

Abstract
The two most common methods used to generate transgenic Xenopus embryos, restriction enzyme mediated insertion and I-SceI meganuclease, take advantage of relatively common but spatially unpredictable double stranded breaks in sperm, egg or early embryo genomes. These methods also tend to insert multimeric copies of the transgene. An alternative is to use bacteriophage or transposon derived integrase or recombinase to mediate more site-specific insertion of the transgene. The use of phiC31 integrase requires a defined sequence for insertion and is compatible with insertion of a single copy of the transgene. We describe the protocol we use to facilitate phiC31 integrase transgene insertion including the use of insulator sequences to reduce position effect disruption of transgene activity.
Keywords: ΦC31 integrase, embryo injection, transgenesis, Xenopus laevis
1. Introduction
Select viruses and bacteriophage are capable of inserting their DNA into the genomes of their hosts. Bacteriophage ΦC31 is equipped to insert its DNA into the genome of Streptomyces species(1, 2).This process has many features that fit well with the goals for making transgenic Xenopus embryos. First, a single serine recombinase encoded by the phage, ΦC31 integrase, is sufficient to catalyze and complete integration (1, 3). Second, the DNA sequences that define the donor and acceptor sites been defined, and once integration occurs the excision of the inserted DNA cannot be accomplished by ΦC31 integrase by itself (1, 4). Third, relatively large pieces of DNA can be integrated, as is evident from the ability to insert the 41,491 base pairs of the ΦC31 phage into the Streptomyces genome.
The practical demonstration that ΦC31 integrase can act in a variety of backgrounds was first described by Calos and colleagues(5). Their work, and that of others have used ΦC31 integrase to insert genetic material in yeast, plants, fruit flies, fish, Xenopus and mammals(6–14).
In order to mediate phage integration, ΦC31 integrase must bind to a 34bp sequence in the bacterial genome (the attB) site and a 39bp sequence in the phage (the attP site)(4, 15). During integration neither site remains complete, rather ΦC31 integrase creates two new hybrid sites as depicted in figure 1. It is useful to note here for readers unfamiliar with this type of nomenclature that attB and attP are used to identify the DNA binding in a variety of phage/host systems, however the sequences are specific for each phage and host. Although ΦC31 integrase mediated integration efficiency is adversely affected when changes in attB or attP occur, integrase is capable of recognizing attP sites, called pseudo-attP sites that have as little as 29–54% similarity to a consensus attP site(5). Tolerance for mismatches does not imply that integration at a pseudo-attP site will occur with the efficiency of a bone fideattP site. In studies in human cells, fruit fly and zebrafish the presence of a true attP site dramatically increases the efficiency of ΦC31 integrase (12, 16).
Figure 1.
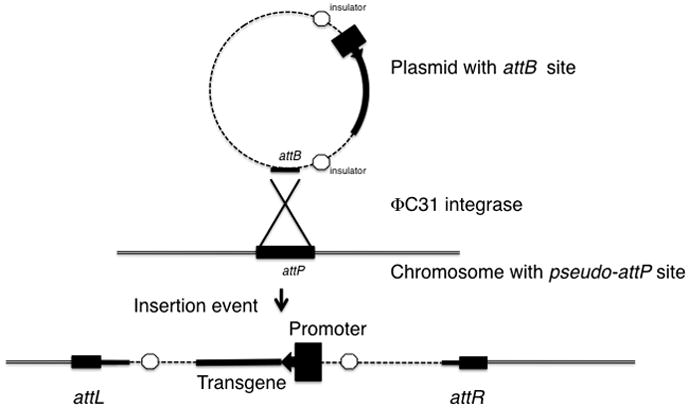
Schematic of ΦC31 integrase mediated insertion of plasmid DNA into a chromosome. ΦC31 integrase recognizes and binds to the attB site in the reporter plasmid. Typically the regions to be expressed would be flanked by insulators. Insulators are represented by octagons. The chromosome (double black lines) with a pseudo-attP site is bound in juxtaposition with the attB site in the plasmid. The attB site in the reporter plasmid and a pseudo-attP site in the embryo genome undergo recombination to form hybrid sites attR and attL that flank the integrated reporter plasmid.
Although there are no consensus attB or attP sites in the Xenopus genome, integrase tolerance for pseudo-attP sites allows recombination to occur. As mentioned above, pseudo-attP sites work with reduced efficiency and frog to frog variation in pseudo-attP site sequence may explain the wide variation (from 1% to as high as almost 50%) in integration efficiency using this method.
Our analysis of transgenic embryos using this method suggests that a single transgene is normally inserted, though we have yet to find a particular genomic location favored for the insertion event. We have observed that expression of genes integrated using this method may be affected by position effects, presumably related to the specific site of integration. By flanking genes to be integrated with insulator elements, like the HS4 insulator found in the chicken s-globin gene, position effects can be significantly reduced(13, 17–19).
We note that this article is an update of previous descriptions of using ΦC31 integrase (13, 20–22).
2. Materials
2.1. Plasmids and Plasmid Preparation Reagents
Plasmids: The integrase plasmid pET11-ΦC31 poly(A) can be obtained from Dr. Michelle P. Calos (calos@stanford.edu) and the attB reporter plasmids, CMV-EGFP-DI-attB and CL-EGFP-DI-attB can be obtained from Dr. Daniel Weeks (daniel-weeks@uiowa.edu). A modified version of the ΦC31 integrase expression vector is (available from DLW, but a MTA for the use of the integrase should still be obtained from Dr. Calos) transcribed using T3 polymerase and containing the β-globin 5′ and 3′ UTRs in a pBluescript (S/K) vector has also been generated. Reporter plasmids, with either the Xenopus γ crystalline lens promoter or the CMV promoter driving GFP flanked by the chicken β-globin HS4 insulators and a ΦC31 attB site can be obtained from the authors (DLW). However, permission to use HS4 insulator sequences and HS4 insulator constructs needs to be obtained from Dr. Gary Felsenfeld (gary.felsenfeld@nih.gov) (and see note 1 and 2).
Bacterial plate and culture reagents: Bacto-YeastExtract, Bacto-Tryptone powder and Bacto-agar (BD Biosciences, Sparks, MD).
Antibiotics: Ampicillin and Kanamycin (Sigma Aldrich, St. Louis, MO).
Plasmid Purification Kits: Qiaprep Spin Miniprep Kit, HiSpeed Plasmid Maxi Kit (Qiagen, Valencia, CA).
In vitro RNA transcription: T7 mMessage machine (Ambion, Austin, TX).
Agarose (RPI, Mt. Prospect, IL).
10X TAE: 0.4 M Tris-acetate, 0.01 M EDTA.
10x non-denaturing DNA loading buffer: 50% glycerol, 60 mM EDTA, 1% SDS and 0.05% bromophenol blue.
1% agarose, 2.2 M formaldehyde MOPS gel.
Denaturing RNA loading buffer (NorthernMax formaldehyde loading buffer, Ambion, Austin, TX or make loading buffer using 5 parts deionized Formamide, 1.5 parts formaldehyde, and 1 part 10X MOPS buffer. Use 3 parts loading buffer to 1 part sample.)
Gel electrophoresis apparatus for flat bed agarose gel electrophoresis (Owl Separation Systems, Portsmouth, NH).
Spectrophotometer: Nanodrop ND1000 (Nanodrop Technologies, Wilmington, DE).
UV-light trans illuminator.
2.2. Xenopus Injection
Xenopus laevis: May be obtained from either Xenopus I (Dexter, MI) or Nasco (Fort Atkinson, WI) and see note 3.
Human Chorionic Gonadotropin (Sigma Aldrich).
Tricaine (3-Aminobenzoic Acid Ethyl Ester) (Sigma Aldrich).
10X Marc’s Modified Ringers Solution (MMR): 1M NaCl, 20 mM KCl, 20 mM CaCl2, 10 mM MgCl2, 50 mM HEPES at pH 7.4.
Injection Buffer: 88 mM NaCl, 10 mM HEPES.
0.3X MMR with 3% Ficoll Type 400 (Sigma Aldrich).
2% Cysteine (RPI) made in dH2O, pH to 7.8–7.9 with NaOH. This solution should be made the day of injection.
Microinjection needle puller.
Micromanipulator: Singer MK-1 (Singer instrument company, Somerset, England) or similar instrument.
Microinjector: inject+matic or similar instrument.
Glass capillary tubes appropriate size for microinjector (Singer). Microinjection needles uses in this technique have an injection tip of approximately 10μm.
18°C Incubator.
2.3. Screening for transgenics
Dissecting microscope: Nikon SMZ (Nikon Instruments, Melville, NY) and Zeiss Stemi SV 11 fluorescent dissecting microscope(Zeiss MicroImaging, Thornwood, NY).
Compound microscope with fluorescence: Zeiss Axioplan 2 (Carl Zeiss MicroImaging) or similar instrument.
Camera for photo documentation: SPOT camera (Diagnostic Instruments, Sterling Heights, MI), Zeiss Axiocam (Carl Zeiss MicroImaging) or similar instrument.
Genomic DNA purification method. Qiagen DNeasy (Qiagen) for example.
PCR primers that will amplify the plasmid across the attB site.
PCR primers that will amplify within the coding region of the gene to be inserted.
A control primer set to compare copy number of an endogenous gene with the transgene.
PCR thermocycler.
RNA purification using an RNA Aqueous kit (Ambion, Austin TX.) or similar protocol.
Reverse transcriptase Polymerase Chain Reaction using OneStep RT-PCR (Qiagen,) or similar protocol.
3. Methods
3.1 Preparation of plasmids and ΦC31 integrase mRNA
Transform the pET11-ΦC31 poly(A) plasmid and the attB reporter plasmids (CMV-EGFP-DI-attB and CL-EGFP-DI-attB) into E. coli and plate onto antibiotic selective plates using standard techniques. The pET11-ΦC31 poly(A) plasmid contains an ampicillin resistance gene and the attB reporter plasmids contain a kanamycin resistance gene. We have found that insulated attB reporter plasmids frequently undergo rearrangements during growth in E. coli and thus recommend propagating these plasmids in E. coli Stbl2 cells (Invitrogen) or similar E. coli strain that are designed to prevent recombination. (see note 2)
Select individual colonies on each plate and grow 3ml cultures at 37°C overnight with agitation. Use 1 ml for mini-preps (Qiaprep mini-prep kit) and save the other 2 mls of culture at 4°C. Using the mini-prep generated DNA, confirm plasmid size and insulator orientation by restriction enzyme digestion and gel analysis. Once plasmid size and insulator orientation are confirmed, inoculate 1 liter of LB broth containing the appropriate antibiotic with the remaining 2 mls of culture. Grow at 37°C with agitation overnight.
Perform maxi-preps on the 1 liter cultures following the manufactures instructions (Hi Speed Plasmid Maxi Kit). Do not add RNase to buffer P1.
Confirm maxi-prep generated plasmid identity by restriction enzyme digest and gel analysis and by DNA sequencing reactions. Store the maxi-prep generated DNA at −20°C.
Linearize 5 μg of pET11-ΦC31 poly(A) maxi-prep DNA with either BamHI or EcoRI restriction enzymes. Run approximately one tenth of the digestion on a 1% agarose gel to ensure linearization. Heat inactivate the restriction enzyme in the remaining portion of digestion by heating reaction to 65°C for 20 minutes. Precipitate the DNA by adding one tenth the volume of 5M NH4 Acetate and 2 volumes of ethanol. Place solution at −20°C for 15 minutes and centrifuge at 10,000g for 15 min. Remove the supernatant and resuspend the pellet in 10 μl of RNase free TE buffer.
Synthesize the ΦC31 integrase mRNA using the T7 mMessage machine following the manufactures instructions. Because the protocol includes a DNase treatment step, the DNase needs to be removed or inactivated. We routinely follow the LiCl precipitation protocol described in the manufactures instructions. Resuspend the integrase mRNA in RNase free water at a concentration of 1 mg/ml.
Run 1 μg of ΦC31 integrase mRNA on a formaldehyde MOPS gel in 1X MOPS buffer to ensure that the transcript is approximately 1.9 kilobases long.
Store ΦC31 integrase mRNA (for up to 1 month) at −80°C until it is needed.
3.2. Injection of Xenopus Embryos
Any experiments with Xenopus should first start with approval of your plans to house and care for Xenopus adult frogs and embryos from your host institution. If you have limited experience using Xenopus other chapters in this volume or the book, Early Development of Xenopuslaevis – a laboratory manual (23), may be a useful reference to consult.
Induce Xenopus females to lay eggs by injecting 1ml (1000 IU) of Human Chorionic Gonadotropin into the dorsal lymph sac the night before desired day of egg collection.
The next morning, the cloaca on the injected females should be swollen and there may be eggs in the water tank holding the frogs.
After eggs are present in the tank, inject a lethal dose of Tricaine (1 ml of a 10% solution) into the dorsal lymph sac of a Xenopus male and then surgically remove the testes. Store the testes in 1X MMR.
Induce egg laying into a dry Petri dish by spreading apart the female’s legs and gently putting pressure on the pelvic region. Immediately fertilize the eggs by rubbing a small piece of testis(~1/6th of a testis) through the eggs. Alternatively, the testis can be crushed in ~1 ml of 0.3X MMR and this solution spread over the eggs. After fertilization (allow about 1 minute) flood the eggs with 0.1X MMR.
Successful fertilization can be determined if the eggs align with the animal pole (pigmented half) facing up. Embryos should be spherical, uniform size and have smooth even pigmentation of the animal hemisphere. 30 minutes after fertilization, remove the 0.1X MMR and place the embryos in 2% cysteine, pH 7.8–7.9 for 2 minutes to remove the jelly coats.
Remove the cysteine and wash the embryos in three 5ml washes of 0.3X MMR.
Place the embryos in 0.3X MMR, 3% ficoll.
Transfer embryos to an injection slide. A variety of injection slide formats can be used. We routinely fasten two 5 cm X 7.5 cm microscope slides together with a 0.5cm offset, forming a ‘stairstep’ like platform. Approximately 50 embryos can be placed in a single line in a cushion of the 3% ficoll 0.3XMMR solution in the step, holding the embryos in place.
Inject single cell embryos into the center of the animal hemisphere with either: 10 nl of injection solution, 10 nl of injection solution containing 5 pg of reporter plasmid or 10 nl of injection solution containing 5 pg of reporter plasmid + 1 ng ofΦC31 integrase mRNA. (see note 4)
3.3. Monitoring developing embryos for transgenesis
Allow injected embryos to develop at 18°C in 0.3X MMR, 0.3% Ficoll for approximately 6–8 hours after injection and then transfer to 0.3X MMR.
Remove delaminating or dead embryos and provide fresh 0.3X MMR at least twice a day for the first 3 days.
On the second day after fertilization, begin to monitor the embryos for GFP expression using a fluorescent microscope optimized to detect green fluorescence. Embryos injected with 1ng integrase mRNA and 5 pg of CMV-EGFP-DI-attB should express GFP uniformly while embryos injected with 1 ng integrase mRNA and 5 pg of CL-EGFP-DI-attB should express GFP only in the lens of the eye (Figure 2). Embryos injected with 5 pg of reporter plasmid alone rarely give GFP expression however we occasionally detect mosaic expression that is less intense than expression from that seen in transgenics (Figure 2A and B show an example of this phenomena in embryos injected with the γ crystalline lens:GFP plasmid). Digital photography with long exposure times may detect fluorescence before it is visible by eye.
Once fluorescence is detected, photograph the embryos using a fluorescent microscope with a digital camera. If embryo movement prevents photography, consider anesthetizing the embryos with 0.02% Tricaine. Embryos may be exposed to the Tricaine solution for approximately 5 minutes; longer exposure times may lead to the demise of the embryos. Embryos should be placed back into 0.3X MMR after photography.
Figure 2.
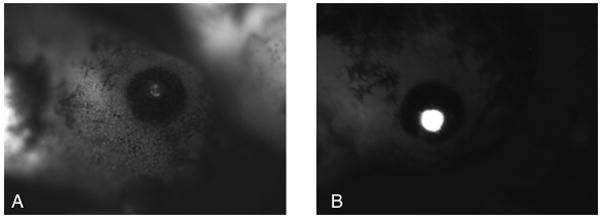
ΦC31integrase mediated transgenesis of insulated reporter plasmids in stage 42(28) Xenopus embryos. Expression of GFP controlled by the γ crystalline lens promoter. (A) Weak mosaic expression derived from extrachromasomal reporter plasmid CL-EGFP-DI-attB. (B) Full lens expression of GFP from integrated pCL-GFP dIattB.
3.4. Molecular analysis to confirm integration
The molecular confirmation of transgenesis depends, in part, on the experimental goal. Genomic DNA can be analyzed by Southern blot analysis as has been described previously(13, 20, 21). Alternatively, genomic DNA or mRNA can be used for PCR analysis of transgene insertion or expression.
Isolate DNA from single embryos using the Qiagen DNeasy kit following the manufactures instructions. In some studies whole embryos can be used for this analysis, while in others the DNA can be isolated from excised tails can be removed. For whole embryos, one stage 46 embryo should yield approximately 2–5 μg of DNA, the tails from a stage 48–50 can easily yield a similar amount of genomic DNA. DNA should be collected from non-injected embryos, embryos injected with reporter plasmid alone and transgenic embryos determined by green fluorescence.
Starting with 250ng of genomic DNA, and primer pairs designed to amplify the inserted plasmid or a single copy gene from Xenopus (in figure 3A the control used was for the Xenopus NKX2-10 gene with primers 5′ CGCTACCTCTACCCCCTTCT3′ and 5′ TGATCAGCTGTGGAGGACTC 3′, the integrated plasmid being tested was being used to express an epitope tagged version of Nkx2-5 and the primers were 5′ CATGACAGGAGGACAGCAGA3′ and 5′ TGTGTGGAGGTGAGCTTCAG3′). If you wish to estimate copy number it is useful to pick control primers that will give the same size of amplified product as the primers used to identify the transgene. Twenty-eight cycles of PCR should easily show a single copy gene. To control for persistent extra-chromosomal (non-integrated plasmid) the isolated genomic DNA should be used for an amplification reaction using primers that generate a product across the attB site. Because the attB site is split apart during insertion, no product will be amplified in integrated copies of the plasmid. If information on insertion copy number is needed identify cycle numbers consistent with a doubling of product production in successive cycles.
Run amplified DNA on a 1% agarose gel. Stain the gel with ethidium bromide and then photograph the gel while on a UV transiluminator.
To estimate the expression of a transgene compared to an endogenous gene isolate mRNA from the embryos (including non-injected and plasmid only injected controls). When using the Qiagen OneStep RT-PCR, between 1ng and 2μg of RNA is needed, with PCR cycle numbers adjusted according to input RNA and target. In the example shown in figure 3B, the endogenous expression of the NKX2-5 gene was compared to a transgene expression GFP under the control of 4.5 Kb NKX2-5 promoter described by Mohun and colleagues. RNA was isolated from stage 46 embryos and 200ng of RNA was used in the RT-PCR reaction. Primers used for this amplification were 5′-ACTGACAGGAGGACAGCAGA-3′ and 5′-GCCGTTTGCATTTGTACCTT-3′ (NKX2-5) 5′-GACATGAAGCAGCACGACTT-3′ and 5′-TGCTCAGGTAGTGGTTGTCG-3′ (GFP).
Figure 3.
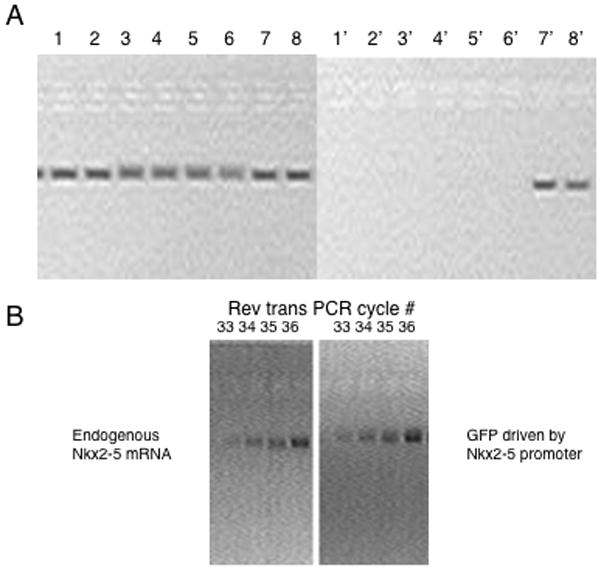
Confirming ΦC31 integrase mediated transgenesis and expression. (A) PCR amplification of an endogenous gene (NKX2.10) compared to the amplification of a transgene encoding a T7epitope tagged version of Nkx2-5. 200ng of DNA from individual stage 47(28)embryos was analyzed. Lane 1–8 is the amplification of DNA from eight different embryos using the primers for an endogenous gene, lanes 1′-8′ the amplification of the DNA from the same eight embryos using primers that query the presence of the transgene. Embryos 7 and 8 were transgenic. (B) Analysis of the endogenous expression of Nkx2-5 compared to the transcription controlled by a transgene expressing GFP under the control of the NKX2-5 promoter. Analysis used 200 ng of mRNA isolated from stage 46 embryos and RT-PCR and sampled sequential cycles from cycle 33–36.
Acknowledgments
The authors would like to thank Michele Calos for providing the pET11ΦC31 poly(A) plasmid, Gary Felsenfeld and colleagues for providing the HS4 insulator sequences, Paul Krieg for providing the γ crystallin lens promoter and Tim Mohun for providing the Nkx2-5 promoter. This work was supported by funding from the NIH (GM069944 and DC007481). Bryan Allen was a student in the Medical Scientist Training Program at the Roy J. and Lucille A Carver College of Medicine, University of Iowa.
Footnotes
MTA agreements are required from Dr. Gary Felsenfeld (NIH) to use the HS4 insulator sequences and Dr. Daniel Weeks (Iowa) for the CMV-EGFP-DI-attB and CL-EGFP-DI-attB plasmids. An MTA is also required to obtain the plasmid pET11ΦC31 poly(A) from Dr. Michele Calos (Stanford).
In studies done by Allen and Weeks (2005), flanking the regulatory region and the reporter in the plasmid to be integrated significantly reduced position effects that lead to reduced expression of the transgene. The chicken β-globin HS4 insulator sequence is effective both as a block to gene silencing and enhancers. Thus positioning the transgene between insulators should help reduce position effects. The Felsenfeld laboratory suggests that tandem copies of the core HS4 insulator, oriented all in the same direction work best. When configured this way the HS4 insulators form direct sequence repeats that can be unstable in many bacterial backgrounds.
We have historically used these suppliers, but other vendors of Xenopus laevis may also provide useable animals. In fact, this method was successful using Xenopus laevis from different breeding facilities (Rennes, France; Labor-Atoire Evolution et Développement, Université Paris Sud, France)(22).It is also important to note that in our hands we see significant variation in integration efficiency that seems to be animal dependent, perhaps due to the degree of mismatches in the pseudo attP sites. In the future we hope determine if animals that carry bonafideattP sites improve the ability to broadly use this method. If animals with bonafideattP sites improve efficacy of this approach they will be made available through the Xenopus stock centers.
The rapid production of ΦC31 integrase seems essential and care should be taken to ensure that the mRNA encoding ΦC31 integrase is stable and capped. Protocols that include RNaseA in the purification of plasmid can prove problematic if that plasmid is used for injection with ΦC31 integrase mRNA. We have also generated insertions using ΦC31 integrase protein made in vitro using a Promega TNT (transcription: translation) extract. However, to date, we have not seen significant increases in transgene generation using in vitro generated ΦC31 integrase protein.
The amount of fluorescence produced from a single copy transgene (ΦC31 integrase most commonly gives a single insertion) may be significantly less than an embryo containing multiple copies of a transgene generated from the restriction enzyme mediated insertion(24, 25)approach or the meganuclease approach(26, 27).
References
- 1.Thorpe HM, Smith MC. In vitro site-specific integration of bacteriophage DNA catalyzed by a recombinase of the resolvase/invertase family. Proc Natl Acad Sci U S A. 1998;95:5505–5510. doi: 10.1073/pnas.95.10.5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith MC, Burns RN, Wilson SE, Gregory MA. The complete genome sequence of the Streptomyces temperate phage straight phiC31: evolutionary relationships to other viruses. Nucleic Acids Res. 1999;27:2145–2155. doi: 10.1093/nar/27.10.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Groth AC, Calos MP. Phage integrases: biology and applications. J Mol Biol. 2004;335:667–678. doi: 10.1016/j.jmb.2003.09.082. [DOI] [PubMed] [Google Scholar]
- 4.Thorpe HM, Wilson SE, Smith MC. Control of directionality in the site-specific recombination system of the Streptomyces phage phiC31. Mol Microbiol. 2000;38:232–241. doi: 10.1046/j.1365-2958.2000.02142.x. [DOI] [PubMed] [Google Scholar]
- 5.Groth AC, Olivares EC, Thyagarajan B, Calos MP. A phage integrase directs efficient site-specific integration in human cells. Proc Natl Acad Sci U S A. 2000;97:5995–6000. doi: 10.1073/pnas.090527097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thyagarajan B, Guimaraes MJ, Groth AC, Calos MP. Mammalian genomes contain active recombinase recognition sites. Gene. 2000;244:47–54. doi: 10.1016/s0378-1119(00)00008-1. [DOI] [PubMed] [Google Scholar]
- 7.Thomason LC, Calendar R, Ow DW. Gene insertion and replacement in Schizosaccharomyces pombe mediated by the Streptomyces bacteriophage phiC31 site-specific recombination system. Mol Genet Genomics. 2001;265:1031–1038. doi: 10.1007/s004380100498. [DOI] [PubMed] [Google Scholar]
- 8.Thyagarajan B, Olivares EC, Hollis RP, Ginsburg DS, Calos MP. Site-specific genomic integration in mammalian cells mediated by phage phiC31 integrase. Mol Cell Biol. 2001;21:3926–3934. doi: 10.1128/MCB.21.12.3926-3934.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Combes P, Till R, Bee S, Smith MC. The streptomyces genome contains multiple pseudo-attB sites for the (phi)C31-encoded site-specific recombination system. J Bacteriol. 2002;184:5746–5752. doi: 10.1128/JB.184.20.5746-5752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olivares EC, Hollis RP, Chalberg TW, Meuse L, Kay MA, Calos MP. Site-specific genomic integration produces therapeutic Factor IX levels in mice. Nat Biotechnol. 2002;20:1124–1128. doi: 10.1038/nbt753. [DOI] [PubMed] [Google Scholar]
- 11.Ortiz-Urda S, Thyagarajan B, Keene DR, Lin Q, Fang M, Calos MP, Khavari PA. Stable nonviral genetic correction of inherited human skin disease. Nat Med. 2002;8:1166–1170. doi: 10.1038/nm766. [DOI] [PubMed] [Google Scholar]
- 12.Groth AC, Fish M, Nusse R, Calos MP. Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics. 2004;166:1775–1782. doi: 10.1534/genetics.166.4.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen BG, Weeks DL. Transgenic Xenopus laevis embryos can be generated using phiC31 integrase. Nat Methods. 2005;2:975–979. doi: 10.1038/nmeth814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Held PK, Olivares EC, Aguilar CP, Finegold M, Calos MP, Grompe M. In vivo correction of murine hereditary tyrosinemia type I by phiC31 integrase-mediated gene delivery. Mol Ther. 2005;11:399–408. doi: 10.1016/j.ymthe.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 15.Smith MC, Till R, Brady K, Soultanas P, Thorpe H. Synapsis and DNA cleavage in phiC31 integrase-mediated site-specific recombination. Nucleic Acids Res. 2004;32:2607–2617. doi: 10.1093/nar/gkh538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lister JA. Transgene excision in zebrafish using the phiC31 integrase. Genesis. 2010;48:137–143. doi: 10.1002/dvg.20595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saitoh N, Bell AC, Recillas-Targa F, West AG, Simpson M, Pikaart M, Felsenfeld G. Structural and functional conservation at the boundaries of the chicken beta-globin domain. Embo J. 2000;19:2315–2322. doi: 10.1093/emboj/19.10.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bell AC, West AG, Felsenfeld G. Insulators and boundaries: versatile regulatory elements in the eukaryotic genome. Science. 2001;291:447–450. doi: 10.1126/science.291.5503.447. [DOI] [PubMed] [Google Scholar]
- 19.West AG, Gaszner M, Felsenfeld G. Insulators: many functions, many mechanisms. Genes Dev. 2002;16:271–288. doi: 10.1101/gad.954702. [DOI] [PubMed] [Google Scholar]
- 20.Allen BG, Weeks DL. Using phiC31 integrase to make transgenic Xenopus laevis embryos. Nat Protoc. 2006;1:1248–1257. doi: 10.1038/nprot.2006.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allen BG, Weeks DL. Bacteriophage phiC31 integrase mediated transgenesis in Xenopus laevis for protein expression at endogenous levels. Methods Mol Biol. 2009;518:113–122. doi: 10.1007/978-1-59745-202-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chesneau A, Sachs LM, Chai N, Chen Y, Du Pasquier L, Loeber J, Pollet N, Reilly M, Weeks DL, Bronchain OJ. Transgenesis procedures in Xenopus. Biol Cell. 2008;100:503–521. doi: 10.1042/BC20070148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sive HL, Grainger RM, Harland RM. Early development of Xenopus laevis : a laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 2000. [Google Scholar]
- 24.Kroll KL, Amaya E. Transgenic Xenopus embryos from sperm nuclear transplantations reveal FGF signaling requirements during gastrulation. Development. 1996;122:3173–3183. doi: 10.1242/dev.122.10.3173. [DOI] [PubMed] [Google Scholar]
- 25.Sparrow DB, Latinkic B, Mohun TJ. A simplified method of generating transgenic Xenopus. Nucleic Acids Research. 2000;28:E12. doi: 10.1093/nar/28.4.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogino H, McConnell WB, Grainger RM. Highly efficient transgenesis in Xenopus tropicalis using I-SceI meganuclease. Mech Dev. 2006;123:103–113. doi: 10.1016/j.mod.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 27.Pan FC, Chen Y, Loeber J, Henningfeld K, Pieler T. I-SceI meganuclease-mediated transgenesis in Xenopus. Dev Dyn. 2006;235:247–252. doi: 10.1002/dvdy.20608. [DOI] [PubMed] [Google Scholar]
- 28.Nieuwkoop PD, Faber J. Normal Table of Xenopus laevis (Daudin) Garland; New York: 1994. [Google Scholar]