. The estimated association of these antibodies with genotype-specific infection risk was similar across the genotypes, such that the sieve effects are not explained by these antibody correlates. However, there is low power to detect differences by genotype because only 34 infected vaccinees could be included in the analysis (see Supplementary Table S2).
Table 1.
Estimated vaccine efficacies (VEs = 1 – Hazard Ratios (HRs)) to prevent infection with specific HIV-1 genotypes, and estimated ratios of HRs measuring the relative protection against given pairs of HIV-1 genotypes.
| Genotype | Number of Infections | VE (95% CI) | p-value | |
|---|---|---|---|---|
| Vaccine | Placebo | |||
| Overall | 44 | 66 | 34% (7.8%,54.7%) | 0.034 |
| K169 | ||||
|
| ||||
| match | 30 | 57 | 48% (18%,66%) | 0.0036 |
| K169X | 14 | 9 | −55% (−258%,33%) | 0.3 |
| I181 | ||||
|
| ||||
| match | 40 | 48 | 17% (−26%,45%) | 0.38 |
| I181X | 4 | 18 | 78% (35%,93%) | 0.0028 |
| K169-I181 | ||||
|
| ||||
| K169-I181 | 27 | 42 | 36% (−4%,61%) | 0.071 |
| K169-I181X | 3 | 15 | 80% (31%,94%) | 0.0046 |
| K169X-I181 | 13 | 6 | −116% (−467%, 19%) | 0.11 |
| K169X-I181X | 1 | 3 | 67% (−219%,97%) | 0.32 |
| Est. HR/HRa(95%CI) | p-value | |||
|---|---|---|---|---|
| K169X/K169 | 2.73 (1.08,6.92) | 0.034 | ||
| I181/I181X | 3.77 (1.19,11.92) | 0.024 | ||
| Elseb/K169-I181 | 1.04 (0.49,2.22) | 0.92 | ||
| Else/K169-I181X | 1.85 (0.79,4.32) | 0.16 | ||
| K169X-I181/Else | 2.76 (1.23,6.20) | 0.014 | ||
| Else/K169X-I181X | 1.06 (0.40,2.86) | 0.9 |
Each HR is the hazard ratio (vaccine vs. placebo) of HIV-1 infections with a particular genotype. For example, for the K169X/K169 entry, the numerator HR measures the vaccine effect to prevent HIV-1 infections with K169X-variants and the denominator HR measures the vaccine effect to prevent HIV-1 infections with K169-variants, and the result 2.73 means that the vaccine lowers the rate of infection 2.73-times more against K169-matched HIV-1 viruses than against K169-mismatched HIV-1 infections, i.e., the level of protection is 2.73 greater against K169-matchedthan against K169-mismatched HIV-1 viruses (that is against all viruses with a residue differing from K at site 169).
Else: all genotypes other than the joint genotype under consideration.
To further evaluate site-specific differences between viruses from vaccine and placebo recipients, we applied three additional site-scanning methods to both the `contact residues' and `EPIMAP' sets of sites: the nonparametric weighted distance comparison test (GWJ)13, the Mismatch Bootstrap method (MMBootstrap)7, and a model-based method that is more sensitive to differences in non-insert AA frequencies14. Based on both the `contact residues' and `EPIMAP'-derived sets of sites, the most general method GWJ identified positions 169 and 181 as significantly distinguishing HIV-1 sequences from vaccine and placebo recipients (Figure 2). These two sites showed significant or borderline significant results with the additional site-scanning methods (MMBootstrap and Model-based) (Supplementary Table S3). These results were corroborated after excluding a subject from the transmission pair: results were consistent although with weaker statistical support. As pre-specified, corrections for multiple testing were performed separately for each analysis method and for each of the three vaccine insert sequences; no correction for multiple tests was applied across the tests because these are considered sensitivity analyses meant to evaluate congruence.
Figure 2.
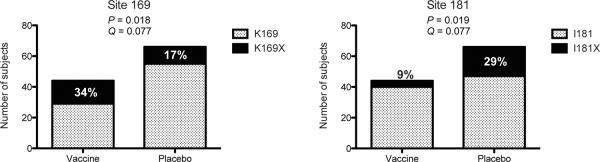
Bar graphs representing the mutations at positions 169 and 181 based on sequences from 44 vaccine and 66 placebo recipients, with p- and q-values corresponding to the site-scanning sieve analysis method GWJ. The values correspond to comparisons against the 92TH023 vaccine insert based on the selected sites identified through the `contact residues' approach. Pre-specified p-value and q-value significance thresholds of .05 and .2, respectively. Full results in Supplementary Table S3.
The fact that our sequence data were obtained through an effectively implemented randomized trial15 reduces the need for phylogenetic corrections as performed in observational studies (Supplementary Note S1). Nonetheless, to potentially assess mechanisms behind sieve effects, we tested the impact of shared ancestry among viruses in our dataset with phylogenetic dependency networks16 and independent contrasts17. Both approaches found that the sieve effect at site 181 was independent of the tree topology, whereas that of site 169 was not (Supplementary Tables S4 and S5); these results must be interpreted with caution because the sequence data were used to both infer the phylogenetic tree and to evaluate sieve effects17.
In agreement with the VE results, the consensus AA K169 was more frequently different from the CRF01_AE vaccine sequence among viruses from vaccinees, whereas site 181, the third position of the putative tri-peptide α4β7 integrin binding motif, was more likely to be similar to the vaccine sequence in vaccinees. When we tested whether sites in the two groups were evolving under different selective pressures along internal tree branches, likelihood ratio test results18 provided evidence that site 169 was under differential pressure across treatment arms (p = 0.043 based on dataset with 110 subjects; p = 0.064 based on 109 subjects) (Supplementary Table S6).
HIV-1 variants with K169X might have had longer V2 loops [Mean = 43.2 (IQR: 40–46) vs 41.9 (IQR: 39–44), p=0.11] and more potential N-linked glycosylation sites (PNGS) [Mean = 2.44 (IQR: 2–3) vs 2.12 (IQR: 2–3), p=0.08] than K169 viruses (Supplementary Table S7). Although the latter p-values were not significant, longer loops and increased PNGS have been associated with reduced sensitivity to neutralization19. The trend toward longer V2 and more PNGS in individuals with K169X was apparently not explained by a longer duration of HIV-1 infection (no difference in the time since the last HIV-1 negative visit used as a proxy for the duration of HIV-1 infection (K169X vs K169: Median = 181 vs 179, Mean = 250 vs 207, p =0.41) (Supplementary Table S7). Furthermore, studies with quaternary-structure-preferring (QSP) antibodies (e.g., PG9, PG16, CH01–04 and PGT141–145) showed that mutations at positions 169 and 181 were associated with significant alterations in neutralization10,20. Some strains became sensitive to QSP antibodies by mutating residue 169 to K21. These results suggest that K169X mutations could be a mechanism for avoidance of QSP antibodies and potentially other V2-specific antibodies. Indeed, the epitope of CH58, an anti-V2 mAb isolated from an RV144 vaccine recipient, spanned AA167-180 and position 169 was critical for binding22.
To explore whether the sieve effects at sites 169 and 181 were linked, we looked at structural data and co-variability. We found no conclusive evidence suggesting that the two sites were contained within the same epitope. The Cα-Cα distance between residues 169 and 181 was 21±7 Å (mean±St.dev) based on the 3000 low-energy all-atom models computed for the three vaccine inserts (Supplementary Table S8), while sites 169 and 181 were more than 30Å apart in the crystal structure of J08 scaffolded HIV-1 gp120 V1/V2 bound to the broadly neutralizing mAb PG923 (Supplementary Figure S3). The two sites could lie in a single antibody epitope because the average diameter of antibody-bound epitopes was predicted to be 30.5 Å (based on 32 pairs of bound antigen structures24). Furthermore, we found no evidence that the sieve effect at site 181 was linked to site 169 by analyzing co-variation on pre-selected V1/V2 sites 25,26. We noted co-variability in the mid-V2 loop, which corresponds to the binding site of the V1/V2-specific mAb from RV1442,22 (Supplementary Table S9).
The unexpected sieve effect at site 181 showing greater VE against mismatched HIV-1, suggests a vaccine-induced constraint that either hindered the establishment of infection with I181× variants or promoted infections with I181 variants. While it is plausible that vaccine-induced antibodies enhanced HIV-1 infections with I181 variants, there was no statistical support for such enhancement because there was no evidence of negative VE against I181 viruses: Est. VE = 17%, 95% CI = −26% to 45%, p=0.38 (Table 1). Other hypotheses include: i) the stricter conservation at site 181 may be a marker of a non-identified sequence characteristic (possibly linked to the variable loop that starts a few AA downstream of 181), and ii) specific variants may be unable to establish infection due to steric/quaternary structure constraints with vaccine-induced antibodies; iii) I181× viruses may be preferentially targeted by vaccine induced antibodies.
Sieve analysis is an important component of assessing immune correlates of protection because it compares vaccine to placebo recipients and could identify selective pressures below the level of detection of standard immune assays7. Our V1/V2-focused analysis leveraged randomized treatment assignments to establish a causal connection between vaccination and a selective filtering of V1/V2 variants. The identification of signatures in V2 provides a corroborative virological determinant to the V1/V2 antibodies correlate of risk2 and highlights the mid-V2 loop as an important target for antibody-mediated prevention of HIV-1 infection, but also suggests that VE is ablated by viral sequences with signature sequence variants, raising the possibility of population-level adaptation to the vaccine. Given the 31% efficacy against infection afforded by the vaccine and the suspected mechanism of action of V1/V2 Ab, our analysis suggests that specific HIV-1 variants were blocked from establishing HIV-1 infection (an acquisition sieve effect); however, viral sequences may also have mutated in response to vaccine-induced immune pressure, corresponding to a post-infection sieve effect. Further studies with infectious molecular clones carrying V1/V2 signature mutations are needed to clarify their role in viral infectivity, fitness, and escape. Together, these results vouch for sieve analysis as an integral part of the search for immune correlates of protection in vaccine studies and show that sieve analysis can be a powerful tool to assess the efficacy of new vaccine candidates.
Methods
Supplementary Figure S1 provides a schematic description of the study.
Study design
The protocol was approved by the Institutional Review Boards of the Ministry of Public Health, the Royal Thai Army, Mahidol University, and the U.S. Army Medical Research and Materiel Command. Written informed consent was obtained from all participants. The vaccine regimen consisted of four injections (at 0, 1, 3, and 6 months) of ALVAC-HIV[vCP1521], which expresses gp120 of CRF01_AE (92TH023), and two injections (at 3 and 6 months) with AIDSVAX B/E which is composed of two gp120 proteins truncated at the N-terminus (start at AA42): MN (subtype B) and CM244 (CRF01_AE).
HIV-1 sequencing
Viral genomes were amplified by endpoint-dilution PCR of viral RNA from plasma specimens collected at the time of HIV-1 diagnosis, and were directly sequenced based on methods adapted from7. Briefly, a first round near-full-length genome Polymerase Chain Reaction (PCR) was done with the Advantage LA Polymerase (50 μl-reaction), followed by a real-Time PCR (TaqMan® Gene Expression Master Mix) on 5 μl of the first round product for the detection of HIV-1 gag (186 bp) and env (232 bp). For first-round products identified as endpoint-positive by real-time PCR, two μl were subjected to a second-round amplification using KAPA LR HS DNA Polymerase. A re-amplification using 1 μl of the first-round product (dilution factor: 1 to 100) with second-round PCR primers is performed to obtain sufficient material for sequencing. PCR and sequencing primers are listed in Supplementary Tables S10 and S11.
EPIMAP
Potential patches, defined by lists of residues and atoms within each putative antibody epitope, were predicted for each vaccine insert using an unbiased, exhaustive, structure-based computational approach taking into account vaccine-specific orientational constraints. Full-length glycosylated vaccine inserts were threaded onto known structures of gp120 and regions not crystallized were built de novo. Each model was interrogated by a full-atom antibody probe placed at multiple rotations and orientation vectors were centered on each amino acid. The proportion of patches containing a given V1/V2 site were calculated for each insert (Supplementary Figure S4) and the top-ranking V1/V2 sites were selected based on the average of the percentages for the prime (92TH023) and the max of the two boost proteins (MN,CM244), yielding 22 sites (Supplementary Figure S5) of which 12 passed the conservation and alignability criteria (described in Supplementary Table S1).
Filtering of V1/V2 sites
Procedure described in Supplementary Methods S1.
Vaccine efficacy
Genotype-specific VE was assessed with the Cox proportional hazards model and score test as described by Prentice and colleagues27, and differential VE by genotype was assessed with the same model as described by Lunn and McNeil28, and Gilbert29. VEs were calculated based on one representative sequence per individual (the `mindist': smallest Hamming distance to the subject's consensus sequence) but using all the RV144 participants including uninfected ones. To test if the proportional hazards assumption was violated for any of the HIV-1 genotypes, we used the Grambsch and Therneau proportional hazards test (based on Schoenfeld residuals)30.
Sieve analyses
Site-scanning sieve analysis methods evaluate each site to identify those that discriminate the vaccine and placebo group. In addition to the differential VE analysis, the pre-selected sites were tested against the three vaccine insert sequences using three other methods: a nonparametric weighted distance comparison test (GWJ)13, a Mismatch Bootstrap method (MMBootstrap) adapted from7, and a model-based Bayesian-frequentist hybrid method that is more sensitive to differences in non-insert AA frequencies14. The GWJ method computes a two-sample pooled-variance t-statistic and compares this statistic to a permutation-derived null distribution. Each subject contributes a weight that is computed as the from-insert-AA-to-subject-AA entry in a (probability-form) substitution matrix using the subject's `mindist' sequence. The MMBootstrap method computes the difference in the fraction of mismatches-to-the-insert-AA using all available sequences, and compares this to a bootstrap-derived null distribution (resampling subject labels, not individual sequence labels). The Model-based method compares the posterior probability of a sieve effect to a null distribution (estimated by permutation) for a Bayesian multinomial model. All of these methods were verified for control of type-I error rate. A q-value multiplicity adjustment procedure was pre-specified to limit the false discovery rate to 20%31; it was conducted on a per analysis basis, i.e. per insert and per method. Code provided in Supplementary S2.
Positive selection
We tested whether the relative rates of synonymous and non-synonymous substitutions in the two groups were significantly different by applying a likelihood ratio test as implemented in Hyphy 18 (http://www.hyphy.org).
Independent contrasts
We used independent contrasts17 to test if the character state at each tip of the tree was correlated with the length of the branch leading to it, with the assumption of evolution via Brownian motion (i.e. neutral evolution). Phylogenetically independent contrasts between the vaccine status and the tip data were calculated in Mesquite (http://mesquiteproject.org/pdap_mesquite/). After the poor fit of the tree to the tip data was verified, contrasts were generated by subtracting one degree of freedom for each polytomy in the tree (n = 1) using vaccine status as the dependent variable (“positivized” contrast).
Phylogenetic dependency networks
We used phylogenetic dependency networks to identify associations between the vaccine status and every amino-acid position and state, while taking into account the shared ancestry in the HIV-1 phylogeny16. A maximum likelihood phylogenetic tree was constructed and a model of conditional adaptation was created for the vaccine status and for every position and state. The null hypothesis is that observations depend on the tree structure; then, adaptation due to each variable is modeled along the tree by an additive process. All results were adjusted for multiple comparisons, using a q-value threshold of ≤0.2 (implying a false-positive proportion of 20% among identified associations).
Co-variation
Associations between residues were detected using the Kullback-Leibler divergence co-variation and differential co-variation tests25, and a Bayesian graphical method, which explicitly models the evolutionary history of the sequences26.
Supplementary Material
Figure 1.

Phylogenetic tree of env-V1/V2 nucleotide sequences with highlights for sequences presenting mutations at either site 169 (in pink) or 181 (in yellow) or at both sites (in grey). Sequences from vaccine recipients are figured in red, those from placebo recipients are in blue.
Acknowledgments
We thank Barton F. Haynes and Frederick A. Matsen for advice and comments, and Ian A. Wilson and Robyn L. Stanfield for assistance. This study was supported in part by an Interagency Agreement Y1-AI-2642-12 between U.S. Army Medical Research and Material Command (USAMRMC) and the National Institutes of Allergy and Infectious Diseases. This work was also supported by a cooperative agreement (W81XWH-07-2-0067) between the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., and the U.S. Department of Defense (DOD). Additional support was provided to P.G. through the NIH grant 2R37AI05465-10. The opinions herein are those of the authors and should not be construed as official or representing the views of the U.S. Department of Defense or the Department of the Army.
Footnotes
Author Contributions M.R. conducted the sequence analysis with contributions from B.B.L., W.D., and B.S.M. P.T.E., and P.B.G. conducted the site-scanning sieve analyses. M.R., S.T., E.S-B., J.I.M. designed the sequencing experiments. B.B.L., L.C., P.K., S.N., J.N.S., K.W., H.Z., M.B., S.H., A.B., M.L., A.O'S., E.L., A.B., G.I., and V.A. performed viral sequencing. T.H., A.C.deC., C.A.M., H.A., and M.J. contributed statistical analyses. C.C., S.M., and W.R.S. developed the EPIMAP approach. J.S.M., I.G., P.D.K. identified antibody contact residues and performed V2 structural analyses. J.M.C. performed phylogenetic dependency network analyses. R.J.O'C., M.S.DeS., S.N., S.R.-N., M.L.R., N.L.M., J.H.K. conducted the RV144 trial. M.R., P.T.E., P.B.G., J.I.M., J.H.K. designed the studies, analyzed data, prepared the manuscript (with contributions from J.M.C., P.D.K., and W.R.S.) and supervised the project.
Sequences are available under GenBank accession numbers JX446645-JX448316. Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of this article at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Rerks-Ngarm S, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 2.Haynes BF, et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med. 2012;366:1275–1286. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Plotkin SA, Gilbert PB. Nomenclature for immune correlates of protection after vaccination. Clin Infect Dis. 2012;54:1615–1617. doi: 10.1093/cid/cis238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rolland M, Gilbert P. Evaluating Immune Correlates in HIV Type 1 Vaccine Efficacy Trials: What RV144 May Provide. AIDS Res Hum Retroviruses. 2011 doi: 10.1089/aid.2011.0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilbert PB, Self SG, Ashby MA. Statistical methods for assessing differential vaccine protection against human immunodeficiency virus types. Biometrics. 1998;54:799–814. [PubMed] [Google Scholar]
- 6.Gilbert P, Self S, Rao M, Naficy A, Clemens J. Sieve analysis: methods for assessing from vaccine trial data how vaccine efficacy varies with genotypic and phenotypic pathogen variation. J Clin Epidemiol. 2001;54:68–85. doi: 10.1016/s0895-4356(00)00258-4. [DOI] [PubMed] [Google Scholar]
- 7.Rolland M, et al. Genetic impact of vaccination on breakthrough HIV-1 sequences from the STEP trial. Nat Med. 2011;17:366–371. doi: 10.1038/nm.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei X, et al. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 9.Moore PL, et al. Limited neutralizing antibody specificities drive neutralization escape in early HIV-1 subtype C infection. PLoS Pathog. 2009;5:e1000598. doi: 10.1371/journal.ppat.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomaras GD, et al. Polyclonal B cell responses to conserved neutralization epitopes in a subset of HIV-1-infected individuals. J Virol. 2011;85:11502–11519. doi: 10.1128/JVI.05363-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch RM, et al. The B cell response is redundant and highly focused on V1V2 during early subtype C infection in a Zambian seroconverter. J Virol. 2011;85:905–915. doi: 10.1128/JVI.02006-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robb ML, et al. Risk behaviour and time as covariates for efficacy of the HIV vaccine regimen ALVAC-HIV (vCP1521) and AIDSVAX B/E: a post-hoc analysis of the Thai phase 3 efficacy trial RV 144. Lancet Infect Dis. 2012;12:531–537. doi: 10.1016/S1473-3099(12)70088-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilbert PB, Wu C, Jobes DV. Genome scanning tests for comparing amino acid sequences between groups. Biometrics. 2008;64:198–207. doi: 10.1111/j.1541-0420.2007.00845.x. [DOI] [PubMed] [Google Scholar]
- 14.Edlefsen PT. Model-Based Sieve Analysis. Available on arXiv: http://arxiv.org/abs/1206.6701.
- 15.Gilbert PB, et al. Statistical interpretation of the RV144 HIV vaccine efficacy trial in Thailand: a case study for statistical issues in efficacy trials. J Infect Dis. 2011;203:969–975. doi: 10.1093/infdis/jiq152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carlson JM, et al. Phylogenetic dependency networks: inferring patterns of CTL escape and codon covariation in HIV-1 Gag. PLoS Comput Biol. 2008;4:e1000225. doi: 10.1371/journal.pcbi.1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felsenstein J. Phylogenies and the Comparative Method. American Naturalist. 1985;125:1–15. doi: 10.1086/703055. [DOI] [PubMed] [Google Scholar]
- 18.Pond SL, Frost SD. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005;21:2531–2533. doi: 10.1093/bioinformatics/bti320. [DOI] [PubMed] [Google Scholar]
- 19.Derdeyn CA, et al. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science. 2004;303:2019–2022. doi: 10.1126/science.1093137. [DOI] [PubMed] [Google Scholar]
- 20.Moore PL, et al. Potent and broad neutralization of HIV-1 subtype C by plasma antibodies targeting a quaternary epitope including residues in the V2 loop. J Virol. 2011;85:3128–3141. doi: 10.1128/JVI.02658-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doria-Rose NA, et al. A Short Segment of the HIV-1 gp120 V1/V2 Region Is a Major Determinant of Resistance to V1/V2 Neutralizing Antibodies. J Virol. 2012;86:8319–8323. doi: 10.1128/JVI.00696-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao H-X, et al. HIV-1 Vaccine Induces Antibodies Against a structurally polymorphic site of V1/V2 Immune Pressure. Nature. Submitted. [Google Scholar]
- 23.McLellan JS, et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature. 2011;480:336–343. doi: 10.1038/nature10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubinstein ND, et al. Computational characterization of B-cell epitopes. Mol Immunol. 2008;45:3477–3489. doi: 10.1016/j.molimm.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 25.Gilbert PB, Novitsky V, Essex M. Covariability of selected amino acid positions for HIV type 1 subtypes C and B. AIDS Res Hum Retroviruses. 2005;21:1016–1030. doi: 10.1089/aid.2005.21.1016. [DOI] [PubMed] [Google Scholar]
- 26.Poon AF, Lewis FI, Pond SL, Frost SD. An evolutionary-network model reveals stratified interactions in the V3 loop of the HIV-1 envelope. PLoS Comput Biol. 2007;3:e231. doi: 10.1371/journal.pcbi.0030231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prentice RL, et al. The analysis of failure times in the presence of competing risks. Biometrics. 1978;34:541–554. [PubMed] [Google Scholar]
- 28.Lunn M, McNeil D. Applying Cox regression to competing risks. Biometrics. 1995;51:524–532. [PubMed] [Google Scholar]
- 29.Gilbert PB. Comparison of competing risks failure time methods and time-independent methods for assessing strain variations in vaccine protection. Stat Med. 2000;19:3065–3086. doi: 10.1002/1097-0258(20001130)19:22<3065::aid-sim600>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 30.Grambsch P, Therneau TM. Proportional Hazards Tests and Diagnostics Based on Weighted Residuals. Biometrika. 1994;81:515–526. [Google Scholar]
- 31.Storey JD. The positive false discovery rate: A Bayesian interpretation and the q-value. Annals of Statistics. 2003;31:2013–2035. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.