

Abstract
The effects of applying clinical versus neuropathological diagnosis and the inclusion of cases with coincident neuropathological diagnoses have not been assessed specifically when studying cerebrospinal fluid (CSF) biomarker classification cutoffs for patients with neurodegenerative diseases that cause dementia. Thus, 142 neuropathologically diagnosed neurodegenerative dementia patients [71 Alzheimer’s disease (AD), 29 frontotemporal lobar degeneration (FTLD), 3 amyotrophic lateral sclerosis, 7 dementia with Lewy bodies, 32 of which cases also had coincident diagnoses] were studied. 96 % had enzyme-linked immunosorbant assay (ELISA) CSF data and 77 % had Luminex CSF data, with 43 and 46 controls for comparison, respectively. Aβ42, total, and phosphorylated tau181 were measured. Clinical and neuropathological diagnoses showed an 81.4 % overall agreement. Both assays showed high sensitivity and specificity to classify AD subjects against FTLD subjects and controls, and moderate sensitivity and specificity for classifying FTLD subjects against controls. However, among the cases with neuropathological diagnoses of AD plus another pathology (26.8 % of the sample), 69.4 % (ELISA) and 96.4 % (Luminex) were classified as AD according to their biomarker profiles. Use of clinical diagnosis instead of neuropathological diagnosis led to a 14–17 % underestimation of the biomarker accuracy. These results show that while CSF Aβ and tau assays are useful for diagnosis of AD and neurodegenerative diseases even at MCI stages, CSF diagnostic analyte panels that establish a positive diagnosis of Lewy body disease and FTLD are also needed, and must be established based on neuropathological rather than clinical diagnoses.
Keywords: Biomarker, Cerebrospinal fluid, Alzheimer’s disease, Frontotemporal lobar degeneration, Amyloid beta, Tau
Introduction
Alzheimer’s disease (AD) is the most common dementia and the most frequent neurodegenerative disease [31]. The neuropathological hallmarks of AD are extracellular deposits of Aβ in senile plaques and intracellular aggregates of tau protein in neurofibrillary tangles [3, 26, 32, 39]. The second most common cause of neurodegenerative disease dementia is frontotemporal lobar degeneration (FTLD), a heterogeneous group of neurodegenerative diseases characterized by deposits of TDP-43 (FTLD-TDP), tau (FTLD-TAU) or FUS (FTLD-FUS) [36]. Deposition of α-synuclein in Lewy bodies defines the pathology of Parkinson’s disease without (PD) or with dementia (PDD) [14, 17] and dementia with Lewy bodies (DLB) [37]. While the signature pathologies of these different forms of dementia are well defined, these pathologies frequently co-occur in patients with a neurodegenerative dementia. However, the presence of coincident neurodegenerative disease pathologies in a given patient classified clinically as having either AD, PD, PDD, DLB or one of the major forms of FTLD often is not anticipated prior to postmortem examination [30,31,53].
The identification of characteristic protein aggregates in the brains of patients with these diseases has lead to the development of promising biochemical [5, 55] and neuro-imaging biomarkers [16, 45]. Cerebrospinal fluid (CSF) and positron emission tomography (PET) Aβ measurements show a strong correlation with each other [60] and with AD-related Aβ senile plaque pathology [27]. CSF phosphorylated tau (p-tau) and total tau (t-tau) are also established AD biomarkers and both CSF tau and Aβ have recently been added to the clinical diagnostic criteria for mild cognitive impairment (MCI) and AD [2, 11, 38]. For FTLD patients there are few established neuroimaging or biochemical markers [24, 25], while biomarkers of PD, PDD and DLB are just emerging [41, 47, 57]. So far, the diagnostic accuracy of biomarkers has been determined primarily based on their correlation with clinical diagnostic criteria [5, 48, 63]. Most studies on autopsy-confirmed cases have included small numbers of patients [4, 21, 56]. There are only three CSF biomarker studies with moderate [15] to large numbers (>100) of subjects with neuropathologically confirmed diagnoses [9,59]. However, none of these studies analyzed how overlapping neurodegenerative disease pathologies in patients with clinical evidence of dementia during life influence antemortem CSF biomarker levels and diagnostic accuracy established by postmortem neuropathology studies. Patients with coincident neuropathological diagnoses can represent an important and challenging part of the recruited subjects in clinical trials involving drugs directed specifically against deposits that are thought to be disease specific. Therefore, if a patient has AD and DLB, immunotherapy against amyloid or tau deposits could improve AD pathology, but would not affect α-synuclein deposits and patients could still show cognitive decline even if there is a response of amyloid and tau pathologies to the treatment.
To enable more accurate diagnosis of co-occurring neurodegenerative diseases prior to death, we compared biomarker studies with post-mortem neuropathology studies in a cohort of patients with clinical dementia who had been followed with CSF sampling until death. First, we defined cases neuropathologically, differentiating those with a single neuropathological disease from those with coincident neuropathological diagnoses. In a second step, we built a classification algorithm using clinical controls and neuropathologically diagnosed cases with a single neuropathological disease, either FTLD or AD. This was done using a training–validation scheme that had three diagnostic categories: clinical controls, AD neuropathology, and FTLD neuropathology. In a third step, we tested our model in the complete sample that included the patients who that had two coincident neuropathological diseases and obtained the different sensitivity and specificity values. Finally, we analyzed which would have been the estimated accuracy measures for the biomarkers if we used clinical diagnosis instead of neuropathological diagnosis and determine if there was a change in the classification algorithm. We included data for ELISA and Luminex platforms in the study so that the results would be useful for researchers that use either one of the platforms, but comparing these platforms was not the purpose of the study.
Materials and methods
Subjects
Subjects included 142 patients with clinical diagnoses of AD, DLB or frontotemporal dementia (FTD), and autopsy confirmation of their neuropathological diagnoses (Table 1). Neuropathological and clinical information [including mini-mental state exam (MMSE) and clinical dementia rating (CDR)], as well as CSF biomarker data, were gathered for these subjects, along with clinical and CSF biomarker data from 66 age-matched cognitively normal living controls. All data were obtained from the Penn Center for Neurodegenerative Disease Research (CNDR) Integrated Neurodegenerative Disease Database (INDD) [69]. CSF samples for the biomarker studies had been obtained at the Penn Memory Center (PMC), which is the clinical home of the AD Core Center (ADCC), and the Penn FTD Center (FTDC) [21]. CSF tau and Aβ were assessed as described below. APOE genotype and general demographic information also were retrieved from the INDD.
Table 1.
Characteristics of the neuropathological and clinical diagnosed subjects included in the study
| Neuropathological
|
Controls
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| “Pure” AD (n = 71) | AD-DLB (n = 24) | AD-FTLD (n = 2) | FTLD-TDP (n = 13) | ALS (n = 3) | FTLD-TAU (n = 13) | FTLD-TDP-TAU (n = 3) | FTLD-AD (n = 6) | DLB-AD (n = 6) DLB (n = 1) |
ELISA (n = 43) | Luminex (n = 46) | |
| ELISA | 66 (93.0 %) | 24 (100 %) | 2 (100 %) | 13 (100 %) | 2 (66.7 %) | 13 (100 %) | 3 (100 %) | 6 (100 %) | 7 (100 %) | 43 | – |
| Luminex | 62 (87.3 %) | 19 (79.2 %) | 2 (100 %) | 9 (69.2 %) | 1 (33.3 %) | 7 (53.8 %) | 2 (66.7 %) | 4 (66.7 %) | 4 (57 %) | – | 46 |
| Neuropathological diagnosis | – | – | 2 AD-PSP | – | 2AGD 5 CBD 1 PGRN 2 PSP 2 TPSD 1 Unclas. T |
3 FTLD-TDP and AGD | 1 AGD-AD 4 FTLD-TDP-AD 1 PSP-AD |
1 DLB 6 DLB-AD |
|||
| CERAD staging | 4 % B 96 % C |
8 % B 92 % C |
50 % B 50 % C |
92 % No pl. 8 % A |
100 % No pl. | 69 % No pl. 8 % A 8 % B 15 % C |
67 % No pl. 33 % C |
33 % B 67 % C |
14 % A 29 % B 57 % C |
– | – |
| Braak staging | III–IV 7 % V–VI 93 % |
100 % V–VI | 50 % III–IV 50 % V–VI |
61 % 0 31 % I–II 8 % III–IV |
67 % 0 33 % I–II |
– | 67 % I–II 33 % III–IV |
50 % I–II 17 % III–IV 33.3 % V–VI |
14 % I–II 72 % III–IV 14 % V–VI |
– | – |
| Age at onset of disease | 65.1 (10.6) | 64.3 (11.5) | 77.0 (1.4) | 60.8 (8.3) | – | 64.2 (9.9) | 63.7 (16.0) | 66.3 (9.9) | 66.7 (6.4) | – | – |
| Age at CSF (years) | 68.9 (9.5) | 67.9 (10.7) | 81.0 (1.4) | 61.7 (8.6) | 55.3 (9.5) | 66.6 (10.6) | 64.7 (14.5) | 68.1 (6.4) | 71.6 (9.0) | 69.7 (10.1) | 69.4 (10.8) |
| Time disease onset-CSF sampling (years) | 3.1 (2.3) | 3.3 (2.0) | 4.0 (2.8) | 1.9 (1.7) | – | 3.5 (2.0) | 1.0 (0) | 2.8 (1.2) | 1.4 (0.8) | – | – |
| Follow up time after CSF sampling (months)a | 66.0 (33.6) | 73.9 (45.3) | 45.5 (46.0) | 42.31 (30.1) | 13.3 (9.0) | 60.9 (40.9) | 31.3 (13.6) | 62.6 (28.5) | 53.3 (31.0) | 48.0 (44.0–57.3) | 35 (23.8–46.3) |
| Closest MMSE to CSF sampling | 23.0 (15.0–26.0) | 19.0(15.0–23.0) | 13.5 (2.0–25.0) | 26.0 (19.5–28.0) | - | 23.0 (13.0–26.0) | 22.0(18.0–23.5) | 19.5 (16.0–25.0) | 26.0 (24.5–28.0) | 30.0 (29.0–30.0) | 30.0 (29.0–30.0) |
| Closest CDR to CSF samplingb | 0.5: 31.7 % 1.0: 56.1 % 2.0: 2.4 % 3.0: 9.8 % |
0: 4.8 % 0.5: 23.8 % 1.0: 38.1 % 2.0: 23.8 % 3.0: 9.5 % |
1.0: 100 % | 0: 14.3 % 0.5: 57.1 % 1.0: 14.3 % 2.0: 14.3 % |
0: 100 % | 1.0: 50 % 2.0: 25 % 3.0: 25 % |
– | 1.0: 100.0% | 0.5: 100 % | – | – |
| APOE4 pos. | 51.4 % | 77.3 % | 100 % | 33 % | 33.3 % | 0 % | 0 % | 66.7 % | 71.4 % | 25.6 % | 17.4 % |
| Aβ42. EL | 287.1 (207.6–356.4) | 254.8 (165.7–10.9) | 335.3 (297.2–373.4) | 338.9 (281.8–399.9) | 421.1 (415.7–426.6) | 438.6 (389.6–580.6) | 425.2 (413.9–428.6) | 268.1 (237.0–297.7) | 304.4 (223.9–501.0) | 554.7 (449.1–668.5) | – |
| t-tauEL. | 547.1 (387.4–876.3) | 494.2 (326.1–788.7) | 540.5 (206.1–875.0) | 157.5 (106.9–313.9) | 136.4 (133.9–138.9) | 216.8 (106.9–313.9) | 260.2 (257.1–347.1) | 297.9 (250.8–421.5) | 229.7 (150.0–437.8) | 226.6 (183.0–350.1) | – |
| p-tauEL | 88.4 (57.3–123.4) | 72.4 (58.9–87.0) | 62.6 (32.5–92.8) | 34.7 (31.8–37.7) | 36.8 (32.0–41.5) | 46.0 (36.6–55.1) | 36.8 (34.2–95.0) | 52.83 (43.7–53.2) | 73.1 (64.1–78.6) | 51.5 (41.6–62.1) | – |
| Aβ42Lum | 128.1 (103.0–164.0) | 142.0(123.9–154.1) | 152.5 (140.0–165.0) | 199.8 (182.0–228.0) | 272 | 212 (204.5–233.1) | 220.4(191.3–249.4) | 131.0(122.0–147.5) | 130.7 (91.3–167.8) | – | 2465 (228.5–278.5) |
| t-tauLum | 113.5(70.0–164.0) | 120.0(73.9–151.5) | 106.0 (49.0–163.0) | 33.0 (27.2–56.3) | 36 | 67.0 (47.4–72.2) | 84.5 (62.4–106.5) | 67.0 (48.0–84.5) | 95.0 (68.7–103.7) | – | 50.4 (38.1–65.2) |
| p-tauLum | 29.5 (21.2–70.0) | 24.2 (12.9–43.6) | 14.3 (6.2–22.5) | 5.5 (4.5–7.3) | 6 | 10.9 (7.8–19.4) | 4.3 (3.6–5.1) | 20.9 (16.4–29.1) | 30.3 (18.2–37.2) | – | 20.3 (13.6–31.7) |
Continuous variables are expressed as mean (SD), except biomarker measurements (Aβ and tau), MMSE scores, and clinical controls follow up time that are shown as median (1st quartile-3rd quartile). Categorical variables are expressed as frequency and percentage. In the groups with two neuropathological diagnoses the first represents the main diagnosis and the second the secondary diagnosis
PGRN Progranulin mutation
For neuropathological cases represents time until death and for the clinical controls time until last follow up (available for 79 % and 83 % of the ELISA and Luminex clinical controls, respectively)
CDR available for: 58 % AD, 54 % FTLD-TDP, 31 % FTLD-TAU, 50 % AD-FTLD, 88 % AD-DLB, 67 % FTLD-AD. 43 % DLB-AD, 0 % FTLD-TDP-TAU, 100 % ALS
The clinical diagnoses were established following clinical diagnostic criteria for AD [38], social/executive FTD (bv-FTD) [50], corticobasal syndrome (CBS) [22], primary progressive aphasia [20] and DLB [37] as previously reported [67, 68]. For the purposes of this study, patients diagnosed as CBS, bv-FTD, FTD-motor neuron disease, progressive non-fluent aphasia (PNFA) and semantic dementia (SD) were classified as FTD, while subjects with AD and logopenic progressive aphasia (LPA) were classified as AD. As per current conventions, the term FTD was used for the clinical diagnosis and the term FTLD for the neuropathologically confirmed diagnoses. The age of onset of the disease was established after reviewing the clinical charts.
Written informed consent had been obtained for all patients using a protocol approved by the Institutional Review Board of the University of Pennsylvania.
Biofluid collection/analysis
CSF samples were obtained as described previously and samples were immediately stored at −80 °C until analysis [21].
Samples had been previously analyzed using the ELISA assay (INNOTEST®, Innogenetics, Ghent, Belgium) or the Luminex xMAP platform (for research-use-only INNO-BIA AlzBio3™ immunoassay reagents, Innogenetics, Ghent, Belgium) [21, 46, 56]. Monoclonal (MAb) capture and reporting antibodies used in the ELISA method for detection of t-tau and p-tau l81 in CSF were AT120/HT7 and BT2, HT7/AT270, respectively. The ELISA values for Aβ1–42 were measured using an “in house” ELISA method [66] with MAb BAN-50 as the capture and MAb BC-05 as the reporting antibody [66]. The xMAP platform utilized the capture MAbs 4D7A3 (Aβ1–42), AT120 (t-tau), and AT270 (p-tau l81) bound to color-specific beads. The biomarker analytes were detected using the reporting MAbs 3D6 (Aβ1–42) and HT7 (t-tau, p-tau l81). Luminex measurements were obtained after dividing the samples into three different runs, two using the same kit lot number, and one with a different kit lot number; whereas ELISA measurements were obtained in 12 different runs between the years 1998 and 2009. 23 controls and 104 patients had measurements with both assays. In case of repeated ELISA measurements values from the largest runs were selected.
Neuropathological analysis
Postmortem neuropathology analyses were performed on 12 central nervous system (CNS) regions: amygdala, hippocampus (CAl/subiculum), entorhinal cortex, midfrontal gyrus, angular gyrus, superior/middle temporal gyrus, cingulate gyrus, motor cortex, caudate nucleus, midbrain, medulla oblongata, and cervical spinal cord.
Sections were fixed and cut into 6–10 μm sections, stained with hematoxylin and eosin to assess neuronal loss and gliosis and Thioflavin S to grade senile plaques semiquantitatively. Immunohistochemistry was performed with antibodies for tau, α-synuclein, ubiquitin, and TDP-43 as previously described [18, 19, 42, 43, 62], as well as antibodies to FUS in FTLD cases as described [61]. Pathologies were then rated according to a semi-quantitative scale [19, 62], Briefly, neurofibrillary tangles and senile plaques were graded according to NIA-Reagan criteria using Braak staging [6, 7] and the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) protocol, without considering subject age [39]. A neuropathological diagnosis of AD was assigned if the probability was intermediate or high [26]. The diagnoses of FTLD-TAU, FTLD-TDP and DLB were based on established criteria [36, 37]. FTLD-TAU cases included those with a diagnosis of argyrophilic grain disease (AGD), progressive supranuclear palsy (PSP), tangle predominant senile dementia (TPSD), corticobasal degeneration (CBD), and unclassifiable tau-positive FTLD (Unclas. T).
For cases with coincident neurodegenerative disease pathologies, i.e., those with more than one neuropathological diagnosis, the following terms were used: FTLD-TDP-Tau for FTLD with both TDP-43 and tau pathology, AD-FTLD for AD cases with a secondary diagnosis of FTLD-Tau or FTLD-TDP, FTLD-AD if the main diagnosis was FTLD with concomitant AD pathology, and AD-DLB for AD cases with a secondary diagnosis of DLB. Six of the seven cases with a clinical diagnosis of DLB also had a secondary neuropathological diagnosis of AD and were classified as DLB-AD.
Statistical analysis
Normal distribution and homoscedasticity were tested when necessary. For normally distributed variables, a oneway ANOVA was used to test group differences and Tu-key’s honest square difference was used for post hoc analyses. For comparison of two groups, Student’s T test or Wilcoxon rank-sum test was used according to the presence or absence of a normal distribution. For categorical variables, Pearson χ2 test was used. However, in cases with low number of expected counts Fisher’s exact test was used. To study the relationship between CSF biomarkers and Braak and CERAD scores, an ordinal logistic regression was applied.
For logistic regression analysis, the sample was divided into training–validation (70 % of the sample) and test sets (30 % of the sample). A stepwise logistic regression model using forward and backward steps with a leave-group-out cross-validation (data were randomly split ten times into a 70 % training and 30 % validation set) was used to select the ideal combination of biomarkers for the different models in the training–validation set, using accuracy and kappa index as performance metrics [33, 65]. The logistic regression model rendered a probability value for AD for each subject ranging from 0 to 1. Age, Aβ42, p-tau, and t-tau were introduced as potential predictors in the model and the best model was compared against the corresponding ratio (t-tau/Aβ42 or p-tau/Aβ42). In no case did the ratio offer equal or better performance than the use of the combination of the single analytes. The obtained logistic regression model was then applied to the test set to obtain sensitivity, specificity, accuracy, kappa index, and the area under the curve (AUC) in the receiver operating characteristic (ROC) curve. ROC curves and density plots were used to illustrate the results [51]. The initial analysis classified neuropathologically confirmed AD cases against neuropathologically confirmed FTLD and clinically diagnosed control cases. A second model was then derived to classify control cases against FTLD cases.
The obtained regression models were then applied in the whole sample, including the cases with coincident neuropathological diagnoses. Thereafter, sensitivity and specificity measures for different settings were obtained (i.e., for the MCI and dementia settings). Taking the original sample of the first analysis again, we then used the clinical diagnoses in a new logistic regression model. Metrics obtained with this model and the probabilities obtained in both models (the neuropathological and clinical) were compared. All statistical tests were two-sided. Statistical significance was set at the 0.05 level. Statistical analyses were performed using IBM SPSS statistics v. 19 and R 2.13.1 [23,33,49,51].
Results
A total of 142 clinically diagnosed neurodegenerative dementia patients with an autopsy-confirmed diagnosis of one or more neurodegenerative diseases and antemortem CSF tau and Aβ measurements were included in this study (Table 1). Of the neuropathologically confirmed cases, 71.1% showed a single type of neurodegenerative disease pathology indicative of a single diagnosis (i.e. AD, FTLD-TDP, FTLD-TAU, or DLB), while 28.9 % of cases presented with an overlap of different neurodegenerative disease pathologies. The group with overlapping neuropathological diagnoses included: 24 AD-DLB, 2 AD-FTLD, 6 FTLD-AD, 3 with both subtypes of FTLD, and 6 DLB-AD. Only 1 out of the 7 cases with a main neuropathological diagnosis of DLB did not have a secondary diagnosis of AD. For further analysis this case was grouped with the DLB-AD cases. In the FTLD-TAU and in the FTLD-TDP groups, there was one subject per group that had a CERAD C classification; however, in both cases the tau pathology corresponding to AD was classified as Braak stage I–II. Additionally, 43 cognitively normal living controls were available that had ELISA Aβ and tau data (median follow-up 48 months) and 46 with Luminex data for these CSF biomarkers (median follow-up 35 months). None of the controls progressed to MCI or AD. Of these, 23 controls were included in both assays. The mean time between onset of cognitive symptoms and time of CSF sampling was 2.8 years (SD 2.1). The median time lapse between the MMSE scoring and CSF sampling was 1.3 months (0.8–3.3 months). The different groups of patients with neurodegenerative diseases did not show differences in MMSE scores at the time of CSF sampling (χ2 = 11.3, p = 0.13), and all this groups had lower MMSE scores than the control group. The overall median MMSE score for patients with a neurodegenerative disease at CSF sampling was 22 (15.0–26.0), and 41.4 % of them had a MMSE higher than 23. At CSF sampling, 36 patients did not meet dementia criteria: Five cases [including the three amyotrophic lateral sclerosis (ALS) cases] were cognitively normal, 27 had an MCI diagnosis, three had a primary progressive aphasia (PPA) diagnoses, and one a behavioral FTD diagnosis at the time of CSF sampling. The other 106 neuropathological cases had a dementia diagnosis. CDR was available for 55 of these patients (40 CDR 1.0, 8 CDR 2.0, and 7 CDR 3.0). Overall, the groups differed in the percentage of APOE4 positive subjects (one or more APOE ε4 allele) [χ2(9, n = 204) = 47.4, p < 0.0001). Subjects with FTLD-TDP had a lower frequency of APOE4 than those with AD and AD-DLB, Controls and FTLD-TAU subjects had a lower APOE4 frequency than AD, AD-FTLD, FTLD-AD, DLB-AD, and AD-DLB.
There was no statistically significant difference regarding the time interval from CSF sampling to death (F(8,134) = 1.8, p = 0.08) or regarding age (F(9,198) = 1.44, p = 0.17) between the groups. The overall agreement between clinical and neuropathological diagnosis was 81.4 % (Supplementary Table 1). The CSF biomarker values for the Luminex and ELISA assays stratified by neuropathological diagnosis are represented in Fig. 1 and supplementary figure 1, respectively. Supplementary figure 2 represents the CSF biomarker values according to Braak and CERAD scoring. Supplementary table 2 summarizes the ordinal logistic regression analyses used to assess the association between the biomarkers and neuropathological scores, adjusting for time.
Fig. 1.

Levels of Aβ42, t- and p-tau as measured by Luminex assay
Classification cutoffs based on subjects with a single neuropathological diagnosis and clinically diagnosed controls
The three measured biomarkers in both assays differed in subjects with “pure” AD, FTLD-TDP, and/or FTLD-TAU and controls (p < 0.0001). The post hoc analysis of FTLD subjects and controls differed in all measurements from AD subjects. However, controls and FTLD subjects only differed in Aβ42 measurements in the ELISA and Luminex assays (Supplementary table 3).
The logistic regression model that best distinguished AD from the group combining controls and FTLD cases showed an AUC of 0.96 for ELISA and for Luminex in the test set (Table 2). For distinguishing FTLD from AD patients, the AUC for these two groups were 0.96 and 0.98 for ELISA and Luminex, respectively. The Luminex and ELISA assays showed moderate sensitivity and specificity for classifying FTLD patients compared to controls. AUC, accuracy, kappa, sensitivity, and specificity scores are summarized in Table 2. The AUC and the density plots obtained in the test set are depicted in Fig. 2. The AUC is represented with the 95 % confidence interval (CI) shaded in blue and the selected cutoff is depicted in the curve, including the 95 % CI for specificity and sensitivity of the cutoffs. The density plots show the distribution for the predicted probabilities for the different groups by the logistic regression model. In addition, the accuracy and AUC results from the models obtained using single analytes and the selected combination in the training-validation sample are summarized in supplementary table 4. The coefficients of the selected logistic regression models are shown in supplementary table 5.
Table 2.
Best biomarker combination results for classification of patients in the different groups
| Assay | AD vs. FTLD & Controls
|
FTLD vs. controls
|
AD vs. FTLD
|
Clinical AD vs. clinical FTLD
|
||||
|---|---|---|---|---|---|---|---|---|
| ELISA | Luminex | ELISA | Luminex | ELISA | Luminex | ELISA | Luminex | |
| Selected biomarkers | t-tau and Aβ42 | p-tau and Aβ42 | Aβ42 | t-tau, p-tau and Aβ42 | t-tau and Aβ42 | p-tau and Aβ42 | t-tau and p-tau | t-tau and p-tau |
| AUC | 0.96 (0.91–1.0) | 0.96 (0.91–1.0) | 0.86 (0.68–1.0) | 0.80 (0.45–1.0) | 0.96 (0.88–1.0) | 0.98 (0.96–1.0) | 0.80 (0.61–0.99) | 0.87 (0.72–1.0) |
| Accuracy | 87.1 % (70.2–96.4 %) | 91.3 % (76.9–98.2 %) | 81.3 % (54.4–95.9 %) | 87.5 % (61.7–98.4 %) | 85.7 % (63.7–97.0 %) | 95.8 % (78.9–99.9 %) | 71.4 % (47.8–88.7 %) | 78.3 % (56.3–92.6 %) |
| Kappa | 0.74 (0.49–0.98) | 0.83 (0.64–1.0) | 0.63 (0.24–1.0) | 0.71 (0.33–1.0) | 0.71 (0.42–1.0) | 0.90 (0.72–1.0) | 0.38 (0.0–0.80) | 0.55 (0.21–0.90) |
| Sensitivity | 85.7 % (57.2–98.2 %) | 94.1 % (71.3–99.9 %) | 87.5 % (47.3–99.7 %) | 80.0 % (28.4–99.5 %) | 90.0 % (55.5–99.7 %) | 100.0 % (79.4–100.0 %) | 68.8 % (41.3–89.0 %) | 78.6 % (49.2–95.3 %) |
| Specificity | 88.2 % (63.6–98.5 %) | 88.9 % (65.3–98.6 %) | 75.0 % (34.9–96.8 %) | 90.9 % (58.7–99.8 %) | 81.8 % (48.2–97.7 %) | 87.5 % (47.3–99.7 %) | 80.0 % (28.4–99.5 %) | 77.8 % (40.0–97.2 %) |
| Probability cutoff | ≤0.50 | ≤0.54 | ≤0.55 | ≤0.40 | ≤0.55 | ≤0.50 | ≤0.50 | ≤0.50 |
The values in parentheses represent the 95 % confidence interval
Fig. 2.

ROC curve and density probabilities of the classification groups based on the probabilities obtained in the regression models. The values inside the parenthesis in the AUC curves indicate the specificity and sensitivity of the selected cutoff. The ROC curves and density plots represent values and distributions for AD versus FTLD and controls (a, b), FTLD versus controls (c, d) and AD versus FTLD versus AD (e, f) using ELISA. For the Luminex assay the ROC curves and density plots represent values and distributions for AD versus FTLD and controls (g, h), FTLD versus controls (i, j) and AD versus FTLD versus AD (k, 1)
Although DLB-AD subjects showed lower t-tau levels in the ELISA assay, no variable was entered in the ELISA regression stepwise model, indicating that none of them was useful for classification of patients.
Cutoffs applied to all cases, including cases with coincident neurodegenerative disease pathologies
We applied the obtained regression models that included control subjects in the whole sample. Based on the previous ELISA cutoffs, 69.4 and 96.4 % of the cases with coincident neuropathological diagnoses (that included AD) were classified as AD by the ELISA and Luminex CSF measurements, respectively (Fig. 3). In both assays, the only DLB case that did not have AD pathology was classified as belonging to the FTLD-control group.
Fig. 3.
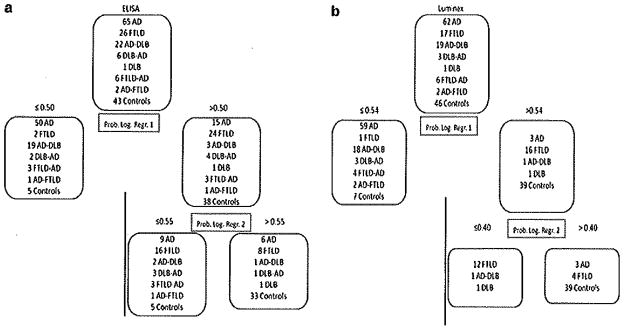
Classification of the cases based on ELISA (a) and Luminex assays (b) using the logistic regression models based on neuropathological diagnoses. The upper box represents all the cases available for the corresponding platform and the boxes below show how cases were classified. Prob. Log. Regr. probability obtained in the logistic regression model
We determined the diagnostic accuracy for distinguishing neuropathologically confirmed cases with concomitant AD pathology versus those cases without concomitant AD pathology. ELISA cutoffs showed a sensitivity of 90.1 % and a specificity of 80.8 % to differentiate subjects with AD pathology (main or secondary) against FTLD cases without an AD diagnosis, while Luminex results were 96.7 and 88.2 %.
We also compared the controls against all the neuropathologically diagnosed cases, finding a sensitivity of 85.6 % and a specificity of 76.7 % to identify subjects with a neurodegenerative disease with ELISA assay; while with the Luminex assay these values were 93.3 and 84.8 %. When classifying AD subjects against FTLD subjects and controls, the sensitivity and specificity were 74.3 and 89.9 % for ELISA assay and 95.6 and 87.3 % for Luminex assay (Table 3).
Table 3.
Sensitivity and specificity of the cutoffs for the different diagnoses (Subjects with a main or secondary AD diagnosis, FTLD without AD, and controls) against other dementias and controls
| ELISA
|
Luminex
|
|||
|---|---|---|---|---|
| Sensitivity | Specificity | Sensitivity | Specificity | |
| Neurodegenerative disease vs. Controlsa | 85.6 % (77.9–91.4 %) | 76.7 % (61.4–88.2 %) | 93.3 % (87.1–97.4 %) | 84.8 % (71.1–93.7 %) |
| AD and AD mixed vs. pure FTLDb | 90.1 % (82.5–95.1 %) | 80.8 % (60.6–93.4 %) | 96.7 % (90.7–99.3 %) | 88.2 % (63.6–98.5 %) |
| AD and AD mixed vs. pure FTLD and Controlsa | 74.3 % (64.6–82.4 %) | 89.9 % (80.2–95.8 %) | 95.6 % (89.0–98.8 %) | 87.3 % (76.5–94.4 %) |
The values in parentheses represent the 95 % confidence interval
Formula derived from comparing AD, FTLD and controls
Formula derived from comparing AD and FTLD
To test if biomarkers showed the same accuracy in subjects with less cognitive impairment, we divided the groups of patients with a main diagnosis of AD or FTLD and no secondary neuropathological diagnosis according to their MMSE scores. Therefore, those subjects with an MMSE below or equal to 24 were compared to those with an MMSE higher than 24. We found that the proportion of patients correctly classified by main diagnosis did not differ in MMSE score, neither in the ELISA [AD: χ2(1,n = 62) = 1.1, p = 0.29; FTLD (n = 25): p = 1,0], nor in the Luminex model [AD: χ2(1, n = 60) = 1.4, p = 0.24; FTLD: p = 1.0].
Classification cutoffs based on clinical diagnosis
As seen in Table 2 the obtained sensitivity and specificity for the biomarker classification formulas based on clinical diagnosis were worse than the ones based on neuropathological diagnosis. When the predicted probability of each subject obtained by the clinical classification model was compared with the predicted probability based on the neuropathological model, there was a median change in the predicted probability of 3.0 % in the ELISA assay (1st and 3rd quartiles: 0.3–22.2 %) and 12.8 % in the Luminex assay (1st and 3rd quartiles: 1.0–35.2 %).
In addition we compared the diagnosis based on clinical, neuropathological, and biomarker criteria using the Luminex assay for the biomarker values. As shown in supplementary table 6 Luminex classification algorithm predicts the neuropathological diagnosis more accurately than the clinical diagnosis in 13 patients, while it incorrectly assigned a diagnosis compared to the clinical and neuropathological diagnosis in two cases.
Discussion
This is the first study to use CSF biomarkers to assess the relevance and classification of coincident neurodegenerative disease pathologies, as determined by autopsy-confirmed diagnoses in a large neuropathological cohort. We show that patients with overlapping diagnoses represent an important subgroup in clinical cohorts, but are often not identified correctly with the current biomarkers. It is also the first study to demonstrate that using samples classified by clinical diagnosis leads to an underestimation of biomarker sensitivity and specificity values and shifts the cutoffs. Our results confirm that Aβ42, p-tau and t-tau as measured by ELISA and ‘Luminex assays enable the differentiation of AD from FTLD in a sample of demented patients and that AD can be distinguished from FTLD and controls in a cohort sample of MCI and early dementia patients, i.e., at early disease stages. In summary, these biomarkers are helpful to distinguish subjects with an underlying neurodegenerative disease (FTLD, AD or both) from cognitively normal controls.
The cases studied here included patients with typical AD pathology overlapping with FTLD-TDP or α-synuclein proteinopathies. In our sample we found that 28.9 % of the cases presented with multiple neurodegenerative disease pathologies, in agreement with previously described samples [13, 31, 44, 52].
Our study demonstrates that the patients studied here were mainly classified as AD by a diagnostic biomarker panel, which includes p-tau, t-tau and Aβ, regardless of the presence of other co-morbid neurodegenerative pathologies. While this may not be surprising since the biomarkers studied here are among the most informative AD biomarkers, it is an important observation that is highly relevant for neurodegenerative disease clinical trials because disease modifying therapies specifically directed against amyloid or tau might reduce the burden of tangles or plaques, but may not affect deposits of other disease proteins. In the absence of cognitive improvement, this could be mistakenly interpreted as a failure of the treatment to ameliorate plaque and tangle pathology rather than a failure to target the appropriate protein aggregate. Therefore, it is critically important to discover and validate biomarkers for non-AD pathologies.
As shown here, the bias introduced by the use of clinical categories when establishing biomarker cutoffs can have an important effect on the calculated sensitivity and specificity of CSF tau and Aβ biomarker-based disease classification. Indeed, this may help explain the variability of published results validating these biomarkers based on clinical diagnosis and the variation in these biomarker cutoffs and estimated group probabilities. Recently, a large clinical sample with a small subset of neuropathological cases found that 34 % of the patients diagnosed as dementia of the non-AD type were found to have an AD CSF biomarker profile, and were thought to be incorrectly classified by these biomarkers and, therefore, the specificity of these biomarker was not deemed to be good [54]. The non-AD dementia clinical diagnoses in this study that showed a higher percentage of AD CSF profile were FTD and CBS patients that in our cohort have a 65.2 and 33 % clinical-pathological agreement, similar to other studies [34], Thus, these clinical diagnoses may not be accurate for establishing CSF biomarker cutoffs or predicting the nature of the underlying neurpathology. Here, we demonstrate that the use of clinical diagnoses instead of neuropathological diagnoses underestimates at about 10–20 % sensitivity and specificity values for CSF tau and Aβ biomarkers and that cohorts with larger numbers of subjects with neuropathological diagnoses are needed to establish the real accuracy of biomarkers as well as to establish cutoffs for classification.
These analyses point to the urgent need for reliable and informative CSF biomarkers that can be used to identify more homogeneous samples of AD or FTD patients with respect to underlying neuropathology for clinical trials, as well as for imaging, genetic and other studies of neurodegenerative dementias. Indeed, this study emphasizes the importance of recognizing that a large subset of patients who are classified as AD by currently available CSF biomarker measurements also may have other concomitant neurodegenerative disorders that may contribute to their cognitive impairments.
We have developed two classification algorithms. The first was designed for a dementia stage that would consist of patients with an underlying AD or FTLD neuropathological diagnosis (and a small subset of DLB cases without AD). The second was designed for earlier stages and offers three diagnostic categories: AD neuropathology, FTLD neuropathology, and a group that potentially has no neurodegenerative disease. This would allow classification of patients in an MCI stage as having or not having an underlying AD neuropathology, which would be useful for clinical trials, or as having or not having any neurodegenerative disease. In our cohort, we observed that 25.4 % of the subjects had an MCI diagnosis and another 28.2 % had a dementia diagnosis with a CDR of 1.0. The diagnostic accuracy did not differ in the group of more impaired patients when compared with the less impaired patients. This is in agreement with other studies that show that CSF Aβ and tau levels are stable in MCI and AD patients [29, 35] and indicates that established CSF tau and Aβ cutoffs are useful not only at the dementia stage but also in the MCI stage of disease. However, recently a study that included MCI patients with a median follow-up of 9.2 years, found that early converters (less than 5 years) had higher t-tau and p-tau levels than late converters (more than 5 years), while Aβ showed no changes [8]. Most of our MCI patients converted to dementia in less than 5 years (data not shown); therefore, our model cannot be inferred to apply to the early stages of MCI without further studies.
This study has several strengths. First it is the largest cohort of subjects with a neuropathologically confirmed diagnosis from whom ante-mortem CSF tau and Aβ measures were obtained. Only two other studies have reported similar data from >100 subjects [9,59]. Second, we present results for the two most commonly used biomarker platforms, i.e. Luminex and ELISA and give the classification algorithm for both assays. Third, the control subjects were closely age matched to the other groups and since the patients had postmortem neuropathological confirmation of their diagnoses, we were able to study a patient cohort that had well established coincident neuropathological diagnoses. Finally, the large sample size here allowed us to divide our cohort into a training–validation and a test set.
All the subjects with a main diagnosis of DLB in our sample, except one, had a secondary diagnosis of AD and were classified as cases with coincident neuropathological diseases. Other studies report DLB cases without an AD diagnosis; however, in our series of 1,240 neuropathological cases in the CNDR INDD, there are 81 cases with a coincident AD diagnosis but only 11 cases (12 % of all DLB cases) with a main diagnosis of pure DLB with no coincident AD diagnosis. The fact that only 3 % of DLB cases did not have a coincident AD neuropathological diagnosis could be due to a selection bias of cases based on a dementia clinic recruitment of the sample. This skewed representation might be due to a referral bias of our patients to a dementia clinic, which may result in a significant overlap between DLB and AD pathology in many patients drawn from such clinics. Finally, based on the small numbers of DLB cases studied here, we cannot make any claims on the overall CSF signature of DLB patients. We confirm previous results of lower CSF t-tau levels in cases with DLB (data not shown) [63], but the overlap of CSF biomarker measurements together with a lower prevalence of DLB present a significant challenge for improving the recognition of DLB based on CSF biomarkers [58]. However, it is possible that measuring other species of Aβ, including Aβ1–40ox, could improve the classification of DLB using CSF biomarkers [40].
One of the weaknesses of this study and previous studies is the lack of neuropathologically diagnosed controls. Previous studies using unsupervised analyses have shown that up to 36 % of cognitively normal subjects have a pathological AD CSF signature [10] and this is in agreement with a long preclinical or prodromal phase of AD during which time AD neuropathology is accumulating [1, 28] and the observation that CSF Aβ measures are most dynamic in the MCI stage of AD but are stable when AD dementia becomes clinically manifest [27, 35, 64]. Thus, it is possible that some of the cognitively normal subjects, classified as AD by the CSF measurements, are asymptomatic subjects at risk for AD [12] and these subjects may have shifted our cutoffs. While we also indicated that biomarkers could improve clinical diagnoses, we acknowledge that for the comparison shown in supplementary table 5 we used some subjects who had been used to train the logistic regression models. Finally, cases with vascular dementia were not included in our study and we therefore have mainly focused on neurodegenerative diseases in this study.
In summary, our study demonstrates the importance of using neuropathological diagnoses for establishing biomarker cutoffs and provides important new insights into the interpretation of CSF biomarkers for the classification of neurodegenerative dementias. We also emphasize the important clinical need to develop specific biomarkers for PD, PDD and DLB as well as other forms of FTLD in order to be able to accurately classify subjects with other coincident neurodegenerative disease pathologies as the basis for their neurodegenerative dementia. In addition, studies analyzing biomarkers in early stages of the disease are needed because of the dynamic characteristics of some biomarkers. Finally, this study underlines the critical importance of CSF biomarker assay standardization to increase accuracy for the early diagnosis of neurodegenerative diseases, especially since many patients with a neurodegenerative dementia will show evidence of coincident neurodegenerative disease pathologies and more than one neuropathologically confirmed neurodegenerative disorder as the underlying basis for their cognitive impairment.
Acknowledgments
This work was supported by the NIH (AG033101, AG17586, AG10124, AG17586, NS053488, NS44266, and AG15I16), the Wyncote Foundation, and the Koller Family Foundation. VMYL is the John H. Ware, 3rd, Professor of Alzheimer’s Disease Research. JQT is the William Maul Measey-Truman G. Schnabel, Jr., Professor of Geriatric Medicine and Gerontology. JBT is supported by a grant of the Fundación Alfonso Martin Escudero. JB is supported by a grant of the Deutsche Forschungsgemeinschaft DFG (AOBJ586910). We acknowledge Christopher Clark’s previous work at the ADCC and PMC. We thank Sue Leight for her work with the ELISA and Young Baek for his work with INDD.
Footnotes
Electronic supplementary material The online version of this article (doi:10 1007/s00401-012-0983-7) contains supplementary material, which is available to authorized users.
Conflict of interest J.Q.T., L.M.S. V.M.Y.L., S.X.X., W.T.H., J.B., M.G., S.E.A. and J.B.T. have no conflicts of interest.
Contributor Information
Jon B. Toledo, Department of Pathology and Laboratory Medicine, Institute on Aging, Center for Neurodegenerative Disease Research, CNDR, University of Pennsylvania School of Medicine, 3rd Floor Maloney Building, 3600 Spruce Street, Philadelphia, PA 19104, USA
Johannes Brettschneider, Department of Pathology and Laboratory Medicine, Institute on Aging, Center for Neurodegenerative Disease Research, CNDR, University of Pennsylvania School of Medicine, 3rd Floor Maloney Building, 3600 Spruce Street, Philadelphia, PA 19104, USA.
Murray Grossman, Department of Neurology, University of Pennsylvania School of Medicine, Philadelphia, PA, USA.
Steven E. Arnold, Department of Psychiatry, University of Pennsylvania School of Medicine, Philadelphia, PA, USA
William T. Hu, Department of Neurology, Center for Neurodegenerative Diseases, Emory University, Atlanta, GA, USA
Sharon X. Xie, Department of Biostatistics and Epidemiology, University of Pennsylvania School of Medicine, Philadelphia, PA 19104, USA
Virginia M.-Y. Lee, Department of Pathology and Laboratory Medicine, Institute on Aging, Center for Neurodegenerative Disease Research, CNDR, University of Pennsylvania School of Medicine, 3rd Floor Maloney Building, 3600 Spruce Street, Philadelphia, PA 19104, USA
Leslie M. Shaw, Department of Pathology and Laboratory Medicine, Institute on Aging, Center for Neurodegenerative Disease Research, CNDR, University of Pennsylvania School of Medicine, 3rd Floor Maloney Building, 3600 Spruce Street, Philadelphia, PA 19104, USA
John Q. Trojanowski, Email: trojanow@mail.med.upenn.edu, Department of Pathology and Laboratory Medicine, Institute on Aging, Center for Neurodegenerative Disease Research, CNDR, University of Pennsylvania School of Medicine, 3rd Floor Maloney Building, 3600 Spruce Street, Philadelphia, PA 19104, USA
References
- 1.Acosta-Baena N, Sepulveda-Falla D, Lopera-Gomez CM, et al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: a retrospective cohort study. Lancet neurology. 2011;10(3):213–220. doi: 10.1016/S1474-4422(10)70323-9. [DOI] [PubMed] [Google Scholar]
- 2.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alzheimer A. Allgemeine Zeitschrift für Psychiatrie und psychischgerichtliche Medizin 64 Band. Verlag von Georg Reimer; 1907. Über eine eigenartige Erkrankung der Hirnrinde. [Google Scholar]
- 4.Bian H, Van Swieten JC, Leight S, et al. CSF biomarkers in frontotemporal lobar degeneration with known pathology. Neurology. 2008;70(19 Pt 2):1827–1835. doi: 10.1212/01.wnl.0000311445.21321.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6(3):131–144. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- 6.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 7.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112(4):389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchhave P, Minthon L, Zetterberg H, et al. Cerebrospinal fluid levels of beta-Amyloid 1–42, but not of Tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry. 2012;69(1):98–106. doi: 10.1001/archgenpsychiatry.2011.155. [DOI] [PubMed] [Google Scholar]
- 9.Clark CM, Xie S, Chittams J, et al. Cerebrospinal fluid tau and beta-amyloid: how well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch Neurol. 2003;60(12):1696–1702. doi: 10.1001/archneur.60.12.1696. [DOI] [PubMed] [Google Scholar]
- 10.De Meyer G, Shapiro F, Vanderstichele H, et al. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch Neurol. 2010;67(8):949–956. doi: 10.1001/archneurol.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDSADRDA criteria. Lancet Neurol. 2007;6(8):734–746. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 12.Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9(11):1118–1127. doi: 10.1016/S1474-4422(10)70223-4. [DOI] [PubMed] [Google Scholar]
- 13.Echavarri C, Caballero MC, Aramendia A, Garcia-Bragado F, Tunon T. Multiprotein deposits in neurodegenerative disorders: our experience in the tissue brain bank of Navarra. Anat Rec (Hoboken) 2011;294(7):1191–1197. doi: 10.1002/ar.21413. [DOI] [PubMed] [Google Scholar]
- 14.Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord. 2007;22(12):1689–1707. doi: 10.1002/mds.21507. (quiz 1837) [DOI] [PubMed] [Google Scholar]
- 15.Engelborghs S, De Vreese K, Van de Casteele T, et al. Diagnostic performance of a CSF-biomarker panel in autopsy-confirmed dementia. Neurobiol Aging. 2008;29(8):1143–1159. doi: 10.1016/j.neurobiolaging.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 16.Frisoni GB, Fox NC, Jack CR, Jr, Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol. 2010;6(2):67–77. doi: 10.1038/nrneurol.2009.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999;56(1):33–39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- 18.Geser F, Martinez-Lage M, Robinson J, et al. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol. 2009;66(2):180–189. doi: 10.1001/archneurol.2008.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geser F, Robinson JL, Malunda JA, et al. Pathological 43-kDa transactivation response DNA-binding protein in older adults with and without severe mental illness. Arch Neurol. 2010;67(10):1238–1250. doi: 10.1001/archneurol.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–1014. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grossman M, Farmer J, Leight S, et al. Cerebrospinal fluid profile in frontotemporal dementia and Alzheimer’s disease. Ann Neurol. 2005;57(5):721–729. doi: 10.1002/ana.20477. [DOI] [PubMed] [Google Scholar]
- 22.Grossman M, Libon DJ, Forman MS, et al. Distinct ante-mortem profiles in patients with pathologically defined frontotemporal dementia. Arch Neurol. 2007;64(11):1601–1609. doi: 10.1001/archneur.64.11.1601. [DOI] [PubMed] [Google Scholar]
- 23.Hothorn T, Hornik K, van de Wiel MA, Zeileis A. A Lego system for conditional inference. Am Stat. 2006;60(3):257–263. [Google Scholar]
- 24.Hu W, Chen-Plotkin A, Arnold S, et al. Biomarker discovery for Alzheimer’s disease, frontotemporal lobar degeneration, and Parkinson’s disease. Acta Neuropathol. 2010;120(3):385–399. doi: 10.1007/s00401-010-0723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu WT, Trojanowski JQ, Shaw LM. Biomarkers in frontotemporal lobar degenerations—progress and challenges. Prog Neurobiol. 2011 doi: 10.1016/j.pneurobio.2011.04.012. (in press, corrected proof) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56(10):1095–1097. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Postmortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131(6):1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jack CR, Jr, Vemuri P, Wiste HJ, et al. Evidence for ordering of Alzheimer disease biomarkers. Arch Neurol. 2011;68(12):1526–1535. doi: 10.1001/archneurol.2011.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jellinger KA. Neuropathological aspects of Alzheimer disease, Parkinson disease and frontotemporal dementia. Neuro-degener Dis. 2008;5(3–4):118–121. doi: 10.1159/000113679. [DOI] [PubMed] [Google Scholar]
- 31.Jellinger KA, Attems J. Prevalence of dementia disorders in the oldest-old: an autopsy study. Acta Neuropathol. 2010;119(4):421–433. doi: 10.1007/s00401-010-0654-5. [DOI] [PubMed] [Google Scholar]
- 32.Khachaturian ZS. Diagnosis of Alzheimer’s disease. Arch Neurol. 1985;42(11):1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 33.Kuhn M. Building Predictive Models in R Using the caret. J Stat Softw. 2008:28. [Google Scholar]
- 34.Ling H, O’Sullivan SS, Holton JL, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain. 2010;133(7):2045–2057. doi: 10.1093/brain/awq123. [DOI] [PubMed] [Google Scholar]
- 35.Lo RY, Hubbard AE, Shaw LM, et al. Longitudinal change of biomarkers in cognitive decline. Arch Neurol. 2011;68(10):1257–1266. doi: 10.1001/archneurol.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mackenzie I, Neumann M, Bigio E, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 38.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41(4):479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 40.Mollenhauer B, Esselmann H, Trenkwalder C, et al. CSF amyloid-beta peptides in neuropathologically diagnosed dementia with Lewy bodies and Alzheimer’s disease. J Alzheimers Dis. 2011;24(2):383–391. doi: 10.3233/JAD-2011-101551. [DOI] [PubMed] [Google Scholar]
- 41.Mollenhauer B, Locascio JJ, Schulz-Schaeffer W, et al. alpha-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol. 2011;10(3):230–240. doi: 10.1016/S1474-4422(11)70014-X. [DOI] [PubMed] [Google Scholar]
- 42.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 43.Neumann M, Kwong LK, Lee EB, et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009;117(2):137–149. doi: 10.1007/s00401-008-0477-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS) Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Lancet. 2001;357(9251):169s–175s. doi: 10.1016/s0140-6736(00)03589-3. [DOI] [PubMed] [Google Scholar]
- 45.Nordberg A, Rhine JO, Kadir A, Langstrom B. The use of PET in Alzheimer disease. Nat Rev Neurol. 2010;6(2):78–87. doi: 10.1038/nrneurol.2009.217. [DOI] [PubMed] [Google Scholar]
- 46.Olsson A, Vanderstichele H, Andreasen N, et al. Simultaneous measurement of beta-amyloid(l–42), total tau, and phosphorylated tau (Thr 181) in cerebrospinal fluid by the xMAP technology. Clin Chem. 2005;51(2):336–345. doi: 10.1373/clinchem.2004.039347. [DOI] [PubMed] [Google Scholar]
- 47.Parnetti L, Chiasserini D, Bellomo G, et al. Cerebrospinal fluid tau/α-synuclein ratio in Parkinson’s disease and degenerative dementias. Mov Disord. 2011;26(8):1428–1435. doi: 10.1002/mds.23670. [DOI] [PubMed] [Google Scholar]
- 48.Prvulovic D, Hampel H. Amyloid beta (Abeta) and phospho-tau (p-tau) as diagnostic biomarkers in Alzheimer’s disease. Clin Chem Lab Med. 2011;49(3):367–374. doi: 10.1515/CCLM.2011.087. [DOI] [PubMed] [Google Scholar]
- 49.R Development Core Team. R Foundation for statistical computing. Vienna, Austria: 2011. R: a language and environment for statistical computing. [Google Scholar]
- 50.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(9):2456–2477. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S + to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69(24):2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- 53.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Aim Neurol. 2009;66(2):200–208. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schoonenboom NS, Reesink FE, Verwey NA, et al. Cerebrospinal fluid markers for differential dementia diagnosis in a large memory clinic cohort. Neurology. 2012;78(1):47–54. doi: 10.1212/WNL.0b013e31823ed0f0. [DOI] [PubMed] [Google Scholar]
- 55.Shaw LM, Korecka M, Clark CM, Lee VMY, Trojanowski JQ. Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nat Rev Drug Discov. 2007;6(4):295–303. doi: 10.1038/nrd2176. [DOI] [PubMed] [Google Scholar]
- 56.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Annals of Neurology. 2009;65(4):403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shi M, Bradner J, Hancock AM, et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol. 2011;69(3):570–580. doi: 10.1002/ana.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sinha N, Firbank M, O’Brien JT. Biomarkers in dementia with Lewy bodies: a review. Int J Geriatr Psychiatry. 2011 doi: 10.1002/gps.2749. [DOI] [PubMed] [Google Scholar]
- 59.Tapiola T, Alafuzoff I, Herukka SK, et al. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol. 2009;66(3):382–389. doi: 10.1001/archneurol.2008.596. [DOI] [PubMed] [Google Scholar]
- 60.Toledo JB, Vanderstichele H, Figtirski M, et al. Factors affecting Abeta plasma levels and their utility as biomarkers in ADNI. Acta Neuropathol. 2011;122(4):401–413. doi: 10.1007/s00401-011-0861-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Urwin H, Josephs KA, Rohrer JD, et al. FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 2011;120(1):33–41. doi: 10.1007/s00401-010-0698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uryu K, Nakashima-Yasuda H, Forman MS, et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol. 2008;67(6):555–564. doi: 10.1097/NEN.0b013e31817713b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Harten AC, Kester MI, Visser PJ, et al. Tau and p-tau as CSF biomarkers in dementia: a meta-analysis. Clin Chem Lab Med. 2011;49(3):353–366. doi: 10.1515/CCLM.2011.086. [DOI] [PubMed] [Google Scholar]
- 64.Vemuri P, Wiste HJ, Weigand SD, et al. Serial MRI and CSF biomarkers in normal aging, MCI, and AD. Neurology. 2010;75(2):143–151. doi: 10.1212/WNL.0b013e3181e7ca82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Venables WN, Ripley BD. In: Modern applied statistics with S. Chambers WEJ, Hardle W, Sheather S, Tierney L, editors. Springer Verlag; New York: 2002. [Google Scholar]
- 66.Winton MJ, Lee EB, Sun E, et al. Intraneuronal APP, not free Abeta peptides in 3xTg-AD mice: implications for tau versus Abeta-mediated Alzheimer neurodegeneration. J Neurosci. 2011;31(21):7691–7699. doi: 10.1523/JNEUROSCI.6637-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Xie SX, Ewbank DC, Chittams J, et al. Rate of decline in Alzheimer disease measured by a Dementia Severity Rating Scale. Alzheimer Dis Assoc Disord. 2009;23(3):268–274. doi: 10.1097/WAD.0b013e318194a324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xie SX, Libon DJ, Wang X, et al. Longitudinal patterns of semantic and episodic memory in frontotemporal lobar degeneration and Alzheimer’s disease. J Int Neuropsychol Soc. 2010;16(2):278–286. doi: 10.1017/S1355617709991317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xie SX, Baek Y, Grossman M, et al. Building an integrated neurodegenerative disease database at an academic health center. Alzheimers Dement. 2011;7(4):e84–e93. doi: 10.1016/j.jalz.2010.08.233. [DOI] [PMC free article] [PubMed] [Google Scholar]