

Abstract
Phosphoinositide 3-kinase enhancer (PIKE) binds and enhances phosphatidylinositol 3-kinase (PI3K)/Akt activities. However, its physiological functions in brain have never been explored. Here we show that PIKE is important in regulating the neuronal survival and development of neocortex. During development, enhanced apoptosis is observed in the ventricular zone of PIKE knock-out (PIKE−/−) cortex. Moreover, PIKE−/− neurons show reduced dendritic complexity, dendritic branch length, and soma size. These defects are due to the reduced PI3K/Akt activities in PIKE−/− neurons, as the impaired dendritic arborization can be rescued when PI3K/Akt cascade is augmented in vitro or in PIKE−/−PTEN−/− double-knock-out mice. Interestingly, PIKE−/− mice display behavioral abnormality in locomotion and spatial navigation. Because of the diminished PI3K/Akt activities, PIKE−/− neurons are more vulnerable to glutamate- or stroke-induced neuronal cell death. Together, our data established the critical role of PIKE in regulating neuronal survival and development by substantiating the PI3K/Akt pathway.
Introduction
Normal cortical development is a highly orchestrated process that involves proper proliferation, migration, and differentiation of newly formed neurons to achieve a functional network (Chan et al., 2002). To control the number of neurons during development, apoptosis regulated by neurotrophic factors is an effective mechanism to eliminate differentiated neurons projected to an inappropriate target (Raff et al., 1993). However, our understanding of the molecular detail of this “survive or death” determination is incomplete. The implication is that the integrity of phosphatidylinositol 3-kinase (PI3K)/Akt pathway during development is necessary for preserving an intact nervous system (Backman et al., 2001; Kwon et al., 2001; Pimentel et al., 2002; Peng et al., 2004; Easton et al., 2005). It is also the central pathway for dendritogenesis that an intact PI3K/Akt pathway is essential for maintaining the complexity of dendritic arbor (Jaworski et al., 2005; Kumar et al., 2005). As such, any defect in molecules mediating the activation of PI3K/Akt might result in dwindled dendritic expansion. For example, conditional brain-derived neurotrophic factor (BDNF) knock-out mice have reduced neuron survival, dendritic arborization, and memory formation (Alcántara et al., 1997; Xu et al., 2000; Gorski et al., 2003a,b).
Phosphatidylinositol 3-kinase enhancers (PIKEs) are a family of GTPases with three isoforms, namely, PIKE-L, PIKE-S, and PIKE-A. In cultured neurons, PIKE-L interacts with various transmembrane receptors to trigger PI3K activation. Activation of mGluR-I by its agonists enhances formation of an mGluR-I-Homer-PIKE-L complex, leading to activation of PI3K and prevention of neuronal apoptosis (Rong et al., 2003). We also showed that netrin-1, a secreted laminin-related protein that plays a major role in mediating chemoattraction and chemorepulsion of axons/neurons, induces the interaction of its receptor UNCB with PIKE-L, which antagonizes the proapoptotic activity of UNC5B by enhancing PI3K activity (Tang et al., 2008). In addition, PIKE-L exerts its neuroprotective actions through protecting neuronal DNase inhibitor SET from degradation by AEP (asparagine endopeptidase, or legumain) during stoke or kainic acid treatment (Liu et al., 2008). On the other hand, PIKE-S (the C-terminal truncate of PIKE-L) is a nuclear PI3K activator in response to nerve growth factor (NGF) to exert its nuclear function (Ye et al., 2000; Ahn et al., 2004c). Hence, these findings support the idea that PIKE GTPases are important signaling molecules in protecting neurons from neurotoxic insults. However, the functions of neuronal PIKE in a physiological context have never been explored.
The aim of the present study is to functionally establish the regulatory role of PIKE on PI3K in vivo. Here, we found the PIKE knock-out (PIKE−/−) mice have smaller brains with reduced neuronal size and density in neocortex. Arborization of cortical neurons is also severely impaired, leading to memory defect and locomotion hyperactivity. These defects could be rescued by amplifying the activity of PI3K/Akt through introducing activated PI3K/Akt in vitro, or depleting PTEN in vivo. Moreover, the reduced PI3K/Akt activities in PIKE−/− lead to enhanced neuronal apoptosis during development or under neurotoxic insults. Therefore, PIKE is a critical factor in mediating neuronal survival and dendritogenesis by substantiating the PI3K/Akt pathway.
Materials and Methods
Materials.
All chemical were purchased from Sigma-Aldrich unless specifically stated. Human recombinant BDNF was obtained from PeproTech. In Situ Cell Death Detection (Fluorescein) Kit was from Roche Diagnostic. Anti-caspase 3 (active), phospho-Akt, phospho-ERK, ERK, phospho-TrkB, TrkB, phospho-mTOR (mammalian target of rapamycin), mTOR, phospho-CREB, and CREB antibodies were purchased from Cell Signaling Technology. Anti-p110α and anti-PTEN antibodies were obtained from Santa Cruz Biotechnology. Ant-Ki67 antibody was from BD Biosciences. Anti-Nestin antibody was from Covance. Anti-BrdU-FITC antibody was from Abcam. The BDNF ELISA kit was purchased from Promega.
Knock-out mice generation.
Heterozygous whole-body PIKE knock-out C57BL/6 mice with a targeted deletion of exon 3–6 of CENTG1 were generated under contract by Ozgene. Mice were then bred to homozygosity by heterozygous mating. PIKE/PTEN double-knock-out mice were generated by crossing the Cre-recombinase overexpressed PIKE−/− mice with PTEN flox/flox mice (The Jackson Laboratory) until homozygosity. Identities of the mice were examined by PCR using genomic DNA extracted from the tail as previously reported (Lesche et al., 2002; Chan et al., 2010b). All animal experiments were performed according to the care of experimental animal guideline and approved by the Institutional Animal Care and Use Committee from Emory University.
DNA and protein content in mouse brain.
Brain DNA and protein content from male mice was deduced as previously described (Easton et al., 2005). Briefly, the brains of age-matched male mutant and control mice were weighed and homogenized in TNE buffer (10 mm Tris base, 10 mm EDTA, 200 mm NaCl, pH 7.4). Samples were then diluted in TNE buffer containing 0.1 μg/ml Hoechst 33258 for measurement of the DNA content using fluorescence plate reader (BMG Labtech) at an excitation of 365 nm and an emission of 460 nm. A standard curve was generated using salmon sperm DNA. NP-40 was added to the remaining brain homogenate to a final concentration of 1%, and the lysate was clarified by centrifugation at 16,000 × g at 4°C. The protein concentration of the supernatant was determined by a BCA protein assay (Bio-Rad).
Cortical neuron culture.
Cortical primary neurons were dissected from an E18 embryo and cultured as described previously (Tang et al., 2008). DIV 3 or 7 neurons were infected with various adenoviruses as indicated. Three days after infection, pictures of the neurons were taken by fluorescence microscopy, and dendritic complexity was scored using computer software ImageJ (National Institutes of Health). For BDNF treatment, neurons of DIV 3 were stimulated with BDNF (100 ng/ml) for 3 d. The neurons were then fixed in 4% formaldehyde, permeabilized, and stained with anti-MAP2 antibody. Dendritic arborization was scored as mentioned previously.
Immunohistochemical/immunofluorescent staining.
E12.5 embryo or brain tissues were fixed in 4% paraformaldehyde, paraffin embedded, and sectioned in standard procedures. After serial rehydration and permeabilization in 0.1% TBST, sections were immunostained using specific antibodies, as indicated, and counterstained with hematoxylin using Zymed Histostain SP kit (Invitrogen) or DAPI. For BrdU incorporation, pregnant mice (E12.5) were injected with BrdU (Sigma) (50 mg/kg, i.p.) to label the dividing neuronal cells and were killed 2 h later. The embryos were fixed in 4% paraformaldehyde, paraffin embedded, and sectioned in standard procedures. DNA was denatured in 2 m HCl and stained using anti-BrdU-FITC antibody.
In vitro PI3K assay.
P110α was immunoprecipitated with anti-p110α antibody from 1 mg lysate (male brain) and washed with the following buffers: 3 times with buffer A (PBS, 1% NP-40, 1 mm DTT); 2 times with Buffer B (PBS, 0.5 m LiCl, 1 mm DTT); and 2 times with Buffer C (10 mm Tris-HCl, pH 7.4, 0.1 m NaCl, 1 mm DTT) at 4°C. PI3K activities were determined using an in vitro PI3K assay as described previously (Ye et al., 2000). The 32P-labeled phosphatidylinositol lipids were resolved on TLC.
Stroke experiment.
The middle cerebral artery occlusion (MCAO)-induced stroke was performed as previously reported (Liu et al., 2008). The animals (male) were anesthetized with 4% chloral hydrate. The rectal and masseter muscle temperatures were controlled at 37°C with a homeothermic blanket. Cerebral perfusion (CP) in the distribution of the middle cerebral artery was monitored throughout the surgical procedure with a laser Doppler (Perimed), and only animals with a >80% decrease in CP were included in this study. After 48 h, MCAO mice were killed, and brains were cut into 5 μm sections and stained with TUNEL assay. TUNEL assay was performed using In Situ Cell Death Detection (Fluorescein) Kit.
Behavioral tests.
The water maze test was performed in a circular swim arena (diameter of 116 cm, height of 75 cm) that was surrounded by extra-maze visual cues that remained in the same position for the duration of training and was filled to cover the platform by 1 cm at 22°C. Water was made opaque with nontoxic, white tempera paint. The escape platform was a circular, nonskid surface (area 127 cm2) placed in the northwest (NW) quadrant of the maze. Acquisition training consisted of 5 test days with four daily trials. Male mice entered the maze facing the wall and began each trial at a different entry point in a semirandom order. Trials lasted 60 s or until the animal mounted the platform, with a 15 min intertrial interval. A probe trial was conducted on day 6 at which time the platform was removed; the animal swam for 60 s and the time spent in the target quadrant (NW) versus the adjacent and opposite quadrants was recorded. A video camera mounted above the swim arena and linked to TopScan software recorded swim distance, swim speed, and time to platform, and was used for tracking and analysis.
The Y-maze trial was performed such that each mouse (male) was placed at the end of one arm and allowed to move freely in the maze for 8 min. The total number of arm entries and the alternation behavior were recorded by video camera and scored. Spontaneous alternation was calculated as follows: total of alternation/total arm entries − 2.
Metabolic cage studies using female mice were performed as previously described (Chan et al., 2010b).
Statistics.
Results are expressed as mean ± SEM. Statistical analysis was performed using computer software Prism (GraphPad Software). Data were considered significant when p < 0.05.
Results
PIKE−/− mice display smaller brain size with reduced cortical thickness
To study the functions of PIKE in a physiological manner, we generated whole-body PIKE−/− mice with all PIKE isoforms deleted using the loxP/Cre system (Chan et al., 2010b). In peripheral tissues, PIKE-A, the third PIKE isoform that binds Akt and elevates its kinase activity (Ahn et al., 2004a,b), regulates mammary gland development and obesity onset (Chan et al., 2010a,b). We also observed a noteworthy reduction in the brain size of adult PIKE−/− mice without alternations in the body length (7.84 ± 0.11 cm in wild-type mice vs 7.72 ± 0.17 cm in knock-out mice). Measurement of the adult brain mass confirmed the weight of PIKE−/− brain was significantly lower (0.47 ± 0.01 g in wild-type mice vs 0.41 ± 0.01 g in knock-out mice; p < 0.001, Student's t test, n = 5). To clarify how PIKE controls the brain size, we measured the total DNA and protein content in wild-type and PIKE−/− mouse brains. Both DNA (60.4 ± 1.6% of wild-type control) and protein contents (74.1 ± 5.6% of wild-type control) (Fig. 1A) were notably decreased in PIKE−/− brain, suggesting that the number of cells in PIKE−/− brain is reduced. Also, a decrease of DNA-to-protein ratio (65.2 ± 8.5% of control mice) was also recorded, which suggests that the decrease of brain size in PIKE−/− mice is a combined result of diminished cell number and the cell size.
Figure 1.
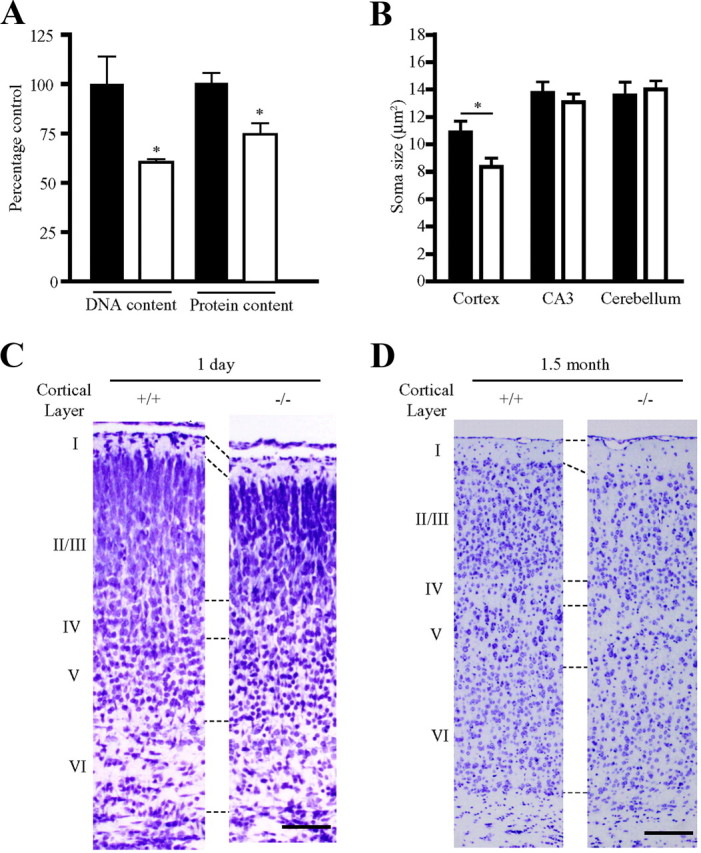
Reduction of soma size, neuronal density, and cortical thickness in PIKE−/− brain. A, DNA and protein contents in brain collected from 1.5-month-old wild-type (solid bar) and PIKE−/− (open bar) mice (n = 5). Results are expressed as a percentage relative to the wild-type control. *p < 0.05, Student's t test. B, Soma size of cortical neurons is significantly decreased in wild-type (solid bar) and PIKE−/− (open bar) mice (1.5 months old). Results are expressed as the mean ± SEM of five mice from each genotype. *p < 0.05, Student's t test. C, Nissl staining of 1-d-old wild-type (+/+) and PIKE−/− (−/−) brains. Representative results from at least three mice of each genotype are shown. Scale bar, 50 μm. D, Nissl staining of 1.5-month-old wild-type (+/+) and PIKE−/− (−/−) brains. Representative results from at least three mice of each genotype are shown. Scale bar, 100 μm.
To further confirm that the diminution of brain mass in PIKE−/− mice is caused by a reduction of neuron size, we quantified the soma area of neurons in somatosensory cortex, hippocampus, and cerebellum (Fig. 1B). We detected no significant changes in morphology and soma area of neurons in hippocampus CA3 region and cerebellum. In contrast, a significant reduction (76.9% of wild-type mice) of cell body size was detected in cortical neurons. To assess whether this defect alters the architecture of the brain, we examined the structure of somatosensory cortex histologically. Compared with the age-matched littermate controls, the cortical layers are not well defined in PIKE−/− mice, with a reduction of layer II/III thickness in both infant (Fig. 1C) and adult brains (Fig. 1D). Moreover, lower neuronal density was scored in layer II/III of PIKE−/− adult brain (169 0.0 ± 10.5 in wild-type vs 138.1 ± 10.2 neuron per field in knock-out mice; p < 0.05, Student's t test, n = 4). These histological findings further demonstrate that the reduced brain size in PIKE−/− mice is attributed to the decreased cell number and soma size in the cortex.
PIKE depletion ablates neuronal survival but not proliferation
To determine whether the decreased neurons in PIKE−/− brain resulted from dysregulation of cell proliferation or excessive cell death during cortical development, we determined the proliferation of cortical neurons of E12.5 embryo using BrdU labeling. A comparable density of proliferating cells was detected between wild-type and PIKE−/− cortexes (Fig. 2A), suggesting that neurogenesis is not affected in PIKE−/− mice. To further examine cell proliferation, we conducted Ki67 and nestin costaining (Fig. 2B). No difference was observed between wild-type control and PIKE−/− mutants, further indicating normal cortical neurogenesis. We next sought to assess the degree of cell death in PIKE−/− embryos. Markedly enhanced apoptosis was observed in the nestin-positive progenitor cells in the ventricular zone of PIKE−/− neocortex at E12.5, as revealed by TUNEL assay (Fig. 2C). The apoptosis in cortical neurons was sustained beyond the developmental stages as we observed positive caspase-3 staining in the somatosensory cortex of adult PIKE−/− mice but not in wild-type controls (Fig. 2D). Together, these data support the idea that PIKE is essential for cortical neuronal survival but dispensable for cortical neurogenesis.
Figure 2.

Enhanced apoptosis in PIKE−/− neurons in vivo. A, PIKE is not required for neurogenesis. Normal neurogenesis was detected in E12.5 neocortex in PIKE−/− (−/−) and wild-type (+/+) littermates as revealed by BrdU labeling. Scale bar, 50 μm. B, Nestin and Ki67 costaining in E12.5 neocortex in PIKE−/− (−/−) and wild-type (+/+) littermates. Scale bar, 20 μm. C, PIKE ablation enhances neuronal apoptosis during brain development. Enhanced TUNEL-positive signals were observed in ventricular zone of PIKE−/− (−/−) brain. TUNEL-positive cells are indicated with white arrows. Scale bar, 50 μm. D, Immunohistochemical staining of active caspase in adult (1.5-month-old) PIKE−/− (−/−) and wild-type (+/+) cortexes. Cells with positive signals are indicated by black arrows. Scale bar, 50 μm.
PIKE is essential for dendritic branching
We next examined whether PIKE ablation causes any morphological defect to neurons. We prepared cortical neuronal cultures from wild-type and PIKE−/− embryos (E18) and infected the neurons with adenovirus expressing GFP at DIV 7 (Fig. 3A). Twenty-four hours after infection, dendritic morphology was monitored by fluorescent microscopy. PIKE−/− neurons displayed significantly reduced dendritic branching as revealed by Sholl analysis (Fig. 3B). Total dendritic branch length (414.6 ± 31.1 μm in wild-type vs 163.8 ± 14.7 μm in knock-out mice; p < 0.001, Student's t test, n = 10) and terminal tip number (23.4 ± 1.7 in wild-type vs 9.9 ± 1.1 in knock-out mice; p < 0.001, Student's t test, n = 10) were also curtailed, underscoring that fact that PIKE plays a critical role in controlling the overall development of dendritic morphology. However, no significant change in the number of primary dendrites was observed in PIKE−/− neurons (data not shown). In agreement with the reduced soma size of cortical neurons in brain sections, the soma of cultured cortical neurons from PIKE−/− embryos was also smaller (47.0 ± 6.0 wild-type vs 29.2 ± 1.5 in knock-out mice; p < 0.05, Student's t test, n = 10). We have performed an identical analysis on hippocampal neurons isolated from PIKE−/− mice, but no significant difference was found (data not shown). Therefore, deletion of PIKE might selectively affect cortical but not hippocampal neuronal morphogenesis.
Figure 3.

Dendritic arborization is reduced in PIKE−/− neurons. A, Overall view of 7 DIV cortical neurons from wild-type (+/+) and PIKE−/− (−/−) mice. The cultured neurons were infected with adenovirus expressing GFP. Scar bar, 10 μm. B, Sholl analysis of wild-type (■) and PIKE−/− (□) cortical neurons (DIV 7). Significant difference of intercrossing begins at ∼20 μm from soma. **p < 0.01; ***p < 0.001, two-way ANOVA, n = 10.
Overexpression of active PI3K/Akt signaling rescue dendritic arborization defects in PIKE−/− neurons
The reduced dendritic arborization in PIKE−/− neurons fits with previous findings that chronic inhibition of PI3K reduces soma size and dendritic complexity (Kumar et al., 2005). Given that PIKE proteins are PI3K/Akt enhancers, it is anticipated that PI3K and Akt activity in PIKE−/− brain might be reduced. As expected, we observed a reduction of PI3K activity in both cytosolic and nuclear fractions of PIKE−/− cortex (Fig. 4A). Conceivably, the decreased PI3K activity in PIKE−/− neurons might account for the reduced dendritogenesis. To test this possibility, we infected PIKE−/− cortical neurons (DIV 3) with adenovirus overexpressing wild-type PIKE-L; dominant-negative PIKE-L KS (PIKE-L K413AS414N), which prevents the stimulation of PI3K (Rong et al., 2003); constitutively active PI3K (PI3K-CA); and Akt (Akt-CA), respectively. Three days after infection, overexpression of PIKE-L, PI3K-CA, or Akt-CA, but not PIKE-L KS, prominently increased dendritic complexity and significantly augmented spine morphology (Fig. 4B,C). Quantitative analysis revealed that the number of terminal tips (Fig. 4D) and total dendritic length (Fig. 4E) were markedly increased in PIKE-L, PI3K-CA, and Akt-CA virus-infected PIKE−/− neurons.
Figure 4.
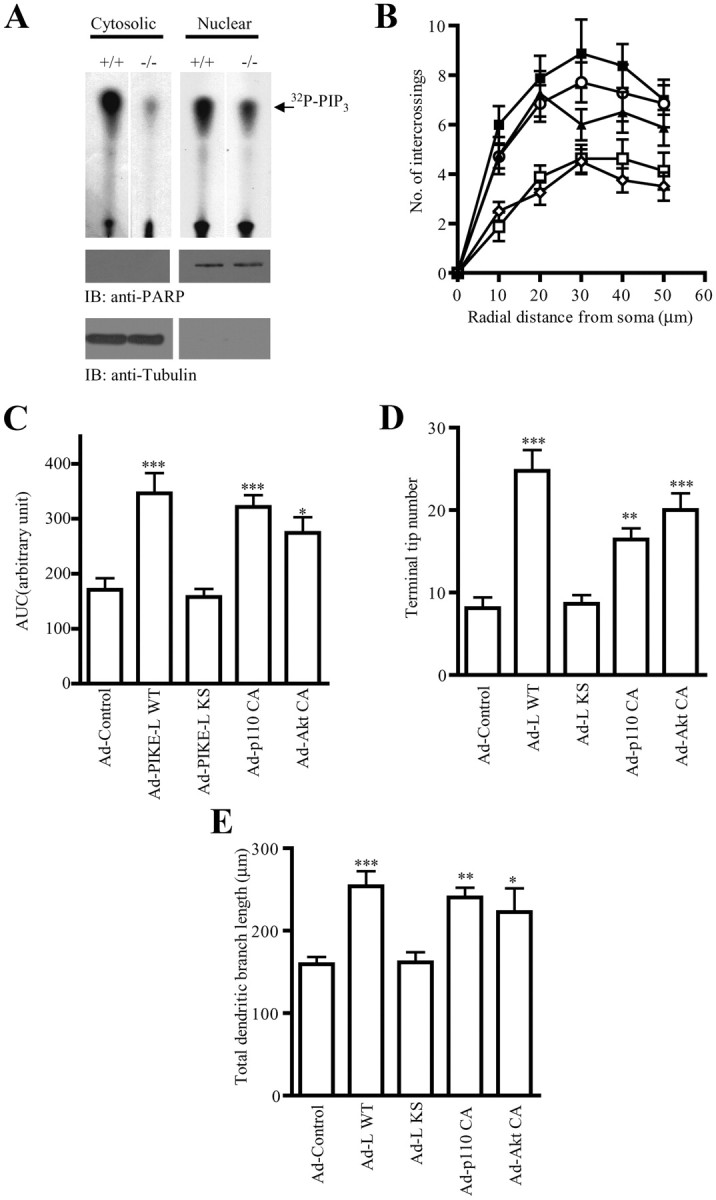
Dendritic arborization in PIKE−/− neurons could be antagonized by overexpression of PI3K/Akt in vitro. A, PI3K activities in cortex of PIKE−/− mice (+/+) and its control (+/+) littermate. Cytosolic and nuclear fractions of mouse brain tissues were isolated and subjected to immunoprecipitation using anti-p110α antibody followed by PI3K assay (top). The purity of each fraction was verified (middle, bottom). B, Sholl analysis of PIKE−/− cortical neurons infected with various adenoviruses including control virus (Ad-control, □), virus overexpressing wild-type PIKE-L (Ad-PIKE-L WT, ■), GTPase-mutated PIKE-L (Ad-PIKE-L KS, ◇), constitutively active PI3K subunit p110 (Ad-p110 CA, ○), or constitutively active Akt (Ad-Akt CA, ▴). C, Area under curve of Sholl analysis showed in B. *p < 0.05, ***p < 0.001 vs Ad-control, Student's t test, n = 8. D, Total terminal tip number of PIKE−/− cortical neurons infected with various adenoviruses as indicated. **p < 0.01, ***p < 0.001 vs Ad-control, Student's t test, n = 8. E, Total dendritic branch length of PIKE−/− cortical neurons infected with various adenoviruses as indicated. **p < 0.01, ***p < 0.001 vs Ad-control, Student's t test, n = 8.
To confirm that the impaired arborization in PIKE−/− neurons is PI3K/Akt dependent in vivo, we generated the whole-body PIKE−/−PTEN−/− double-knock-out mice and examined the morphology of the cultured cortical neurons. PTEN is a phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase that negatively regulates the PI3K/Akt pathway (Myers et al., 1998). Therefore, it is anticipated that PTEN ablation in PIKE−/− neurons should restore the PI3K/Akt function, thus rescuing the defective arborization. As expected, Akt phosphorylation was increased in PIKE−/−PTEN−/− neurons as revealed by immunofluorescence staining (Fig. 5A) and immunoblotting (Fig. 5B, first panel). Arbor complexity (Fig. 5C), total dendritic length (165.3 ± 12.3 μm in PIKE−/− vs 333.4 ± 37.7 μm in PIKE−/− PTEN−/−, p < 0.001, Student's t test, n = 10), and the number of total terminal tips (16.9 ± 0.8 in PIKE−/− vs 29.3 ± 3.5 in PIKE−/− PTEN−/− neurons; p < 0.01, Student's t test, n = 10) were also significantly increased in PIKE−/−PTEN−/− neurons than in PIKE−/− mutants. Most importantly, dendritic arborization was rescued in PIKE−/−PTEN−/− mice in vivo, as demonstrated by the Golgi staining of neurons in the somatosensory cortex (Fig. 5D). Hence, PIKE regulates neuronal dendritic branching through mediating PI3K signaling cascade.
Figure 5.

PTEN ablation enhances dendritogenesis in PIKE−/− brain. A, Overall view of cortical neurons from PIKE knock-out (PIKE−/−) and PIKE/PTEN double-knock-out (PIKE−/−PTEN−/−) mice. The cultured neurons were visualized by infecting with adenovirus expressing GFP. Phosphorylation of Akt was also examined by immunofluorescence staining. Scale bars, 20 μm. B, Verification of PTEN ablation in PIKE−/−PTEN−/− cortical neurons. Total cell lysates were collected from PIKE−/− and PIKE−/− PTEN−/− neurons, and were subjected to immunoblotting analysis using antibodies, as indicated. C, Sholl analysis of PIKE−/− (■) and PIKE−/−PTEN−/− (□) cortical neurons (7 DIV). **p < 0.01, ***p < 0.001, two-way ANOVA, n = 10. D, Camera lucida drawings of cortical neurons in layer II/III of 1.5-month-old wild-type (PIKE+/+PTEN+/+), PIKE−/− and PIKE−/−PTEN−/− brain after Golgi staining. Scale bar, 20 μm.
PIKE−/− mice display impaired spatial navigation and locomotor activity
Cortex is involved in navigation and working memory formation (Hoh et al., 2003). To test whether PIKE depletion causes any behavioral or learning ability change, we performed T-maze and Morris water maze tests in PIKE−/− mice. PIKE−/− mice demonstrated comparable learning ability with the wild-type control, as similar latencies to find the submerged platform in the water maze were recorded between these two genotypes (Fig. 6A). In the probe test, however, the mutant animals spent significantly less time (47.6 ± 4.2% in wild-type vs 35.8 ± 4.9% in PIKE−/− mice) in the quadrant where the platform used to be (Fig. 6B), suggesting that PIKE−/− mice are disabled to perform tasks involving long-term memory (Gunnersen et al., 2007). A similar observation was made in another working memory test. PIKE−/− mice also exhibited a reduction in the percentage of alternation in the Y-maze test (61.1 ± 1.9% in wild-type vs 54.0 ± 1.7% in knock-out mice; p < 0.05, Student's t test, n = 7), which further suggests that mutant mice have impaired short-term memory (Ma et al., 2007).
Figure 6.

Behavioral deficits of PIKE−/− mice. A, PIKE−/− mice possess comparable learning ability. Latency of platform finding during acquisition phase of the Morris water maze test performed by 2-month-old wild-type (■) and PIKE−/− (□) male mice (n = 7). B, Percentage time spent in each quadrant of the water tank during the probe trial of the Morris water maze test. The tank was divided into four quadrants: northeast (NE), southeast (SE), southwest (SW), and the platform. The time that the animals spent on each quadrant was recorded. *p < 0.05, Student's t test, n = 7; wild type, solid bars; PIKE−/−, open bars. C, Swim speed of the wild-type (■) and PIKE−/− (□) male mice during the acquisition phase of Morris water maze test. ***p < 0.001, two-way ANOVA, n = 7. D, Swim distance of the wild-type (■) and PIKE−/− (□) male mice during the acquisition phase of Morris water maze test. *p < 0.05, two-way ANOVA, n = 7. E, Locomotor activity of single-housed wild-type (■) and PIKE−/− (□) mice on the computerized metabolic cage over a 24 h period. Inset, Comparison of total activity between the genotypes. *p < 0.05, Student's t test, n = 6.
Interestingly, we found that PIKE−/− mice are hyperactive in both water maze and Y-maze tests. In the water maze test, both swim speed and swim distance are substantially higher in PIKE−/− mice (Fig. 6C,D). Similarly, significantly faster speed (48.4 ± 2.6 mm/s in wild-type vs 61.0 ± 4.8 mm/s in knock-out mice; p < 0.05, Student's t test, n = 7) and longer distance traveled (23,227 ± 1,264 mm in wild-type vs 29,276 ± 2,304 mm in knock-out mice; p < 0.05, Student's t test, n = 7) in the Y-maze were recorded in the PIKE−/− animals. Open field testing also revealed a higher activity in PIKE−/− mice (Chan et al., 2010b). To eliminate the possibility that the mutant animals have higher curiosity about the novel environment that causes the enhanced activity, we single housed the animals in computerized metabolic cages for 3 d. At day 4, the activity was measured over a 24 h period. As anticipated, enhanced activity was observed in PIKE−/− mice during dark phase with normal circadian rhythm (Fig. 6E).
BDNF-induced dendritogenesis is impaired in PIKE−/− neurons
Dendritic arborization is regulated by numerous external clues and cooperation of various intracellular signaling molecules. BDNF has been well recognized as a critical factor in controlling cortex development and dendritic patterning (Cohen-Cory and Fraser, 1995; Alcántara et al., 1997; Xu et al., 2000; Gorski et al., 2003b). Given that PIKE-S is a downstream mediator of another neurotrophin, NGF (Ye et al., 2000, 2002), we hypothesized that PIKE might be involved in the BDNF signaling. To test this hypothesis, we cultured cortical neurons from wild-type and PIKE−/− embryos (E18), and examined the signaling pathways induced by BDNF stimulation. As shown in Figure 7A, BDNF triggers comparable TrkB phosphorylation in both genotypes (Fig. 7A, first panel). However, both BDNF-induced PI3K activities (Fig. 7A, third panel), Akt and mTOR phosphorylation (Fig. 7A, fourth and eighth panels) were markedly decreased in PIKE−/− neurons. On the other hand, ERK and CREB phosphorylations were not affected in the absence of PIKE (Fig. 7A, sixth and tenth panels), suggesting that PIKE selectively mediates BDNF-triggered PI3K/Akt signaling but not the Ras/ERK cascades.
Figure 7.

PIKE mediates BDNF-provoked dendritic arborization. A, Signaling cascade examination in wild-type (+/+) and PIKE−/− (−/−) cortical neurons after BDNF stimulation. Cortical neurons (7 DIV) were stimulated by BDNF (10 ng/ml) for 15 min. Total cell lysates were prepared, and phospho-TrkB (first panel), phospho-Akt (fourth panel), phospho-EKR (sixth panel), phospho-mTOR (eighth panel), and phospho-CREB (10th panel) were examined by immunoblotting analysis. Total proteins of TrkB (second panel), Akt (fifth panel), ERK (seventh panel), mTOR (ninth panel), and CREB (11th panel) were determined to show equal loading. PI3K activity (third panel) was also examined by in vitro kinase assay. PIKE-L expression was checked to verify the genotype (12th panel). B, Sholl analysis of wild-type and PIKE−/− cortical neurons (3 DIV) before (■: wild-type; □: knock-out) and after (•: wild-type; ○: knock-out) BDNF (10 ng/ml, 3 d) stimulation. C, Area under curve of Sholl analysis shown in B. **p < 0.01, ***p < 0.001 vs different genotypes of the same treatment; cp < 0.001 vs the same genotype under different treatment, Student's t test, n = 15. D, Number of primary dendrite in wild-type (solid bar) and PIKE−/− (open bar) cortical neurons (3 DIV) before and after BDNF treatment. *p < 0.001 vs different genotype of the same treatment; ap < 0.05 vs the same genotype under different treatment, Student's t test, n = 15. E, Total dendritic branch length in wild-type (solid bar) and PIKE−/− (open bar) cortical neurons (3 DIV) before and after BDNF treatment. *p < 0.05, **p < 0.01 vs different genotype of the same treatment; ap < 0.05, bp < 0.01 vs the same genotype under different treatment, Student's t test, n = 15. F, Terminal tip number in wild-type (solid bar) and PIKE−/− (open bar) cortical neurons (3 DIV) before and after BDNF treatment. *p < 0.05, **p < 0.01 vs different genotype of the same treatment; ap < 0.05, bp < 0.01 vs the same genotype under different treatment, Student's t test, n = 15.
Next, we tested whether PIKE is critical for BDNF-induced dendritogenesis. Stimulation of wild-type neurons with BDNF increased the number of primary dendrites and dendritic arborization (Fig. 7B–D). However, no significant increase of these parameters occurred in BDNF-stimulated PIKE−/− neurons. On the other hand, BDNF was able to increase the total dendritic length and terminal tip number in both wild-type and PIKE−/− neurons, but the absolute value of the increment was significantly smaller in PIKE−/− neurons (Fig. 7E,F).
PIKE is necessary for protecting neurons from glutamate and ischemia attacks
BDNF plays an essential role in promoting neuronal survival in addition to dendritogenesis. Interestingly, we found that the neuroprotective function of BDNF in PIKE−/− neurons was also compromised. In the cultured wild-type cortical neurons (DIV 7), glutamate stimulation caused 20.6 ± 2.0% of wild-type neurons to undergo apoptosis. Preincubation of BDNF reduced the percentage of cell deaths to 12.0 ± 1.2%. In contrast, glutamate challenge resulted in 31.2 ± 2.1% cell death in PIKE−/− neurons, which is significantly higher than in the wild-type control neurons. Although BDNF stimulation reduced the percentage of cell deaths to 25.1 ± 2.4% in PIKE-ablated neurons, it is still significantly higher than that in the wild-type cells under the same treatment (Fig. 8A). Thus, the neuroprotective function of BDNF in excitotoxicity challenge is partially impaired in the crippled PIKE proteins.
Figure 8.
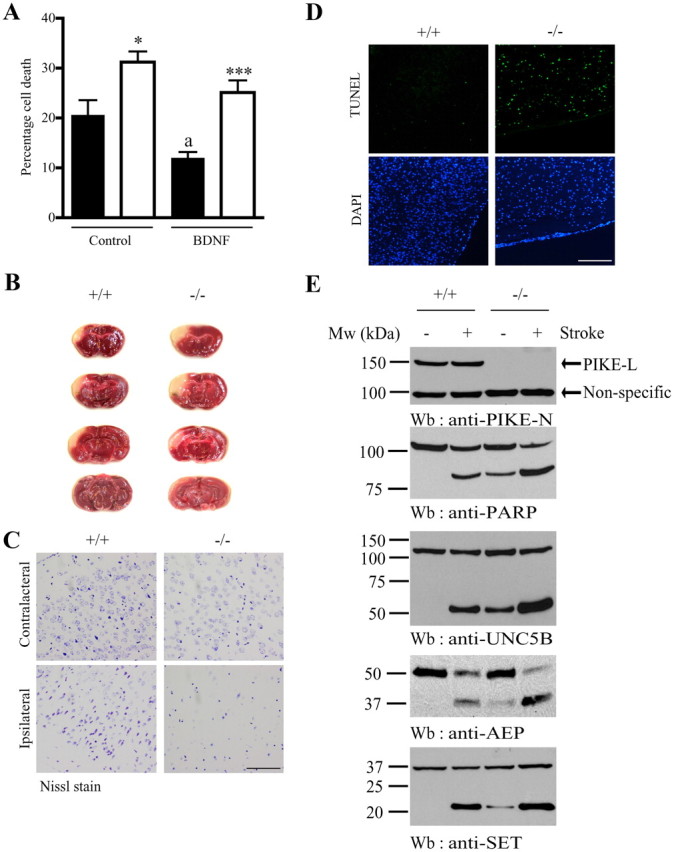
Enhanced apoptosis in PIKE−/− neurons in vitro and in vivo. A, TUNEL analysis of cortical neurons from wild-type (solid bar) and PIKE−/− (open bar) mice (7 DIV) after glutamate challenge. Neurons were pretreated with either PBS or BDNF (100 ng/ml) for 24 h followed by glutamate (50 μm) for 16 h. The number of apoptotic cells was scored under fluorescent microscope. *p < 0.05, **p < 0.01 vs different genotype of the same treatment; ap < 0.05 vs the same genotype under different treatment, Student's t test, n = 3. B, PIKE−/− mice were vulnerable to stroke attack. Triphenyl-tetrazolium chloride stained coronal brain sections from wild-type (+/+) and PIKE−/− (−/−) mice killed after MCAO. Results are representative pictures of four mice from each genotype. C, More profound cell death was observed in infarcted areas of PIKE−/− brain. Notably, more densely stained cells were found in the wild-type (+/+) sample but not in the mutant (−/−) sample. Scale bar, 50 μm. D, TUNEL assay of infarct area after MCAO. Enhanced cell death was found in PIKE-null brain. Scale bar, 100 μm. E, Enhanced apoptotic signaling in PIKE−/− brain after stroke. Thirty-six hours after MCAO, the mice were killed and the apoptotic markers in the brain were detected by immunoblotting. Increased level of apoptotic markers (PARP, second panel) was detected in PIKE−/− brain. Cleavage of PIKE-mediated neuronal effectors including UNC5B (third panel), AEP (fourth panel), and SET (fifth panel) were also examined. The expression of PIKE-L was confirmed (first panel).
To test whether PIKE is also critical in protecting cells from neurotoxic insult in vivo, we conducted ischemic experiments using the transient MCAO model. Ischemic stroke is one of the major causes leading to neuronal cell death and represents a common model of apoptotic insult to neurons. It is reported that cortical BDNF expression is increased to protect the cells from further damage after MCAO (Kokaia et al., 1995, 1998; Ferrer et al., 2001). Thus, it is anticipated that brain tissue with PIKE depletion would be more vulnerable to cell death in the affected region. After ischemic insult, damage in cortical areas was observed in both wild-type and knock-out animals (Fig. 8B). The infarct volume, however, was significantly larger in PIKE−/− brain (22.0 ± 2.1 in wild-type vs 32.5 ± 1.3 mm3 in knock-out mice; p < 0.01, Student's t test, n = 4), suggesting that PIKE−/− brains are more vulnerable to the ischemic damage. Nissl staining of the infarcted cortical area revealed extensive neuronal cell loss in PIKE−/− mice (Fig. 8C). Necrosis is the major cause of neuronal cell death after stroke, but apoptosis also plays a role in triggering cell death. More DNA degradation in infarcted brain areas was detected by TUNEL assay in PIKE−/− brain than that in wild-type control brain (Fig. 8D), implying that PIKE is indispensable for suppressing stroke-induced apoptosis. Immunoblotting analysis revealed that fragmentation of PARP spontaneously occurred in PIKE knock-out brain and was strongly increased after stroke (Fig. 8E, second panel). As expected, more pronounced SET degradation, which coupled to AEP activation, was observed in PIKE−/− mice than in control mice (Fig. 8E, fourth and fifth panels). Furthermore, degradation of UNC5B was substantially enhanced in PIKE knock-out mouse brains after stroke compared with wild-type mouse brains (Fig. 8F, third panel). Hence, our results support that PIKE is critical for preventing neuronal cell death upon glutamate challenge or ischemic stroke.
Discussion
In the present study, we report that PIKE GTPases are important in controlling neuronal survival and dendritic arborization by maintaining the integrity of the PI3K/Akt cascade. PIKE−/− brain has a thinner cortex, less neuronal density, and smaller neuronal soma size (Fig. 1). This reduction in neuronal number is a result of enhanced apoptosis during development as apoptotic neurons were readily detectable in both infant and adult brains (Fig. 2). Moreover, dendritic arborization is impaired in PIKE−/− neurons (Fig. 3). All these morphological defects in PIKE−/− neurons are caused by reduced PI3K/Akt signaling (Figs. 4, 5), which is partially incited by BDNF resistance (Fig. 6). Consequently, mice lacking PIKE are retarded in performing tasks that involve working memory (Fig. 7) and are more vulnerable to neurotoxic insults like stroke-induced cell death (Fig. 8). Therefore, our results suggest that PIKE is an important factor for neuronal development, survival, and function by substantiating the PI3K/Akt cascade both in vitro and in vivo.
PI3K/Akt signaling has been implicated in essential aspects of axonal and dendritic morphogenesis during nervous system development, including axon/dendrite elongation, guidance, and branching (Cosker et al., 2008; Luikart et al., 2008). It is also fundamental for the maintenance of appropriate architecture of individual neurons and thus influences connectivity in the adult brain (Kwon et al., 2006; van Diepen and Eickholt, 2008). Here, we show that dendritic morphology is prominently attenuated in PIKE−/− neurons (Fig. 3). Introduction of wild-type PIKE-L but not GTPase-deficient mutant PIKE-L-KS evidently increased the dendritic complexity (Fig. 4), indicating that the GTPase activity of PIKE is critical for this process and fitting our previous studies that PIKE upregulates PI3K/Akt signaling in a GTPase-dependent manner (Ye et al., 2000; Rong et al., 2003; Ahn et al., 2004b). Further, we show that overexpression of PI3K-CA or Akt-CA in vitro or deleting PTEN in vivo notably augments the dendritic patterning in PIKE−/− neurons (Fig. 5), supporting the idea that reduced dendritic morphology is mainly attributed to the diminished PI3K/Akt activity in PIKE−/− neurons. We also hypothesized that PIKE might be a downstream effector of extracellular clues for dendritogenesis via PI3K/Akt pathway. As expected, we find that PIKE−/− neurons are partially irresponsive toward BDNF-induced dendritic arborization but not dendritic elongation (Fig. 7). Intriguingly, PIKE−/− neurons are selectively resistant toward BDNF-stimulated PI3K but not ERK activation. It is reported that dendritic growth and arborization are controlled by PI3K/Akt and Raf/ERK pathways and that the PI3K/Akt cascade is mainly responsible for dendritic complexity increase, whereas the dendritic length growth is coregulated by PI3K/Akt and Raf/ERK signaling (Jaworski et al., 2005; Kumar et al., 2005). Conceivably, it is the BDNF-induced ERK activation that causes the slight increase of dendritic length in the PI3K/Akt-defective PIKE−/− neurons.
Neurotrophins also regulate the survival of several types of neurons by provoking the PI3K/Akt pathway. We have shown previously that PIKE-S is a downstream effector of NGF signaling to mediate the antiapoptotic actions (Ye et al., 2000, 2002; Ahn et al., 2004c). We show here that PIKEs are also mediators of BDNF signaling. Indeed, PIKE−/− mice shared striking phenotypic similarities with cortex-specific BDNF/TrkB knock-out mice. Both of them have compressed cortical thickness in layer II/II, diminished soma size of cortical neurons, decreased cortical neuron number, reduced dendritic complexity, enhanced locomotor activity, and impaired memory (Xu et al., 2000; Gorski et al., 2003a). Because a detailed study on cortical cell survival in the embryonic cortex has not been performed in BDNF−/− or TrkB−/− mice, it is unknown whether enhanced apoptosis, as detected in the neocortex of PIKE−/− embryos, will be found in these animals. In contrast with the severe loss of sympathetic neurons in dorsal root and superior cervical ganglia in BDNF or TrkB−/− mice (Klein et al., 1993; Jones et al., 1994), we found no such significant defects in PIKE−/− mice (data not shown), suggesting that BDNF-induced PIKE functions are mainly restricted in the CNS.
We observed a significant number of neurons in the cortex of adult PIKE−/− brains undergoing apoptosis in both basal and neurotoxic conditions (Figs. 2, 8), although the cortical BDNF content is comparable between wild-type and PIKE−/− mice as determined by BDNF ELISA (178.8 ± 16.9 in wild-type vs 163.9 ± 24.6 pg/mg lysate in knock-out mice; n = 4). BDNF is well known for its neuroprotective functions during various neurotoxic insults. It has been reported that BDNF protects against glutamate-induced apoptotic cell death via PI3K and ERK pathways in vitro (Almeida et al., 2005). Moreover, in pathological conditions like forebrain ischemia, infusion of BDNF shortly after stroke can effectively reduce total infarct volume (Yamashita et al., 1997). Indeed, increased BDNF expression after ischemia and seizure induction is suggested as a protective mechanism against excessive neuronal death (Tsukahara et al., 1994; Kokaia et al., 1995, 1998). Therefore, it is logical to find that PIKE−/− neurons, which are partially defective in BDNF signaling, are more vulnerable to both glutamate- and stroke-induced cell death.
Dendrites are the primary sites in which neurons receive, process, and integrate inputs from multiple presynaptic partners. The functions of dendrites are dependent on the branching pattern and specialization (Cosker and Eickholt, 2007). The development of a highly branched dendritic tree is thus essential for the establishment of functional neuronal connections. In fact, several studies have shown that learning processes alter the dendritic morphology of pyramidal cells in the cerebral cortex (Turner et al., 2003; Kolb et al., 2008; Gelfo et al., 2009). In general, stimulation induces an area-dependent dendritic growth related to the experience. One of these important learning experiences is spatial navigation. Although most of the effort to study the mechanisms of spatial navigation has focused on the hippocampus, many studies suggest that cortex also plays a role in this process. For example, rats with lesions in a variety of cortical areas show impaired water maze task performance (Hoh et al., 2003). It has also been shown that spatial task learning increases dendritic length and branching of neurons in layer III of occipital cortex (Kolb et al., 2008). BDNF/PIKE signaling is also involved in the spatial learning process as forebrain-specific BDNF conditional knock-out mice or PIKE−/− mice are defective in the water maze test (Fig. 6) (Gorski et al., 2003a). Presumably, wiring of the nervous system and the neuronal circuit in BDNF−/− or PIKE−/− brains is impaired, leading to a reduced ability in performing memory task.
The energy metabolism is altered in PIKE−/− mice that the knock-out animals preferentially use lipid as the energy source without changing the amount of food intake (Chan et al., 2010b). It has been reported that perinatal malnutrition reduces the brain mass, cortical and hippocampal BDNF expression in cortex, and the ability of memory formation (Wang and Xu, 2007; Hernández et al., 2008). Surprisingly, overnutrition during fetal development also reduces BDNF expression and dendritic arborization in hippocampus (Tozuka et al., 2010), suggesting that a balanced energy supply is necessary for proper brain development. Although we have provided evidence in the current study that the TrkB signaling defect is not caused by reduced BDNF concentration but by an intrinsic impairment of the intracellular signaling component, we could not completely exclude the possibility that the change of energy metabolism in PIKE−/− brain, if any, contributes to the reduction of brain mass, behavioral abnormalities, and neuronal vulnerability to insults. A detailed examination of neuronal metabolic profile of PIKE−/− mice is thus necessary to address the issue.
Collectively, our data support that the idea that PIKEs are critical neurotrophin-mediated regulators of neuronal development and survival under both physiological and pathological conditions.
Footnotes
This work is supported by NIH Grants RO1 (NS45627) to K.Y. and R01 (NS49478) to M.Y. We thank the Rodent Behavioral Core Facility of Emory University for performing the Morris water maze and Y-maze, and the Histology and EM Laboratory of Yerkes National Primate Research Center, Emory University for the preparation of the tissue sections.
The authors declare no competing financial interests.
References
- Ahn JY, Rong R, Kroll TG, Van Meir EG, Snyder SH, Ye K. PIKE (phosphatidylinositol 3-kinase enhancer)-A GTPase stimulates Akt activity and mediates cellular invasion. J Biol Chem. 2004a;279:16441–16451. doi: 10.1074/jbc.M312175200. [DOI] [PubMed] [Google Scholar]
- Ahn JY, Hu Y, Kroll TG, Allard P, Ye K. PIKE-A is amplified in human cancers and prevents apoptosis by up-regulating Akt. Proc Natl Acad Sci U S A. 2004b;101:6993–6998. doi: 10.1073/pnas.0400921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn JY, Rong R, Liu X, Ye K. PIKE/nuclear PI 3-kinase signaling mediates the antiapoptotic actions of NGF in the nucleus. EMBO J. 2004c;23:3995–4006. doi: 10.1038/sj.emboj.7600392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcántara S, Frisén J, del Río JA, Soriano E, Barbacid M, Silos-Santiago I. TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell death. J Neurosci. 1997;17:3623–3633. doi: 10.1523/JNEUROSCI.17-10-03623.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida RD, Manadas BJ, Melo CV, Gomes JR, Mendes CS, Grãos MM, Carvalho RF, Carvalho AP, Duarte CB. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 2005;12:1329–1343. doi: 10.1038/sj.cdd.4401662. [DOI] [PubMed] [Google Scholar]
- Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- Chan CB, Liu X, Ensslin MA, Dillehay DL, Ormandy CJ, Sohn P, Serra R, Ye K. PIKE-A is required for prolactin-mediated STAT5a activation in mammary gland development. EMBO J. 2010a;29:956–968. doi: 10.1038/emboj.2009.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CB, Liu X, Jung DY, Jun JY, Luo HR, Kim JK, Ye K. Deficiency of phosphoinositide 3-kinase enhancer protects mice from diet-induced obesity and insulin resistance. Diabetes. 2010b;59:883–893. doi: 10.2337/db09-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WY, Lorke DE, Tiu SC, Yew DT. Proliferation and apoptosis in the developing human neocortex. Anat Rec. 2002;267:261–276. doi: 10.1002/ar.10100. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S, Fraser SE. Effects of brain-derived neurotrophic factor on optic axon branching and remodelling in vivo. Nature. 1995;378:192–196. doi: 10.1038/378192a0. [DOI] [PubMed] [Google Scholar]
- Cosker KE, Eickholt BJ. Phosphoinositide 3-kinase signalling events controlling axonal morphogenesis. Biochem Soc Trans. 2007;35:207–210. doi: 10.1042/BST0350207. [DOI] [PubMed] [Google Scholar]
- Cosker KE, Shadan S, van Diepen M, Morgan C, Li M, Allen-Baume V, Hobbs C, Doherty P, Cockcroft S, Eickholt BJ. Regulation of PI3K signalling by the phosphatidylinositol transfer protein PITPalpha during axonal extension in hippocampal neurons. J Cell Sci. 2008;121:796–803. doi: 10.1242/jcs.019166. [DOI] [PubMed] [Google Scholar]
- Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Krupinski J, Goutan E, Martí E, Ambrosio S, Arenas E. Brain-derived neurotrophic factor reduces cortical cell death by ischemia after middle cerebral artery occlusion in the rat. Acta Neuropathol. 2001;101:229–238. doi: 10.1007/s004010000268. [DOI] [PubMed] [Google Scholar]
- Gelfo F, De Bartolo P, Giovine A, Petrosini L, Leggio MG. Layer and regional effects of environmental enrichment on the pyramidal neuron morphology of the rat. Neurobiol Learn Mem. 2009;91:353–365. doi: 10.1016/j.nlm.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Balogh SA, Wehner JM, Jones KR. Learning deficits in forebrain-restricted brain-derived neurotrophic factor mutant mice. Neuroscience. 2003a;121:341–354. doi: 10.1016/s0306-4522(03)00426-3. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Zeiler SR, Tamowski S, Jones KR. Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J Neurosci. 2003b;23:6856–6865. doi: 10.1523/JNEUROSCI.23-17-06856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnersen JM, Kim MH, Fuller SJ, De Silva M, Britto JM, Hammond VE, Davies PJ, Petrou S, Faber ES, Sah P, Tan SS. Sez-6 proteins affect dendritic arborization patterns and excitability of cortical pyramidal neurons. Neuron. 2007;56:621–639. doi: 10.1016/j.neuron.2007.09.018. [DOI] [PubMed] [Google Scholar]
- Hernández A, Burgos H, Mondaca M, Barra R, Núñez H, Pérez H, Soto-Moyano R, Sierralta W, Fernández V, Olivares R, Valladares L. Effect of prenatal protein malnutrition on long-term potentiation and BDNF protein expression in the rat entorhinal cortex after neocortical and hippocampal tetanization. Neural Plast. 2008;2008:646919. doi: 10.1155/2008/646919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoh TE, Kolb B, Eppel A, Vanderwolf CH, Cain DP. Role of the neocortex in the water maze task in the rat: a detailed behavioral and Golgi-Cox analysis. Behav Brain Res. 2003;138:81–94. doi: 10.1016/s0166-4328(02)00237-1. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KR, Fariñas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–122. [PubMed] [Google Scholar]
- Kokaia Z, Zhao Q, Kokaia M, Elmér E, Metsis M, Smith ML, Siesjö BK, Lindvall O. Regulation of brain-derived neurotrophic factor gene expression after transient middle cerebral artery occlusion with and without brain damage. Exp Neurol. 1995;136:73–88. doi: 10.1006/exnr.1995.1085. [DOI] [PubMed] [Google Scholar]
- Kokaia Z, Andsberg G, Yan Q, Lindvall O. Rapid alterations of BDNF protein levels in the rat brain after focal ischemia: evidence for increased synthesis and anterograde axonal transport. Exp Neurol. 1998;154:289–301. doi: 10.1006/exnr.1998.6888. [DOI] [PubMed] [Google Scholar]
- Kolb B, Cioe J, Comeau W. Contrasting effects of motor and visual spatial learning tasks on dendritic arborization and spine density in rats. Neurobiol Learn Mem. 2008;90:295–300. doi: 10.1016/j.nlm.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Kumar V, Zhang MX, Swank MW, Kunz J, Wu GY. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci. 2005;25:11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29:404–411. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, Wu H. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32:148–149. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- Liu Z, Jang SW, Liu X, Cheng D, Peng J, Yepes M, Li XJ, Matthews S, Watts C, Asano M, Hara-Nishimura I, Luo HR, Ye K. Neuroprotective actions of PIKE-L by inhibition of SET proteolytic degradation by asparagine endopeptidase. Mol Cell. 2008;29:665–678. doi: 10.1016/j.molcel.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Zhang W, Wayman GA, Kwon CH, Westbrook GL, Parada LF. Neurotrophin-dependent dendritic filopodial motility: a convergence on PI3K signaling. J Neurosci. 2008;28:7006–7012. doi: 10.1523/JNEUROSCI.0195-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma MX, Chen YM, He J, Zeng T, Wang JH. Effects of morphine and its withdrawal on Y-maze spatial recognition memory in mice. Neuroscience. 2007;147:1059–1065. doi: 10.1016/j.neuroscience.2007.05.020. [DOI] [PubMed] [Google Scholar]
- Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Jiang BH, Yang PH, Cao Z, Shi X, Lin MC, He ML, Kung HF. Phosphatidylinositol 3-kinase signaling is involved in neurogenesis during Xenopus embryonic development. J Biol Chem. 2004;279:28509–28514. doi: 10.1074/jbc.M402294200. [DOI] [PubMed] [Google Scholar]
- Pimentel B, Rodríguez-Borlado L, Hernández C, Carrera AC. A role for phosphoinositide 3-kinase in the control of cell division and survival during retinal development. Dev Biol. 2002;247:295–306. doi: 10.1006/dbio.2002.0703. [DOI] [PubMed] [Google Scholar]
- Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA, Tu J, Worley PF, Snyder SH, Ye K. PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci. 2003;6:1153–1161. doi: 10.1038/nn1134. [DOI] [PubMed] [Google Scholar]
- Tang X, Jang SW, Okada M, Chan CB, Feng Y, Liu Y, Luo SW, Hong Y, Rama N, Xiong WC, Mehlen P, Ye K. Netrin-1 mediates neuronal survival through PIKE-L interaction with the dependence receptor UNC5B. Nat Cell Biol. 2008;10:698–706. doi: 10.1038/ncb1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozuka Y, Kumon M, Wada E, Onodera M, Mochizuki H, Wada K. Maternal obesity impairs hippocampal BDNF production and spatial learning performance in young mouse offspring. Neurochem Int. 2010;57:235–247. doi: 10.1016/j.neuint.2010.05.015. [DOI] [PubMed] [Google Scholar]
- Tsukahara T, Yonekawa Y, Tanaka K, Ohara O, Wantanabe S, Kimura T, Nishijima T, Taniguchi T. The role of brain-derived neurotrophic factor in transient forebrain ischemia in the rat brain. Neurosurgery. 1994;34:323–331. doi: 10.1227/00006123-199402000-00016. [DOI] [PubMed] [Google Scholar]
- Turner CA, Lewis MH, King MA. Environmental enrichment: effects on stereotyped behavior and dendritic morphology. Dev Psychobiol. 2003;43:20–27. doi: 10.1002/dev.10116. [DOI] [PubMed] [Google Scholar]
- van Diepen MT, Eickholt BJ. Function of PTEN during the formation and maintenance of neuronal circuits in the brain. Dev Neurosci. 2008;30:59–64. doi: 10.1159/000109852. [DOI] [PubMed] [Google Scholar]
- Wang L, Xu RJ. The effects of perinatal protein malnutrition on spatial learning and memory behaviour and brain-derived neurotrophic factor concentration in the brain tissue in young rats. Asia Pac J Clin Nutr. 2007;16(Suppl 1):467–472. [PubMed] [Google Scholar]
- Xu B, Zang K, Ruff NL, Zhang YA, McConnell SK, Stryker MP, Reichardt LF. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron. 2000;26:233–245. doi: 10.1016/s0896-6273(00)81153-8. [DOI] [PubMed] [Google Scholar]
- Yamashita K, Wiessner C, Lindholm D, Thoenen H, Hossmann KA. Post-occlusion treatment with BDNF reduces infarct size in a model of permanent occlusion of the middle cerebral artery in rat. Metab Brain Dis. 1997;12:271–280. doi: 10.1007/BF02674671. [DOI] [PubMed] [Google Scholar]
- Ye K, Hurt KJ, Wu FY, Fang M, Luo HR, Hong JJ, Blackshaw S, Ferris CD, Snyder SH. Pike. A nuclear gtpase that enhances PI3kinase activity and is regulated by protein 4.1N. Cell. 2000;103:919–930. doi: 10.1016/s0092-8674(00)00195-1. [DOI] [PubMed] [Google Scholar]
- Ye K, Aghdasi B, Luo HR, Moriarity JL, Wu FY, Hong JJ, Hurt KJ, Bae SS, Suh PG, Snyder SH. Phospholipase C gamma 1 is a physiological guanine nucleotide exchange factor for the nuclear GTPase PIKE. Nature. 2002;415:541–544. doi: 10.1038/415541a. [DOI] [PubMed] [Google Scholar]