

ABSTRACT
The large outbreak of diarrhea and hemolytic uremic syndrome (HUS) caused by Shiga toxin-producing Escherichia coli O104:H4 in Europe from May to July 2011 highlighted the potential of a rarely identified E. coli serogroup to cause severe disease. Prior to the outbreak, there were very few reports of disease caused by this pathogen and thus little known of its diversity and evolution. The identification of cases of HUS caused by E. coli O104:H4 in France and Turkey after the outbreak and with no clear epidemiological links raises questions about whether these sporadic cases are derived from the outbreak. Here, we report genome sequences of five independent isolates from these cases and results of a comparative analysis with historical and 2011 outbreak isolates. These analyses revealed that the five isolates are not derived from the outbreak strain; however, they are more closely related to the outbreak strain and each other than to isolates identified prior to the 2011 outbreak. Over the short time scale represented by these closely related organisms, the majority of genome variation is found within their mobile genetic elements: none of the nine O104:H4 isolates compared here contain the same set of plasmids, and their prophages and genomic islands also differ. Moreover, the presence of closely related HUS-associated E. coli O104:H4 isolates supports the contention that fully virulent O104:H4 isolates are widespread and emphasizes the possibility of future food-borne E. coli O104:H4 outbreaks.
IMPORTANCE
In the summer of 2011, a large outbreak of bloody diarrhea with a high rate of severe complications took place in Europe, caused by a previously rarely seen Escherichia coli strain of serogroup O104:H4. Identification of subsequent infections caused by E. coli O104:H4 raised questions about whether these new cases represented ongoing transmission of the outbreak strain. In this study, we sequenced the genomes of isolates from five recent cases and compared them with historical isolates. The analyses reveal that, in the very short term, evolution of the bacterial genome takes place in parts of the genome that are exchanged among bacteria, and these regions contain genes involved in adaptation to local environments. We show that these recent isolates are not derived from the outbreak strain but are very closely related and share many of the same disease-causing genes, emphasizing the concern that these bacteria may cause future severe outbreaks.
Introduction
The large outbreak of diarrhea and hemolytic uremic syndrome (HUS) caused by Escherichia coli O104:H4 (1, 2) in May through early July 2011 focused considerable attention on this previously rarely identified serogroup (1, 3–7). Over 3,800 cases of gastroenteritis were recorded in Germany and among individuals from other countries who had traveled to Germany (2), and a small outbreak took place in France (3, 7). The fraction of patients who developed HUS (>20%) was considerably higher than observed in prior outbreaks of Shiga toxin-producing E. coli, such as E. coli O157:H7 (2). Epidemiological investigations suggested that the outbreak was caused predominantly by contaminated sprouts produced by a farm in Lower Saxony (8). Besides the magnitude of the clinical complications caused by this pathogen, the outbreak was also notable because it highlighted the potential contributions of rapid whole-genome sequencing for understanding the phylogenetic origins of a new pathogen, its transmission and epidemiology, and the genetic basis for its pathogenicity (9–15).
Initial molecular and phenotypic studies revealed that the Shiga toxin-producing outbreak strain (the prototype of which is TY2482, an isolate derived from an early case in the German outbreak) had an enteroaggregative E. coli (EAEC) background because its genome contained certain characteristic genes, such as a plasmid-encoded aggregative adherence fimbriae (in this case, AAF/I), and because of its pattern of adherence to cultured cells (16). Diarrheagenic EAEC strains display marked heterogeneity in the sets of virulence factors they encode, and the TY2482 genome contained an unusual set of putative virulence genes, including long polar fimbriae, IrgA homologue adhesion (iha), serine protease autotransporters of the Enterobacteriaceae (SPATEs), and genes involved in iron uptake and tellurium resistance, as well as broad-spectrum resistance to antibiotics (12–14). Furthermore, unlike most EAEC strains, the outbreak strain was lysogenized by a Shiga toxin 2-encoding lambda-like prophage and produced this potent HUS-associated toxin.
TY2482 also differed from previous E. coli O104:H4 isolates. One of the first characterized O104:H4 strains, 55989, was isolated from an HIV-infected individual in The Central African Republic with persistent diarrhea in the late 1990s (17, 18). This strain does not produce Shiga toxin; furthermore, 55989, like the Shiga toxin-producing 2001 German O104:H4 isolate 01-09591 (HUSEC041) (12), harbors plasmids conferring the enteroaggregative phenotype through a different set of fimbriae genes (AAF/III) (12). Additional O104:H4 isolates from 2004 and 2009 identified in France share with the 2011 outbreak strain the presence of an Stx2-encoding prophage but carry an AAF/III-containing plasmid like the one of 55989 (11, 19). While differences between the TY2482 and 55989 (17) genomes and TY2482 and 01-09591 genomes have been reported (14), comprehensive comparisons of the outbreak genome with additional O104:H4 isolates from other time points can lead to an improved understanding of the fine-scale evolution of this emerging pathogen’s genome over the very short term, as well as the ongoing gene flux mediated by mobile genetic elements (MGEs), including phages, plasmids, and transposons, all of which can carry pathogenicity factors.
After the O104:H4 outbreak ended in early July 2011, sporadic diarrhea/HUS cases linked to E. coli O104:H4 have been reported (15). It is unknown whether these sporadic cases are derived from the outbreak strain and indicate continued transmission. Similarly, the relationship of the E. coli O104:H4 sporadic cases to each other is unknown. Moreover, as noted above, even closely related E. coli can show marked variation in the panel of virulence factors they possess, and it is unclear to what extent the virulence factors that contributed to the pathogenicity of the O104:H4 outbreak strain are shared by these sporadic isolates. To describe the relationship of these isolates to the outbreak and to deepen our understanding of the diversity and evolution of this emerging pathogen, we sequenced the genomes of 5 additional O104:H4 strains isolated from HUS patients whose illness occurred after the German and French outbreaks and were not known to be linked to them epidemiologically. Detailed comparative analyses of these 5 genomes with that of a representative outbreak strain and several recent historical O104:H4 genomes revealed the nature of the diversity of O104:H4 associated with severe infection, shed light on the relationships among the sporadic isolates and between the sporadic and outbreak isolates, and emphasized the importance of the mobile genome in genomic variation at the time scale reflected by these closely related isolates.
RESULTS AND DISCUSSION
Overview.
We determined the genome sequences of 5 E. coli O104:H4 isolates that were derived from sporadic HUS cases that occurred in the late summer through winter of 2011 (Table 1), following the outbreaks in Germany and France. While these patients were all cared for in France, one of the patients became infected in Turkey (isolate Ec11-9450), and another patient had recently traveled in North Africa (isolate Ec12-0466). These 5 new E. coli O104:H4 genomes were compared to each other as well as to 5 previously reported O104:H4 genomes derived from patients with diarrhea and/or HUS, including 55989 (17), TY2482 (representative of the 2011 outbreak) (14), Ec04-8351 (11), Ec09-7901 (11), and 01-09591 (12) (Table 1). Since the latter genome consists of a large number of contigs, it was not possible to generate an assembly of its chromosome sufficient for synteny-based analyses, but the finished plasmid sequences (20) were included in our comparisons.
TABLE 1 .
E. coli O104:H4 analyzed in this study
| Isolate namea | Date of isolation | Location of isolation (additional epidemiological information, if available) |
Antibiotic resistance profileb |
Clinical syndrome |
|---|---|---|---|---|
| 55989 (17, 18) | Late 1990s | Bangui, Central African Republic | TET | Diarrhea |
| 01-09591 (12) | 2001 | Germany | Not available | HUS |
| Ec04-8351 (11, 52) | 2004 | Lille, France | NAL | Not available |
| Ec09-7901 (11, 52) | 2009 | Lyon, France | NAL | HUS |
| TY2482 (14) | 2011 | Germany (2011 outbreak) | AMX CAZ CRO STR SSS TMP SXT TET NAL |
HUS |
| Ec11-9941 | 6 Sept 2011 | Angers, France (sporadic) | AMX STR SSS TMP SXT NAL |
HUS |
| Ec11-9990 | 24 Aug 2011 | Besançon, France (sporadic) | AMX TMP NAL | HUS |
| Ec11-9450 | 3 Oct 2011 | France (sporadic; patient became ill in Turkey, returned to France) |
AMX STR SSS TMP SXT TET NAL |
HUS |
| Ec12-0465 | 4 Nov 2011 | Marseille, France (sporadic) | AMX STR SSS TMP SXT TET NAL |
HUS |
| Ec12-0466 | 9 Dec 2011 | Bry-sur-Marne, France (sporadic; recent travel to North Africa) |
AMX STR SSS TMP SXT TET NAL |
HUS |
Bolded names refer to isolates sequenced in this study. Numbers in parentheses indicate the bibliographic reference of previously sequenced isolates.
Abbreviations are as follows. AMX, amoxicillin; CAZ, ceftazidime; CRO, ceftriaxone; STR, streptomycin; SSS, sulfonamides; TMP, trimethoprim; SXT, sulfamethoxazole; TET, tetracycline; NAL, nalidixic acid.
The chromosomes of the assembled 9 E. coli O104:H4 genomes exhibit extensive similarity to each other (Fig. 1 and 2). The circular chromosome is approximately 5.2 Mbp in each isolate. Of the 4,977 open reading frames (ORFs) we predicted in the TY2482 chromosome, 4,496 (90.3%) have homologs in all of the isolates, and 4,756 (95.6%) have homologs in at least 8 of the 9 other isolates (see Table S1 in the supplemental material).
FIG 1 .

Genome alignment of E. coli O104:H4 isolates highlighting mobile genetic elements and variable regions. The chromosomal sequences of E. coli O104:H4 isolates 55989 (17), Ec04-8351, Ec09-7901, Ec11-9450, Ec11-9941, Ec11-9990, Ec12-0465, Ec12-0466, and TY2482 (14) were aligned by progressiveMauve (45). The background color gradient indicates homologous regions across strains. Prophage predicted by PHAST (48) are designated by rectangles and labeled based on similarity of encoded phage gene content. Genomic islands are designated by black bars. Gray blocks between genomes denote homologous regions at least 5 kb in size. Phylogenetic reconstruction was based on the maximum likelihood method using core SNPs predicted from the progressiveMauve genome alignments and supported by 500 bootstraps.
FIG 2 .

Circular representation of the TY2482 genome and orthologous genes and SNPs on O104:H4 genomes. The circle is divided into arcs representing the sequence of the chromosome and the three plasmids of the reference isolate TY2482, as labeled. The outer track displays the ideogram of the reference genome TY2482, and chromosome and plasmids are labeled. The outer ring shows coordinates of reference sequences, with the yellow-shaded regions representing the plasmid scaffolds. Prophages are indicated in colored boxes (color code matches that in Fig. 1), and predicted rRNAs are in light green. Orthologs for each of the genomes with respect to TY2482 (outermost green track) are show in the order (outside-in) of the legend in the center of the figure (2011 outbreak and sporadic isolates in green tracks; historical isolates in blue tracks). SNPs are identified as red and yellow ticks, with red representing coding and yellow noncoding. The set of SNPs represented here was derived from mapping reads from each of these genomes to TY2482. No SNPs were reported for 55989, as no reads for this genome were available.
The most salient differences in the genomes of these isolates reside in their mobile elements. There are marked variations in the numbers and gene contents of the plasmids (Fig. 2 and 3). For example, only TY2482 harbors pTY1, a large plasmid encoding a blaCTX-M-15 gene conferring extended-spectrum beta-lactamase (ESBL) activity (10, 12–14) (Fig. 3). Besides variation in plasmid content, there is also substantial variation in the number and content of the prophages and genomic islands (GIs) present in these strains (Fig. 1 and 2). For example, TY2482 harbors 7 prophages (depicted as colored rectangles in Fig. 1 and 2), but only 2 additional isolates harbor the same set of prophages, and there is evidence for variation in gene content and for recombination compared to TY2482. Overall, the predicted prophages and genomic islands identified here comprise roughly 14% of the TY2482 chromosome.
FIG 3 .

Plasmid content of E. coli O104:H4 isolates. Heat map showing the degree of sequence identity between nonchromosomal scaffolds from each isolates and a reference set of plasmids associated with E. coli O104:H4 isolates. The shade of green indicates the degree of sequence identity (graded white to dark green, as per the legend). Plasmids considered present in the isolate on the basis of extent of sequence identity are outlined with a thick black border. Key contents of the plasmids, including the CTX-M-15 extended-spectrum beta-lactamase, AAF/I, and AAF/III, are denoted in representative sites. pHUSEC41-2 and p55989 are nearly identical (see Fig. S6C in the supplemental material), and pHUSEC41-3 (not shown) is not present in the isolates analyzed in this study. pTY3, pHUSEC41-4, pEc09-7901-c, and pEc12-0466-c represent cryptic plasmids. Of note, pTY2 was not in the sequenced genome of Ec11-9450; however, PCR analysis of multiple colonies from the original sample demonstrates that it was lost during laboratory culture steps. Its presence is therefore denoted by a dotted box.
Phylogeny of E. coli O104:H4.
Single nucleotide polymorphisms (SNPs) present in the core genome shared among the E. coli O104:H4 isolates analyzed here (see Materials and Methods) were used to analyze their phylogenetic relationships. This analysis demonstrated that the 5 postoutbreak sporadic isolates were very closely related to each other and to the outbreak isolate. Importantly, the phylogeny revealed that these 5 sporadic isolates are not derived from the 2011 outbreak strain; instead, they share a recent common ancestor with TY2482 (Fig. 1). Furthermore, the 5 sporadic isolates and the 2011 outbreak isolate are much more closely related to one another than to the historical E. coli O104:H4 isolates. We refer to these 6 strains as clade 1 and the 2004 and 2009 isolates as clade 2. Linear regression of genetic distance on year of isolation yields estimates of the rate of divergence over time and suggests the most recent common ancestor of these clades and 55989 existed approximately 30 years ago, with a substitution rate of 2.5 × 10−6 to 3.0 × 10−6 substitutions per site per year (see Fig. S2 in the supplemental material). This is similar to a recent estimate for Staphylococcus aureus (21), but approximately twice the rate recently reported for Shigella (22), which may reflect a biased clock rate due to the shorter time period separating the isolates studied here (23). With approximately 60 SNPs (see Materials and Methods) separating each of the sporadic isolates from TY2482, this suggests that the most recent common ancestor of the 2011 isolates, both outbreak and sporadic, existed around 2008 to 2009.
Prophages.
Our conclusions regarding the evolutionary relationships among these strains are supported and extended by analyses of the number, insertion sites, and sequences of the prophages in the E. coli O104:H4 isolates. The number of predicted prophages varies across the O104:H4 isolates, illustrating the dynamics of phage gain and loss over the relatively short evolutionary time separating them. The TY2482 genome has a total of 7 predicted intact prophages (designated O104H4-A through -G), one of which (O104H4-G) carries the stx2 genes conferring Shiga toxin production (Fig. 1 and 2; see also Fig. S3A to S3I in the supplemental material). 55989, the most divergent of the isolates we analyzed, does not harbor the B or G prophages, which are present in all the other isolates, and harbors 2 prophages (H and J) not present in any other strains. However, 55989 contains prophages A, E, and F at the same sites as TY2482, suggesting that these phages were present in their common ancestor. Comparing only the isolates in clade 1, there is far greater similarity in their phage content: all seven TY2482 prophages (O104H4-A through -G) were likely present in the common ancestor of these isolates, although the E phage was apparently lost from the common ancestor of Ec11-9450 and Ec12-0465. Six of these phages (O104H4-A, -B, -D, -E, -F, and -G) were likely acquired prior to the divergence of clade 1 and 2 lineages (with subsequent loss of phage O104H4-D from Ec09-7901). Interestingly, variants of phage O104H4-C are also present in each genome, but at distinct sites in 55989 and clades 1 and 2 (Fig. 1) and with distinct sets of SNPs (see Fig. S3C), suggesting three independent acquisitions of related phages. Finally, O104H4-I-like prophages are present only in the two clade 2 isolates and in a single clade 1 strain (Ec11-9941). These prophages are present at a different site and with slightly variable gene contents (see Fig. S3H) and therefore also likely represent independent acquisitions. Thus, the apparent relatedness of O104:H4 strains based on analyses of their phage content is similar to that resulting from genomic SNP analysis but also reveals differences between the strains due to phage gain and loss.
Besides providing clues regarding the evolution of E. coli O104:H4, comparison of the prophages also illustrates the dynamic nature of phage genomes. Across the phylogeny described by this set of isolates, each prophage family demonstrates variability, including in their gene contents and SNPs (see Fig. S3A to 3I in the supplemental material). For example, although the O104H4-C-related phages all contain a conserved set of syntenic genes, the prophages in 55989 and clades 1 and 2 exhibit significant nucleotide divergence among the conserved genes, as well as blocks of genes that are restricted to one of the clades (see Fig. S3C); these findings are consistent with the idea that these phages were independently acquired by 55989 and clades 1 and 2.
A key event in the evolution of fully virulent E. coli O104H4 capable of causing HUS was acquisition of O104H4-G (the stx2-containing prophage). This group of prophages exhibits relatively minimal variation (see Fig. S3G in the supplemental material), consistent with the idea that lysogenization of the ancestor of clades 1 and 2 with a Shiga toxin-encoding phage occurred relatively recently. The clusters of SNPs within O104H4-G in isolates Ec11-9450 and Ec12-0465 (Fig. 2; see also Fig. S3G) likely reflect one or more recombination events that introduced these SNPs at some point after divergence of this clade.
The mechanisms that account for the variation in gene content and clustering of SNPs are not known, but the patterns of variation suggest recombination. These observations are consistent with previous understanding of phage mosaicism and evolution (24) and provide among the most detailed descriptions of phage variation over a short time period. Moreover, the multiple distinct integration events of phage O104H4-C and O104H4-I suggested by these examples likely reflect cocirculation of these E. coli isolates and phages.
Variation in genomic islands.
Comparative analysis based on genome alignments and read coverage identifies seven nonprophage regions along the chromosome that are absent in one or more genomes (Table 2). Six of these seven regions are adjacent to tRNAs and contain mobility genes such as transposases, integrases, insertion sequence (IS) elements, and toxin-antitoxin (TA) systems; most also have altered GC contents compared to the rest of the genome (see Fig. S1 in the supplemental material). These regions are consequently described here as GIs (see Fig. S4A to S4F). The remaining region appears to be an IS element-associated deletion in 55989 of an approximately 13-kb region (see Fig. S4G).
TABLE 2 .
Genomic islands in the E. coli O104:H4 isolates
| Genomic region | Position in the genome | Selected gene content | Variants |
|---|---|---|---|
| GI-1 | 5.26–0.06 Mbp,a,c by serX | Microcin locus, iha, tellurium resistance locus, antigen 43, yeeV-yeeU TA system | Locus absent in 55989; deletion of multiple microcin locus genes in Ec11-9990 and -9451 |
| GI-2 | 2.26–2.36 Mbp,a by pheV | T6SS, pic, antigen 43 | Expanded T6SS locus in 55989 |
| GI-3 | 3.11–3.16 Mbp,a by selC | Sulfonamide resistance (sul1, sul2), trimethoprim resistance (dhfr7), ethidium bromide resistance protein (qacEΔ1), beta lactamase (blaTEM), mercuric resistance operon, tetracycline resistance (tetA), antigen 43, yeeV-yeeU TA system |
Not present in 55989, Ec04-8351, Ec09-7901; internal ~18-kb deletion in Ec11-9941 and Ec11-9990 |
| GI-4 | 3.7–3.77 Mbp,a by pheU | Aerobactin locus, pic, sigA, yeeV-yeeU TA system, entericidin TA system | Insertion of iha in this locus in 55989 |
| GI-5 | 4.94–5.00 Mbp,b by leuX | Tetracycline resistance, DNA phosphorothioation locus, yeeV-yeeU TA system | Present only in 55989 |
| GI-6 | 4.27–4.35 Mbp,a by aspV and thrW | T6SS, YafQ/DinJ TA system | Predicted ORF region 1 not present in 55989; region 2 not present in Ec04-8351 and Ec11-9450 |
Indexed according to the TY2482 assembly.
Indexed according to the 55989 assembly.
This locus crosses the break in the linearization of the TY2482 genome.
As with the prophage, the pattern of variation of GIs is consistent with the core genome-based phylogeny. Clades 1 and 2 contain a large genomic island, GI-1, not present in 55989; clade 1 alone contains another, GI-3, that contains a Tn21-like multidrug-resistant (MDR) transposon (25); and 55989 harbors GI-5 and an expansion in GI-2 not present in the other isolates. The common ancestor of Ec11-9941 and Ec11-9990 also underwent two deletions within GIs, with a deletion of several genes critical to the function of the microcin locus in GI-1 (see Fig. S4A in the supplemental material) and deletion of an ~18-kb region of GI-3 that contains antibiotic resistance and mercury reductase elements (see Fig. S4C).
In addition to containing toxin-antitoxin systems, presumably functioning as addiction factors for the GIs (26), these regions are enriched for genes that encode protection from antibiotics and toxins in the environment, that target other organisms in the microenvironment, and that allow for continued growth under low-resource settings. These include many loci associated with virulence during infection: multiple antibiotic and toxin resistance elements, such as resistance determinants to sulfonamides, mercury, ethidium bromide, beta lactams (GI-3), tetracyclines (GI-3 and GI-5), and tellurium (GI-1); iha (27, 28), which is involved in cellular adhesion; the microcin locus (29, 30), involved in bacterial competition; the type 6 secretion system (30, 32), which encodes a contact-dependent toxin delivery system that injects effector proteins into host eukaryotic cells or other bacteria; SPATEs (33), involved in serum resistance and hemagglutination; the aerobactin locus (34, 35), which encodes a siderophore used to sequester iron in low-iron environments; and ag43, which is involved in biofilm formation (36). Several of these loci appear in multiple GIs. For example, ag43 appears in GI-1, -2, and -3, and pic, a member of the SPATE family, appears in GI-2 and GI-4. The presence of several factors in multiple copies in multiple GIs within the same strain and the appearance of factors in alternative GIs (e.g., iha in GI-1 in clades 1 and 2 but in GI-4 in 55989) support the hypothesis that GIs have a modular structure, as has been suggested (37).
The MGEs identified here likely do not represent the full complement of MGEs within this set of genomes, especially as our analysis focuses on variable regions, and other regions suggestive of MGEs, such as transposon sequences with proximity to virulence factors and TA systems, are conserved in all of the genomes analyzed here but may differ in more distant members of the O104:H4 lineage (see Fig. S5 in the supplemental material).
Variation in plasmids.
There is marked variability in the number and gene content of the plasmids in the O104:H4 lineage (Fig. 3). Remarkably, none of the nine E. coli O104:H4 isolates compared here contain the same set of plasmids. Moreover, even when related plasmids are present in more than one isolate, they show evidence of gene variation (see Fig. S6A to S6C in the supplemental material). TY2482 harbors three plasmids: pTY1, an 89-kb plasmid that encodes the ESBL CTX-M-15 and the beta-lactamase TEM; pTY2 (also referred to as pAA), a 73-kb plasmid that encodes the AAF/I fimbriae that confers the enteroaggregative phenotype and which has previously been linked to EAEC virulence; and pTY3, a 1.5-kb cryptic plasmid that appears in high copy number. Notably, besides TY2482, none of the isolates we analyzed harbored a pTY1-like plasmid, suggesting that this replicon bearing several antibiotic resistance genes was acquired very recently and is a marker that distinguishes the outbreak strain from the sporadic isolates in clade 1. Since all clade 1 isolates were from patients with HUS, pTY1 is not required for the development of this severe complication of E. coli O104:H4 infection.
pHUSEC41-1 is present in many of the other clade 1 isolates and mediates resistance to amoxicillin, streptomycin, and sulfonamides through a trbC, sul2, strA, blaTEM-1, and strB array that appears adjacent to a Tn21-like transposase (see Fig. S6A in the supplemental material). The region carrying antibiotic resistance is variable and appears to have been deleted in several isolates, including Ec04-8351, Ec11-9450, and Ec11-9990, while maintained in Ec11-9941 and at least partially in Ec12-0466, suggesting that this region has been lost several times in different lineages, although multiple independent acquisitions cannot be entirely ruled out. The presence of sul2 in Ec11-9941 and its absence in Ec11-9990 may also explain the differential susceptibility to sulfonamides observed on antibiotic susceptibility testing (Table 1) and suggests that the copy of sul1 in GI-3 in both Ec11-9941 and Ec11-9990 is nonfunctional.
All of the strains harbor a pAA-related virulence plasmid of one of two types: either a pTY2-related plasmid in all clade 1 isolates or a p55989-related plasmid, which encodes AAF/III fimbriae in clade 2 and 55989 (note that Ec11-9450 had the AAF/I genes in PCR tests of the initial isolate, but the pTY2 plasmid was not observed in the sequenced genome, indicating it was lost during the culture process). Besides the differences in the type of fimbriae they encode, there are many other differences between these two types of virulence plasmids. pHUSEC41-related plasmids (20) and four different small plasmids (pTY3, pHUSEC41-4, pEc09-7901-c, and pEc12-0466-c) are present in some but not all of either clade 1 and/or 2 isolates (Fig. 3).
Variation in gyrA.
As all the E. coli O104:H4 isolates have been resistant to quinolones at least since 2004, we searched for mutations in the sequence of the gyrA gene encoding a subunit of DNA gyrase, a target of the quinolone class of antibiotics, for the presence of mutations. The isolate 55989 has the wild-type genotype. The isolates 01-09591 (HUSEC041), Ec04-8351, and Ec09-7901 all share the S83L mutation, whereas all of the 2011 sporadic and outbreak isolates have the S83A mutation. Both mutations are known to be associated with resistance to quinolones. The level of resistance was higher (MICs of nalidixic acid, 128 to >256 mg/liter, and ciprofloxacin, 0.125 mg/liter) among isolates with the S83L mutation than among those having the S83A mutation (MICs of nalidixic acid, 24 to 48 mg/liter, and ciprofloxacin, 0.03 to 0.05 mg/liter).
Summary and conclusions.
Using genome assemblies of multiple O104:H4 isolates, including 55989, TY2482, sporadic clinical isolates identified in France in 2004, 2009, and 2011, and a sporadic isolate associated with disease acquired in Turkey, we characterized the phylogenetic relationships and variability seen in this closely related set of genomes.
The observed variation in the O104:H4 genomes demonstrates rapid gain, loss, and variation of genomic islands, prophages, and plasmids and could reflect adaptation to or interaction with local environments. Using the observed variation, we can construct a model of the emergence of these closely related E. coli O104:H4 isolates (Fig. 4). Their common ancestor was likely susceptible to quinolones (given the wild-type gyrA in 55989), lacked GI-3, and had an enteroaggregative phenotype conferred by AAF/III. This ancestor may also have lacked pHUSEC41-1, with its assortment of antibiotic resistance elements, given the absence of this plasmid from 55989. The plasmid’s presence in the 2001, 2004, and 2009 isolates as well as the 2011 sporadic and outbreak isolates suggest that it was acquired before these two lineages split. Subsequently, the two lineages independently acquired distinct gyrA mutations; the split of these lineages into different environments is also supported by the exchange of p55989, which confers the enteroaggregative phenotype via AAF/III, for pTY2, which confers the enteroaggregative phenotype via AAF/I. The acquisition of the antibiotic resistance elements on GI-3 and pHUSEC41-1 in the lineage leading to the outbreak and 2011 isolates suggests that this lineage underwent strong antibiotic selective pressure. As a final recent step leading to the outbreak strain, the pTY1 plasmid encoding resistance to the CTX-M-15 ESBL was acquired, displacing pHUSEC41-1 through plasmid incompatibility. The deletion of some resistance determinants in the Ec11-9941 and Ec11-9990 isolates suggests that these bacterial populations likely entered an environment no longer under pressure from tetracycline. Similarly, the likely abrogation of microcin function through the deletion in this locus indicates a change in competition among bacteria. We estimate that the diversification of this lineage took place over a short time span; approximately 30 years. Moreover, the variation observed among the 2011 outbreak and sporadic isolates has likely taken place much more recently, probably within the past 2 to 3 years.
FIG 4 .
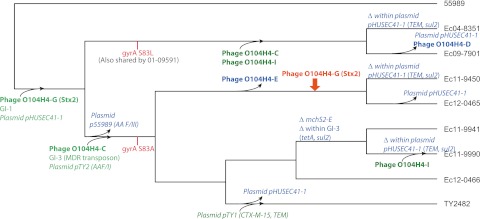
Evolutionary model of E. coli O104:H4 emergence. Events in the emergence of E. coli O104:H4 are represented on the phylogeny of the isolates, including selected gain (green), loss (blue), recombination (orange), and SNP (red) events. Prophages are denoted in bold and plasmids in italic. See text for details and support for hypothetical gain, loss, recombination, and SNP events. Note that not all differences among genomes are represented here; most differences among isolates for which the ancestral state is not certain (for example, phage H on 55989) are not shown.
By comparing the number of differences in MGE content and SNPs with respect to TY2482, we can estimate the ratio of changes in MGE content to SNPs as 0.05 to 0.1 for the sporadic isolates, 0.03 for the 2004 and 2009 isolates, and 0.04 for 55989 (isolated in the late 1990s; see Table S2 in the supplemental material). Although the sample size is small, the trend toward a higher ratio of differences in MGE content per SNP in closely related isolates merits speculation. A parsimonious hypothesis to explain this observation is that only a fraction of the many changes that happen over the short term are preserved by selection over longer periods. This may reflect rapidly changing ecology, such that the only elements that are preserved are those that have consistent adaptive value, and this hypothesis should be further explored in future studies with larger numbers of isolates.
The shared elements among many of the genomic islands (such as the multiple appearances of ag43, pic, and T6SS) suggest convergent evolution, in which independent mobile elements have allowed adaptation to similar environments. That many loci appear multiple times within the same strain (being present in multiple mobile elements, whether through multiple acquisition or gene duplication) raises questions over their function and how this is regulated. It is reasonable to suggest that genes of unknown function in these loci may also be involved in adaptation to local environments and for interaction with other bacteria and hosts. Similarly, the contributions of phage to survival of the host E. coli cell are, if present, often obscure or unproven. For example, even the function of Shiga toxin, cargo of O104H4-G, in natural environments is uncertain, though it has been speculated to benefit E. coli by increasing survival from protozoan predation (38).
A critical conclusion from this work is that the isolates from cases that took place after the summer 2011 outbreaks are not derived from those outbreaks but instead share a close common ancestor, a conclusion facilitated by whole-genome sequencing and analysis. Although the assortment of virulence factors appears to be in flux based on the analysis described here, the key pathogenicity elements, namely, Shiga toxin and aggregative adherence fimbriae, are maintained in each of the five isolates from sporadic clinical cases of HUS in 2011. These factors support the contention that similarly virulent O104:H4 isolates are widespread and emphasize the possibility of future food-borne E. coli O104:H4 outbreaks.
MATERIALS AND METHODS
Isolates.
The E. coli O104:H4 isolates sequenced in this study were provided by the French National Reference Center for E. coli and Shigella (Institut Pasteur and Hôpital Robert Debré, Paris, France; Table 1). These include (i) an isolate from a patient infected in Turkey (15) who was among a group of travelers, a number of whom developed bloody and nonbloody diarrhea, and (ii) four sporadic isolates, previously unreported, from children in France with HUS and without known epidemiological links to described outbreaks (GenBank accession no. for Escherichia coli Ec12-0465, AIPQ01000000; Escherichia coli Ec12-0466, AIPR01000000; Escherichia coli Ec11-9450, AGWF01000000; Escherichia coli Ec11-9941, AGWH01000000; Escherichia coli Ec11-9990,AGWG01000000). Besides the genomes of these 5 isolates, we included previously reported E. coli O104:H4 isolates in our comparative analyses: 55989, isolated from an individual in The Central African Republic in the late 1990s (17); 01-09591 (HUSEC041), isolated from an individual in Germany in 2001 (12); Ec04-8351 and Ec09-7901, isolated from individuals in 2004 and 2009, respectively, in France (11), and TY2482, the prototype isolate from the 2011 German outbreak (14).
Library preparation and sequencing.
Fragment libraries were generated and quantified as previously described (11). Flow cells were sequenced with 101 base-paired-end reads on an Illumina HiSeq2000 instrument, using V3 TruSeq sequencing-by-synthesis kits and analyzed with the Illumina RTA version 1.12 pipeline.
Assembly.
Assemblies were performed by ALLPATHS-LG (39) using default options with the following three exceptions: MIN_CONTIG = 500, ASSISTED_PATCHING = True, EVALUATION = FULL; the last two used TY2482 as the reference sequence file. Initially, the assembly of Ec11-9941 erroneously incorporated a 55,565-nucleotide-long plasmid fragment into the chromosome. For this genome, assembly was redone using the ASSISTED_PATCHING = True parameter, which corrected the plasmid fragment misjoin. In this same assembly run, a different plasmid, the TY2482_pTY2-like plasmid, was captured in two scaffolds. These two scaffolds were merged to capture the plasmid in a single scaffold by use of the reference sequence of the homologous plasmid in TY2482 and read pairs from large-insert “jumping” libraries as linking evidence. Assembly data and assemblies are available at http://www.broadinstitute.org/annotation/genome/Ecoli_O104_H4/MultiHome.html.
Genome comparisons.
Protein-coding gene predictions were made by Prodigal (40). The gene product names were assigned based on top blast hits to an in-house curated set of E. coli K-12 and virulence proteins (parameters: E < 1 × 10−10, ≥60% query coverage, and ≥60% protein identity). Genome sequences and annotations are available from the Broad Institute website specified above. Predicted genes were grouped into putative ortholog clusters using OrthoMCL 1.0 (41) using an inflation value of 1.5 and an E value cutoff of 1 × 10−5. rRNA sites were predicted by RNAmmer (42), and tRNA by tRNAscan-SE (43). The circular map of genes based on presence/absence of TY2482 genes (as defined by existence of an OrthoMCL-determined homolog) in the genomes of 55989, Ec04-8351, Ec09-7901, Ec11-9450, Ec11-9941, Ec11-9990, Ec12-0465, and Ec12-0466 was generated using Circos (44). Read-based SNPs for genomes with compatible reads available (thereby excluding 55989 and 01-09591) were predicted by alignment to the TY2482 genomic sequence as previously reported (11) and rendered on the circular map of the genome. Genomic scaffolds of the isolates except for 01-09591 (excluded because of the prohibitively large number of scaffolds) were ordered and oriented based on progressiveMauve (45) and Nucmer (46) alignments against the chromosome sequence of reference strain TY2482. The reference-ordered and oriented scaffolds were concatenated into a single sequence per circular chromosome. The genome was linearized such that the start of the concatenated genomic sequence was set to the same start as TY2482. A preliminary alignment of concatenated chromosomal sequences suggested a putative misassembly (validated by analysis of mate-pair reads) in the assembled genome of Ec09-7901, which was rectified by inserting the scaffold 1.1 (accession no. JH378062.1) into the coordinate 1,567,752 of scaffold 1.2 (accession no. JH378063.1). A final alignment was performed by progressiveMauve using default parameters. A diagrammatic representation of this alignment and genomic features of interest was prepared using GenoPlotR (Fig. 1) (47). SNPs with respect to TY2482 were output by progressiveMauve, and downstream analysis was based on this assembly-based SNP set.
Phage analysis.
The assembled genome sequences were analyzed by the prophage-predicting PHAST (48) Web server. Regions identified algorithmically as “intact” by PHAST, as well as regions sharing a high degree of sequence similarity and conserved synteny with predicted “intact” prophages, were identified as prophages. These predicted prophage sequences were then aligned by progressiveMauve (45) and grouped according to the extent of sequence similarity and synteny.
Identification of genomic islands.
The genome alignments generated using progressiveMauve (45) were filtered for blocks of 5 kb or more in length that are absent in at least one genome; regions in which reference-based mapping could not confirm absence (such as in duplicate regions, including rRNA genes) were not included. GC content was plotted using DNAPlotter (49) (see Fig. S1 in the supplemental material).
Phylogenetic analysis.
After whole-genome assemblies of 55989, Ec04-8351, Ec09-7901, Ec11-9450, Ec11-9941, Ec11-9990, Ec12-0465, Ec12-0466, and TY2482 were aligned using progressiveMauve (45), the set of SNPs generated by the progressiveMauve alignment was filtered for core SNPs, defined by unambiguous base call in all genomes and exclusion of SNPs in regions of recombination (50). The maximum likelihood tree was generated from the core SNPs using the HKY85 model (51) and rooted on isolate 55989 (Fig. 2); the cladogram was then generated from this tree (Fig. 1). The year of isolation of 55989 was reported as between 1996 and 1999 (17), and the relationship between root-to-tip distance and year of isolation was plotted using each of these dates (Path-O-Gen version 1.3; see Fig. S2 in the supplemental material).
Microsynteny analysis.
Chromosomal regions encompassing prophages and genomic islands and plasmid sequences were visualized based on whole-genome alignment and conserved gene order of predicted orthologs. Phage genes were identified by BLAST against the downloaded PHAST (48) database using an E value cutoff of 1e−10. Regions of specific interest were manually annotated to improve the automated gene prediction and annotation.
Plasmid analysis.
Scaffolds less than 200 kb in size were aligned to reference plasmids by Nucmer (46) to determine plasmid content and extent of identity. The plasmids from 01-09591 (also referred to as HUSEC041) were designated pHUSEC41-1 to -4 (20). The set of reference plasmids included pTY1, pTY2, pTY3, p55989, pHUSEC41-1, pHUSEC41-4, pEc09-7901-c, and pEc12-0466-c. The heat map was rendered in R (52).
SUPPLEMENTAL MATERIAL
Plot of GC content for TY2482 and 55989. Prophage and genomic islands are indicated in the TY2482 genome; GI-5, which is present only in 55989, is indicated in the representation of that genome. Light blue blocks in the outer circle denote genes. In the intermediate circle, red blocks are genomic islands and blue blocks are prophages. In the inner circle, purple indicates GC content below the genome average. Download
Temporal signal in DNA sequences. Plot of root-to-tip distance by year of isolation with an isolation year for 55989 of 1996 (A) and 1999 (B). Download
Genes are denoted by block arrows; predicted phage genes were identified by BLAST against the PHAST (48) database and marked in yellow; genes that appear in only one genome are marked in red, and predicted phage genes that appear in only one genome are marked in brown; red rectangles indicate scaffold breaks; triangles denote the attL and attR sites; blue lines indicate the location of predicted SNPs based on progressiveMauve alignments with respect to TY2482; black bars indicate contig gaps, with the size of the gap determined in the assembly algorithm by the distance between paired reads from large-insert “jumping” libraries. Deletions near scaffold breaks may be artifactual, reflecting difficulties in the assembly of that region. For clarity of presentation, gene annotations are limited to one set of orthologous genes. (A) O104H4-A prophage. Representation of predicted genes and their homology in the region of the predicted O104H4-A prophage. Variation is notable in 55989, with distinct gene content in the 5′ and central regions; the Ec04-8351/Ec09-7901 lineage, with an expansion upstream of the predicted attL sites; and an insertion in Ec12-0466 of capsid and tail proteins. (B) O104H4-B and -H prophages. Representation of predicted genes and their homology in the region of the predicted O104H4-B and -H prophages. The suggestion of deletions at the sites of scaffold breaks in Ec09-7901 and Ec11-9450 instead may be artifacts caused by scaffold breaks. Overall, there is a high degree of identity in the O104H4-B prophage, with only slight variation in gene content between the Ec04-8351/Ec09-7901 branch of the phylogeny and the sporadic and outbreak isolates. (C) O104H4-C prophage. This predicted prophage is very well conserved among the O104:H4 isolates, with some gene content variation between the Ec04-8351/Ec09-7901 lineage and 55989. It has integrated in three separate locations, as indicated by the differences in the flanking regions between 55989, the clade of Ec04-8351 and Ec09-7901, and the clade defined by sporadic and outbreak isolates. (D) O104H4-D prophage. This prophage is missing in 55989 and Ec09-7901. The shared SNP pattern indicates that a region near the gene encoding the antitermination protein was conserved from the Ec04-8351 isolate through all the sporadic isolates, with the exception of the branch leading to Ec12-0466 and the outbreak isolate TY2482. (E) O104H4-E prophage. The predicted prophage appears to have integrated adjacent to the argW site and shows greater gene content divergence between 55989 and the other O104:H4 isolates. The predicted prophage O104H4-E appears to have been deleted in the branch of the phylogeny leading to Ec11-9450 and Ec12-0465, with an additional deletion of the flanking sucrose operon, d-serine permease and dehydrase, and the multidrug resistance proteins K and Y in Ec12-0465. Additionally, Ec11-9990 has a deletion of four upstream genes, of which several are predicted to be involved in fatty acid transport and metabolism. (F) O104H4-F prophage. This prophage is very well conserved across all the O104:H4 isolates. On evaluation of the reads, the duplication represented in Ec11-9941 in the tail component genes and containing two contig breaks appears to be an assembly error. (G) O104H4-G prophage. The Shiga toxin prophage is present in all isolates except 55989, and the gene content is well conserved. SNP level variation with respect to TY2482 indicates that the lineage leading to Ec11-9450 and Ec12-0465 underwent likely recombination with closely related prophage, introducing clusters of SNPs in several areas, notably in phage antirepressor proteins, the adenine methylase, endolysin, and endopeptidase. (H) O104H4-I prophage. This predicted prophage appears only in the Ec04-8351/Ec09-7901 lineage and Ec11-9941, in which it appears to have integrated at a different site, and with small variations in gene content. (I) O104H4-J prophage. While this predicted prophage appears only in the 55989 isolate, it has homology to the O104H4-A family of prophages, here represented by the predicted prophages from the Ec04-8351 and TY2482 genomes. Download
Genes are denoted by block arrows; predicted phage genes were identified by BLAST against the PHAST (48) database and marked in yellow; genes that appear in only one genome are marked in red, and predicted phage genes that appear in only one genome are marked in brown; red rectangles indicate scaffold breaks; triangles denote the attL and attR sites; blue lines indicate the location of predicted SNPs based on progressiveMauve alignments with respect to TY2482; black bars indicate contig gaps, with the size of the gap determined in the assembly algorithm by the distance between paired reads from large-insert “jumping” libraries. Deletions near scaffold breaks may be artifactual, reflecting difficulties in the assembly of that region. For clarity of presentation, gene annotations are limited to one set of orthologous genes. (A) GI-1. This region contains the iha element, the microcin operon, and a tellurium resistance operon, as well as ag43 and yeeV-yeeU TA system. This large genomic island, which has integrated by a serine tRNA, is absent in 55989. In the Ec11-9941 and Ec11-9990 isolates, there is a deletion within the microcin region. (B) GI-2. Multiple genes involved in virulence are present in this locus, including Pic serine protease, type VI secretion system, including a large expansion of this locus in 55989, and ag43. The region has many transposases and insertion sequence elements, which also makes the region difficult to resolve with automated assembly. (C) GI-3. This region, missing from the historical O104:H4 isolates, is similar to the MDR transposon Tn21, with sulfonamide resistance genes, the mercuric reductase operon, an ethidium bromide resistance element, as well as a nearby tetracycline resistance operon, ag43 and yeeV-yeeU TA, and blaTEM that appears to have been introduced via flanking transposon sequences. In the Ec11-9990 and Ec11-9941 branch, much of the transposon sequence was deleted, including the mercuric reductase and the tetracycline resistance operons. Although the Ec11-9941 genome has a contig gap in the region of the TEM beta lactamase, reads map to this region, indicating that it is likely present. (D) GI-4. This region contains multiple loci associated with virulence, including the aerobactin locus and the SPATEs sigA and pic. In 55989, iha appears as part of this locus, whereas it appears in GI-1 in the other O104:H4 isolates together with the microcin and tellurium resistance operons. The region also contains a large number of transposons, complicating automated assembly of this region and yielding the multiple scaffold and contig breaks visible here. (E) GI-5. A transposon similar to Tn10 appears only in 55989. This region contains a tetracycline resistance operon as well as yeeV-yeeU TA. (F) GI-6. This region contains a type VI secretion system that is well conserved across all the genomes. Downstream, near a Thr tRNA, is a region of approximately 15 kb containing predicted ORFs of uncertain function that is absent in the Ec11-9450/Ec12-0465 branch of the lineage and also in Ec04-8351. Additionally, there is a 14-kb region not present in 55989 that is conserved in the other O104:H4 isolates. (G) Deletion. A 14-kb deletion is present in 55989 relative to the other O104:H4 isolates. Insertion sequence elements in 55989 at this site suggest that they are associated with the deletion of this region. Most of the predicted ORFs in this region are of unknown function, with one containing a von Willebrand factor type A domain. IS elements appear in the Ec11-9450/Ec12-0465 lineage slightly upstream. Download
Nonvariable region containing TA system and multiple transposases, suggestive of a genomic island. Genes are denoted by block arrows; predicted phage genes were identified by BLAST against the PHAST (48) database and marked in yellow; genes that appear in only one genome are marked in red, and predicted phage genes that appear in only one genome are marked in brown; red rectangles indicate scaffold breaks; triangles denote the attL and attR sites; blue lines indicate the location of predicted SNPs based on progressiveMauve alignments with respect to TY2482; black bars indicate contig gaps, with the size of the gap determined in the assembly algorithm by the distance between paired reads from large-insert “jumping” libraries. Deletions near scaffold breaks may be artifactual, reflecting difficulties in the assembly of that region. For clarity of presentation, gene annotations are limited to one set of orthologous genes. The presence of yeeV-yeeU may represent a chromosomal instance of the TA system; however, the close proximity of multiple transposases and genes with homology to yersiniabactin biosynthetic protein and pesticin receptor raises the possibility that this region may also represent a mobile genetic element that is conserved among the isolates compared here. Download
Genes are denoted by block arrows; predicted phage genes were identified by BLAST against the PHAST (48) database and marked in yellow; genes that appear in only one genome are marked in red, and predicted phage genes that appear in only one genome are marked in brown; red rectangles indicate scaffold breaks; triangles denote the attL and attR sites; blue lines indicate the location of predicted SNPs based on progressiveMauve alignments with respect to TY2482; black bars indicate contig gaps, with the size of the gap determined in the assembly algorithm by the distance between paired reads from large-insert “jumping” libraries. Deletions near scaffold breaks may be artifactual, reflecting difficulties in the assembly of that region. For clarity of presentation, gene annotations are limited to one set of orthologous genes. (A) Plasmid pTY2. Scaffolds that show high levels of homology to the pTY2 plasmid were included in this representation of the linearized plasmids based on BLAST-based gene homology. (B) Plasmid pHUSEC41-1. Scaffolds that show high levels of homology to the pHUSEC41-1 plasmid were included in this representation of the linearized plasmids based on BLAST-based gene homology. (C) Plasmid p55989/pHUSEC41-2. Scaffolds that show high levels of homology to the p55989 (which is similar to pHUSEC41-2) plasmid were included in this representation of the linearized plasmids based on BLAST-based gene homology. Download
Orthologous genes based on OrthoMCL analysis with respect to TY2482. In the cluster type column, “core” refers to clusters with genes present in at least one copy in every genome; “auxiliary” clusters are missing a gene from at least one genome but contain genes from at least two genomes; “inparalog” clusters contain more than one gene, but all of the genes in the cluster are contained in a single genome; and “ref” identifies genes from clusters with two or more genes in TY2482. “gcMin” is the ortholog cluster gene with the shortest length, and “gcMax” is the ortholog cluster gene with the longest length
SNPs and mobile genetic elements (MGE) variations with respect to TY2482.
ACKNOWLEDGMENTS
This project has been funded in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract number HHSN272200900018C (Broad Institute). M.L. and W.P.H. received support from award number U54 GM088558 from the National Institute of General Medical Sciences. Y.H.G. received support from grant number U54 AI057159. This work was partially supported by the Institut Pasteur and by the Institut de veille sanitaire (Saint-Maurice, France).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We thank all corresponding laboratories of the French National Reference Center for E. coli and Shigella. We gratefully acknowledge Charlotte Balière, Isabelle Carles, Monique Lejay-Colin, and Corinne Ruckly for their expert technical assistance. We thank the Broad Biological Samples Platform and the Broad Genomics Platform. We are grateful to Lucia Alvarado-Balderrama for GenBank data deposition. We thank Nick Croucher and Bruce Birren for comments and input on the analyses and Cheryl Murphy and Chad Nusbaum for their helpful comments on the manuscript.
Footnotes
Citation Grad YH et al. 2013. Comparative genomics of recent Shiga toxin-producing Escherichia coli O104:H4: short-term evolution of an emerging pathogen. mBio 4(1):e00452-12. doi:10.1128/mBio.00452-12
REFERENCES
- 1. Frank C, et al. 2011. Large and ongoing outbreak of haemolytic uraemic syndrome, Germany. Euro. Surveill. 16:pii: 19878 [PubMed] [Google Scholar]
- 2. Frank C, et al. 2011. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N. Engl. J. Med. 365:1771–1780 [DOI] [PubMed] [Google Scholar]
- 3. Gault G, et al. 2011. Outbreak of haemolytic uraemic syndrome and bloody diarrhoea due to Escherichia coli O104:H4, south-west France, June 2011. Euro. Surveill. 16:pii: 19905 [DOI] [PubMed] [Google Scholar]
- 4. Jansen A, Kielstein JT. 2011. The new face of enterohaemorrhagic Escherichia coli infections. Euro. Surveill. 16:pii: 19898 [PubMed] [Google Scholar]
- 5. Struelens MJ, Palm D, Takkinen J. 2011. Enteroaggregative, Shiga toxin-producing Escherichia coli O104:H4 outbreak: new microbiological findings boost coordinated investigations by European public health laboratories. Euro. Surveill. 16:pii: 19898 [DOI] [PubMed] [Google Scholar]
- 6. King LA, et al. 2012. Outbreak of Shiga toxin-producing Escherichia coli O104:H4 associated with organic fenugreek sprouts, France, June 2011. Clin. Infect. Dis. 54:1588–1594 [DOI] [PubMed] [Google Scholar]
- 7. Mariani-Kurkdjian P, Bingen E, Gault G, Silva NJ, Weill FX. 2011. Escherichia coli O104:H4 south-west France. Lancet Infect. Dis. 11:732–733 [DOI] [PubMed] [Google Scholar]
- 8. Buchholz U, et al. 2011. German outbreak of Escherichia coli O104:H4 associated with sprouts. N. Engl. J. Med. 365:1763–1770 [DOI] [PubMed] [Google Scholar]
- 9. Alexander DC, et al. 2012. Escherichia coli O104:H4 infections and international travel. Emerg. Infect. Dis. 18:473–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brzuszkiewicz E, et al. 2011. Genome sequence analyses of two isolates from the recent Escherichia coli outbreak in Germany reveal the emergence of a new pathotype: entero-aggregative-haemorrhagic Escherichia coli (EAHEC). Arch. Microbiol. 193:883–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grad YH, et al. 2012. Genomic epidemiology of the Escherichia coli O104:H4 outbreaks in Europe. Proc. Natl. Acad. Sci. U. S. A. 2011:109:3065-3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mellmann A, et al. 2011. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One 6:e22751 http://dx.doi.org/10.1371/journal.pone.0022751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rasko DA, et al. 2011. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N. Engl. J. Med. 365:709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rohde H, et al. 2011. Open-source genomic analysis of Shiga-toxin-producing E. coli O104:H4. N. Engl. J. Med. 365:718–724 [DOI] [PubMed] [Google Scholar]
- 15. Jourdan-da Silva N, et al. 2012. Outbreak of haemolytic uraemic syndrome due to Shiga toxin-producing Escherichia coli O104:H4 among French tourists returning from Turkey, September 2011. Euro. Surveill. 17:pii:20065. [DOI] [PubMed] [Google Scholar]
- 16. Bielaszewska M, et al. 2011. Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: a microbiological study. Lancet Infect. Dis. 11:671–676 [DOI] [PubMed] [Google Scholar]
- 17. Mossoro C, et al. 2002. Chronic diarrhea, hemorrhagic colitis, and hemolytic-uremic syndrome associated with HEp-2 adherent Escherichia coli in adults infected with human immunodeficiency virus in Bangui, Central African Republic. J. Clin. Microbiol. 40:3086–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Touchon M, et al. 2009. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet. 5:e1000344 http://dx.doi.org/10.1371/journal.pgen.1000344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Monecke S, et al. 2011. Presence of enterohemorrhagic Escherichia coli ST678/O104:H4 in France prior to 2011. Appl. Environ. Microbiol. 77:8784–8786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Künne C, et al. 2012. Complete sequences of plasmids from the hemolytic-uremic syndrome-associated Escherichia coli strain HUSEC41. J. Bacteriol. 194:532–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harris SR, et al. 2010. Evolution of MRSA during hospital transmission and intercontinental spread. Science 327:469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Holt KE, et al. 2012. Shigella sonnei genome sequencing and phylogenetic analysis indicate recent global dissemination from Europe. Nat. Genet. 44:1056–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ho SY, Larson G. 2006. Molecular clocks: when times are a-changin. Trends Genet. 22:79–83 [DOI] [PubMed] [Google Scholar]
- 24. Juhala RJ, et al. 2000. Genomic sequences of bacteriophages HK97 and HK022: pervasive genetic mosaicism in the lambdoid bacteriophages. J. Mol. Biol. 299:27–51 [DOI] [PubMed] [Google Scholar]
- 25. Liebert CA, Hall RM, Summers AO. 1999. Transposon Tn21, flagship of the floating genome. Microbiol. Mol. Biol. Rev. 63:507–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Magnuson RD. 2007. Hypothetical functions of toxin-antitoxin systems. J. Bacteriol. 189:6089–6092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnson JR, et al. 2000. Molecular epidemiological and phylogenetic associations of two novel putative virulence genes, iha and iroN (E. coli), among Escherichia coli isolates from patients with urosepsis. Infect. Immun. 68:3040–3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tarr PI, et al. 2000. Iha: a novel Escherichia coli O157:H7 adherence-conferring molecule encoded on a recently acquired chromosomal island of conserved structure. Infect. Immun. 68:1400–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Azpiroz MF, Bascuas T, Laviña M. 2011. Microcin H47 system: an Escherichia coli small genomic island with novel features. PLoS One 6:e26179 http://dx.doi.org/10.1371/journal.pone.0026179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Patzer SI, Baquero MR, Bravo D, Moreno F, Hantke K. 2003. The colicin G, H and X determinants encode microcins M and H47, which might utilize the catecholate siderophore receptors FepA, cir, Fiu and IroN. Microbiology 149(Part 9):2557–2570 [DOI] [PubMed] [Google Scholar]
- 31. Russell AB, et al. 2011. Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475:343–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Basler M, Pilhofer M, Henderson GP, Jensen GJ, Mekalanos JJ. 2012. Type VI secretion requires a dynamic contractile phage tail-like structure. Nature 483:182–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Henderson IR, Czeczulin J, Eslava C, Noriega F, Nataro JP. 1999. Characterization of pic, a secreted protease of Shigella flexneri and enteroaggregative Escherichia coli. Infect. Immun. 67:5587–5596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andrews SC, Robinson AK, Rodríguez-Quiñones F. 2003. Bacterial iron homeostasis. FEMS Microbiol. Rev. 27:215–237 [DOI] [PubMed] [Google Scholar]
- 35. Vokes SA, Reeves SA, Torres AG, Payne SM. 1999. The aerobactin iron transport system genes in Shigella flexneri are present within a pathogenicity island. Mol. Microbiol. 33:63–73 [DOI] [PubMed] [Google Scholar]
- 36. Danese PN, Pratt LA, Dove SL, Kolter R. 2000. The outer membrane protein, antigen 43, mediates cell-to-cell interactions within Escherichia coli biofilms. Mol. Microbiol. 37:424–432 [DOI] [PubMed] [Google Scholar]
- 37. Juhas M, et al. 2009. Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33:376–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Steinberg KM, Levin BR. 2007. Grazing protozoa and the evolution of the Escherichia coli O157:H7 Shiga toxin-encoding prophage. Proc. Biol. Sci. 274:1921–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gnerre S, et al. 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U. S. A. 108:1513–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hyatt D, et al. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li L, Stoeckert CJ, Jr, Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13:2178–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lagesen K, et al. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35:3100–3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Krzywinski M, et al. 2009. Circos: an information aesthetic for comparative genomics. Genome Res. 19:1639–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Darling AE, Mau B, Perna NT. 2010. ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147 http://dx.doi.org/10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Delcher AL, Phillippy A, Carlton J, Salzberg SL. 2002. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 30:2478–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guy L, Kultima JR, Andersson SG. 2010. genoPlotR: comparative gene and genome visualization in R. Bioinformatics 26:2334–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. 2011. PHAST: a fast phage search tool. Nucleic Acids Res. 39:W347–W352 http://dx.doi.org/10.1093/nar/gkq1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Carver T, Thomson N, Bleasby A, Berriman M, Parkhill J. 2009. DNAPlotter: circular and linear interactive genome visualization. Bioinformatics 25:119–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Croucher NJ, et al. 2011. Rapid pneumococcal evolution in response to clinical interventions. Science 331:430–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gouy M, Guindon S, Gascuel O. 2010. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27:221–224 [DOI] [PubMed] [Google Scholar]
- 52. R Development Core Team 2012. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Plot of GC content for TY2482 and 55989. Prophage and genomic islands are indicated in the TY2482 genome; GI-5, which is present only in 55989, is indicated in the representation of that genome. Light blue blocks in the outer circle denote genes. In the intermediate circle, red blocks are genomic islands and blue blocks are prophages. In the inner circle, purple indicates GC content below the genome average. Download
Temporal signal in DNA sequences. Plot of root-to-tip distance by year of isolation with an isolation year for 55989 of 1996 (A) and 1999 (B). Download
Genes are denoted by block arrows; predicted phage genes were identified by BLAST against the PHAST (48) database and marked in yellow; genes that appear in only one genome are marked in red, and predicted phage genes that appear in only one genome are marked in brown; red rectangles indicate scaffold breaks; triangles denote the attL and attR sites; blue lines indicate the location of predicted SNPs based on progressiveMauve alignments with respect to TY2482; black bars indicate contig gaps, with the size of the gap determined in the assembly algorithm by the distance between paired reads from large-insert “jumping” libraries. Deletions near scaffold breaks may be artifactual, reflecting difficulties in the assembly of that region. For clarity of presentation, gene annotations are limited to one set of orthologous genes. (A) O104H4-A prophage. Representation of predicted genes and their homology in the region of the predicted O104H4-A prophage. Variation is notable in 55989, with distinct gene content in the 5′ and central regions; the Ec04-8351/Ec09-7901 lineage, with an expansion upstream of the predicted attL sites; and an insertion in Ec12-0466 of capsid and tail proteins. (B) O104H4-B and -H prophages. Representation of predicted genes and their homology in the region of the predicted O104H4-B and -H prophages. The suggestion of deletions at the sites of scaffold breaks in Ec09-7901 and Ec11-9450 instead may be artifacts caused by scaffold breaks. Overall, there is a high degree of identity in the O104H4-B prophage, with only slight variation in gene content between the Ec04-8351/Ec09-7901 branch of the phylogeny and the sporadic and outbreak isolates. (C) O104H4-C prophage. This predicted prophage is very well conserved among the O104:H4 isolates, with some gene content variation between the Ec04-8351/Ec09-7901 lineage and 55989. It has integrated in three separate locations, as indicated by the differences in the flanking regions between 55989, the clade of Ec04-8351 and Ec09-7901, and the clade defined by sporadic and outbreak isolates. (D) O104H4-D prophage. This prophage is missing in 55989 and Ec09-7901. The shared SNP pattern indicates that a region near the gene encoding the antitermination protein was conserved from the Ec04-8351 isolate through all the sporadic isolates, with the exception of the branch leading to Ec12-0466 and the outbreak isolate TY2482. (E) O104H4-E prophage. The predicted prophage appears to have integrated adjacent to the argW site and shows greater gene content divergence between 55989 and the other O104:H4 isolates. The predicted prophage O104H4-E appears to have been deleted in the branch of the phylogeny leading to Ec11-9450 and Ec12-0465, with an additional deletion of the flanking sucrose operon, d-serine permease and dehydrase, and the multidrug resistance proteins K and Y in Ec12-0465. Additionally, Ec11-9990 has a deletion of four upstream genes, of which several are predicted to be involved in fatty acid transport and metabolism. (F) O104H4-F prophage. This prophage is very well conserved across all the O104:H4 isolates. On evaluation of the reads, the duplication represented in Ec11-9941 in the tail component genes and containing two contig breaks appears to be an assembly error. (G) O104H4-G prophage. The Shiga toxin prophage is present in all isolates except 55989, and the gene content is well conserved. SNP level variation with respect to TY2482 indicates that the lineage leading to Ec11-9450 and Ec12-0465 underwent likely recombination with closely related prophage, introducing clusters of SNPs in several areas, notably in phage antirepressor proteins, the adenine methylase, endolysin, and endopeptidase. (H) O104H4-I prophage. This predicted prophage appears only in the Ec04-8351/Ec09-7901 lineage and Ec11-9941, in which it appears to have integrated at a different site, and with small variations in gene content. (I) O104H4-J prophage. While this predicted prophage appears only in the 55989 isolate, it has homology to the O104H4-A family of prophages, here represented by the predicted prophages from the Ec04-8351 and TY2482 genomes. Download
Genes are denoted by block arrows; predicted phage genes were identified by BLAST against the PHAST (48) database and marked in yellow; genes that appear in only one genome are marked in red, and predicted phage genes that appear in only one genome are marked in brown; red rectangles indicate scaffold breaks; triangles denote the attL and attR sites; blue lines indicate the location of predicted SNPs based on progressiveMauve alignments with respect to TY2482; black bars indicate contig gaps, with the size of the gap determined in the assembly algorithm by the distance between paired reads from large-insert “jumping” libraries. Deletions near scaffold breaks may be artifactual, reflecting difficulties in the assembly of that region. For clarity of presentation, gene annotations are limited to one set of orthologous genes. (A) GI-1. This region contains the iha element, the microcin operon, and a tellurium resistance operon, as well as ag43 and yeeV-yeeU TA system. This large genomic island, which has integrated by a serine tRNA, is absent in 55989. In the Ec11-9941 and Ec11-9990 isolates, there is a deletion within the microcin region. (B) GI-2. Multiple genes involved in virulence are present in this locus, including Pic serine protease, type VI secretion system, including a large expansion of this locus in 55989, and ag43. The region has many transposases and insertion sequence elements, which also makes the region difficult to resolve with automated assembly. (C) GI-3. This region, missing from the historical O104:H4 isolates, is similar to the MDR transposon Tn21, with sulfonamide resistance genes, the mercuric reductase operon, an ethidium bromide resistance element, as well as a nearby tetracycline resistance operon, ag43 and yeeV-yeeU TA, and blaTEM that appears to have been introduced via flanking transposon sequences. In the Ec11-9990 and Ec11-9941 branch, much of the transposon sequence was deleted, including the mercuric reductase and the tetracycline resistance operons. Although the Ec11-9941 genome has a contig gap in the region of the TEM beta lactamase, reads map to this region, indicating that it is likely present. (D) GI-4. This region contains multiple loci associated with virulence, including the aerobactin locus and the SPATEs sigA and pic. In 55989, iha appears as part of this locus, whereas it appears in GI-1 in the other O104:H4 isolates together with the microcin and tellurium resistance operons. The region also contains a large number of transposons, complicating automated assembly of this region and yielding the multiple scaffold and contig breaks visible here. (E) GI-5. A transposon similar to Tn10 appears only in 55989. This region contains a tetracycline resistance operon as well as yeeV-yeeU TA. (F) GI-6. This region contains a type VI secretion system that is well conserved across all the genomes. Downstream, near a Thr tRNA, is a region of approximately 15 kb containing predicted ORFs of uncertain function that is absent in the Ec11-9450/Ec12-0465 branch of the lineage and also in Ec04-8351. Additionally, there is a 14-kb region not present in 55989 that is conserved in the other O104:H4 isolates. (G) Deletion. A 14-kb deletion is present in 55989 relative to the other O104:H4 isolates. Insertion sequence elements in 55989 at this site suggest that they are associated with the deletion of this region. Most of the predicted ORFs in this region are of unknown function, with one containing a von Willebrand factor type A domain. IS elements appear in the Ec11-9450/Ec12-0465 lineage slightly upstream. Download
Nonvariable region containing TA system and multiple transposases, suggestive of a genomic island. Genes are denoted by block arrows; predicted phage genes were identified by BLAST against the PHAST (48) database and marked in yellow; genes that appear in only one genome are marked in red, and predicted phage genes that appear in only one genome are marked in brown; red rectangles indicate scaffold breaks; triangles denote the attL and attR sites; blue lines indicate the location of predicted SNPs based on progressiveMauve alignments with respect to TY2482; black bars indicate contig gaps, with the size of the gap determined in the assembly algorithm by the distance between paired reads from large-insert “jumping” libraries. Deletions near scaffold breaks may be artifactual, reflecting difficulties in the assembly of that region. For clarity of presentation, gene annotations are limited to one set of orthologous genes. The presence of yeeV-yeeU may represent a chromosomal instance of the TA system; however, the close proximity of multiple transposases and genes with homology to yersiniabactin biosynthetic protein and pesticin receptor raises the possibility that this region may also represent a mobile genetic element that is conserved among the isolates compared here. Download
Genes are denoted by block arrows; predicted phage genes were identified by BLAST against the PHAST (48) database and marked in yellow; genes that appear in only one genome are marked in red, and predicted phage genes that appear in only one genome are marked in brown; red rectangles indicate scaffold breaks; triangles denote the attL and attR sites; blue lines indicate the location of predicted SNPs based on progressiveMauve alignments with respect to TY2482; black bars indicate contig gaps, with the size of the gap determined in the assembly algorithm by the distance between paired reads from large-insert “jumping” libraries. Deletions near scaffold breaks may be artifactual, reflecting difficulties in the assembly of that region. For clarity of presentation, gene annotations are limited to one set of orthologous genes. (A) Plasmid pTY2. Scaffolds that show high levels of homology to the pTY2 plasmid were included in this representation of the linearized plasmids based on BLAST-based gene homology. (B) Plasmid pHUSEC41-1. Scaffolds that show high levels of homology to the pHUSEC41-1 plasmid were included in this representation of the linearized plasmids based on BLAST-based gene homology. (C) Plasmid p55989/pHUSEC41-2. Scaffolds that show high levels of homology to the p55989 (which is similar to pHUSEC41-2) plasmid were included in this representation of the linearized plasmids based on BLAST-based gene homology. Download
Orthologous genes based on OrthoMCL analysis with respect to TY2482. In the cluster type column, “core” refers to clusters with genes present in at least one copy in every genome; “auxiliary” clusters are missing a gene from at least one genome but contain genes from at least two genomes; “inparalog” clusters contain more than one gene, but all of the genes in the cluster are contained in a single genome; and “ref” identifies genes from clusters with two or more genes in TY2482. “gcMin” is the ortholog cluster gene with the shortest length, and “gcMax” is the ortholog cluster gene with the longest length
SNPs and mobile genetic elements (MGE) variations with respect to TY2482.