

ABSTRACT
The herpes simplex virus (HSV) genome is associated with heterochromatic histone modifications, including trimethylation of the lysine 27 residue of histone H3 (H3K27me3), during latent infection of neurons. Here we have examined the kinetics of general chromatin and H3K27me3 association with the viral genome during establishment of latent infection. Using both wild-type virus and a mutant virus that is unable to undergo replication in neurons, we found that histone H3 associates with viral gene promoters by 7 days postinfection (dpi). Levels of H3K27me3 were low at 7 dpi but increased dramatically by 14 dpi. Hence, general chromatin association and/or other factors may play a key role(s) in the initial silencing of lytic genes, and H3K27me3 may play a role in further suppression of the genome and/or the maintenance of latency. A component of Polycomb repressive complex 2 (PRC2), which mediates the addition of K27me3 to histone H3 (Suz12), was also recruited by 14 dpi. We have shown previously that the levels of H3K27me3 during latent infection are increased in the presence of the latency-associated transcript (LAT). However, the initial targeting of PRC2 was not found to be dependent on the LAT. We found that a component of the PRC1 complex (Bmi1), which binds to H3K27me3, was not enriched at promoters found previously to be enriched for H3K27me3. Our results are consistent with (i) chromatinization of viral DNA or other mechanisms causing the initial silencing of HSV lytic genes and (ii) facultative heterochromatin maintaining that silencing during latent infection of neurons.
IMPORTANCE
The human pathogen herpes simplex virus (HSV) hides for the lifetime of the host in peripheral neurons. The mechanism by which HSV is able to shut off its gene expression and persist in neurons is not known. Here we show that the HSV DNA first associates with histone H3, with later recruitment of Polycomb repressor complex 2 (PRC2) and trimethylation of the lysine 27 residue of histone H3 (H3K27me3), a modification associated with heterochromatin. This work indicates that the initial silencing of HSV gene expression is not correlated with enrichment of H3K27me3 and that PRC2 may be recruited to already-silenced genes to further silence gene expression and/or maintain gene silencing. We demonstrate that recruitment of PRC2 is not dependent upon expression of the noncoding HSV latency-associated transcripts, indicating the presence of unknown triggers for PRC2 recruitment during the establishment of latent infection.
Introduction
Herpesviruses persist for the lifetime of an individual in the form of a latent infection. Latent herpes simplex virus 1 (HSV-1) DNA persists as an episome (1) assembled in nucleosomal chromatin (2) in sensory neurons within peripheral ganglia, where lytic cycle protein expression is restricted, and the only viral transcripts expressed to high levels are noncoding RNAs known as the latency-associated transcripts (LATs) (3) and microRNAs (miRNAs) (reviewed in reference 4). Periodic reactivation from latency allows transmission of HSV to a new host. Because all antivirals currently in use target only the lytic stage of infection, latent infection presents a major challenge to the treatment of HSV.
The mechanism(s) by which HSV-1 lytic gene expression is silenced within neurons is not fully understood, but a number of mechanisms have been proposed. First, the inability of VP16, the viral immediate early (IE) gene transcriptional activator, to localize to the neuronal nucleus (5) and/or the cytoplasmic localization of HCF-1, the cellular activator for IE genes, in sensory neurons (6) may lead to the inability of the virus to form the transactivator complex that promotes IE gene expression (7). Second, cellular repressors, such as the restriction element 1 silencing transcription (REST) factor - cofactor of restriction element 1 silencing transcription (Co-REST) complex, have been shown to reduce viral replication in neurons (8). Furthermore, at least two mechanisms by which the RNAs expressed from the LAT locus actively limit lytic protein expression in neurons have been proposed. First, expression of the LATs has been associated with reduced expression of lytic gene transcripts during acute infection (9) or latent infection (10). In latent infection, the viral lytic gene promoters are associated with heterochromatin, and expression of the LATs is associated with increased heterochromatin on the viral lytic gene promoters (11, 12). The viral genome is specifically enriched for chromatin containing the histone H3 lysine 9 trimethyl (H3K9me3), H3K9 dimethyl (H3K9me2), and H3 lysine 27 trimethyl (H3K27me3) modifications (11–13), and H3K27me3 was found to be a major form of heterochromatin on the viral genome (12). Interestingly, the association of H3K9me2 and H3K27me3 with lytic gene promoters was decreased in neurons latently infected with a LAT deletion mutant virus relative to their association in neurons infected with its rescued virus (11, 12). Therefore, a role for the LAT in promoting or maintaining heterochromatin levels is consistent with previous observations that LAT down-regulates lytic gene expression during the establishment and maintenance of latent infection. Second, miRNAs that are expressed from the LAT locus during lytic and latent infection can reduce the expression of certain HSV-1 lytic proteins in cotransfected cells (14). Expression of these miRNAs depends on the LAT promoter (15), so these miRNAs may contribute to the phenotype of a LAT promoter mutant virus.
H3K27me3 is a hallmark of cellular facultative heterochromatin (fHC). fHC is found on different regions of the cellular genome in different cell types and during different developmental stages (16). Because of its differential distribution on developmentally regulated genes, fHC is thought to be able to convert readily to euchromatin to allow gene expression to occur. The cellular proteins involved in the initiation and maintenance of fHC are known as Polycomb group proteins, and they form two complexes, Polycomb repressor complex 1 and 2 (PRC1 and -2) (reviewed in reference 17). PRC2 contains the proteins Ezh2, Suz12, Eed, and RdAp46/48 and is responsible for trimethylation of histone H3. PRC1 binds H3K27me3 and is responsible for maintaining and further compacting fHC. The Bmi1 protein, a component of PRC1, has previously been found on the LAT region of the HSV-1 genome during latent infection and at low levels on certain lytic genes (13).
Following corneal infection and spread of the virus to the trigeminal ganglia (TG) in the mouse model system, there is an initial period of acute replication in the ganglia (18). During the period of ongoing acute infection, a subpopulation of infected neurons does not appear to express lytic genes, and they are thought to enter quiescence and contribute to the pool of latently infected neurons (19, 20). However, recent evidence indicates that prior lytic gene expression in an individual neuron is also compatible with latent infection (20), and certain neurons express both lytic and latent transcripts during the establishment of latent infection (19, 21). Therefore, the pool of neurons latently infected with HSV-1 may originate by two pathways, one in which the genomes become silenced rapidly upon infection of the neuron and one in which lytic genes become silenced following an initial period of lytic gene expression.
Given the potential for two pathways leading to the establishment of latent infection, we were interested in determining the kinetics of total chromatin, H3K27me3, and PRC2 recruitment to the HSV-1 genome during the establishment and maintenance of latent infection. We found that histone H3 is associated with the viral genome at early times (7 days postinfection [dpi]) and that H3K27me3 becomes deposited on viral lytic gene promoters to high levels following the resolution of the acute infection (14 dpi). We also found that recruitment of the Suz12 protein to lytic gene promoters also increased following the resolution of acute infection. Surprisingly, the association of Suz12 with lytic gene promoters was not affected by LAT expression, indicating that during the establishment of latency, the LAT is not required to target PRC2 to lytic gene promoters. Finally, we also investigated whether the PRC1 complex was recruited to viral promoters following the establishment of latent infection. However, we were unable to detect Bmi1, a component of PRC1, on lytic gene promoters during latency.
RESULTS
Kinetics of association of chromatin during the establishment of latent infection with wild-type (WT) HSV-1.
We and others have found that HSV-1 lytic gene promoters are associated with H3K27me3 during latent infection (12, 13). We have also found that H3K27me3 association with the viral lytic gene promoters was decreased following infection with two independent LAT promoter deletion mutant viruses (12). We were interested in determining the mechanism by which the LAT either promotes or maintains the H3K27me3 modification on latent genomes. To this end, we first aimed to establish when H3K27me3 is targeted to the viral genome during the establishment of latent infection.
Previously, we found that H3 association with HSV DNA in TG was apparent by 7 dpi and that the H3K9me2 modification was apparent by 15 dpi (11). Therefore, we looked at this time period for the H3K27me3 modification. We infected mice with HSV-1, harvested the TG at 7, 10, and 14 dpi, and carried out chromatin immunoprecipitation (ChIP) using antibodies specific for histone H3 and H3K27me3. In previous experiments, we noticed that the efficiencies of histone H3 ChIP assays varied depending on the number of input viral genomes (results not shown). To control for this, we first quantified the numbers of copies of viral genomes in the chromatin extracts and used chromatin amounts representing equal numbers of viral genomes. The samples were made up to equivalent amounts of total chromatin by adding chromatin derived from TG of mice that were not infected with HSV.
To validate the ChIP for H3K27me3, we analyzed the Rasgfr1-imprinted locus, for which one copy of the gene is enriched for H3K27me3 (22). The association of H3K27me3 with the GAPDH promoter served as a cellular negative control. There was little immunoprecipitation of either Rasgrf1 or GAPDH DNA with the IgG antibody control (see Fig. S1A in the supplemental material). In contrast, both cellular genes were associated with histone H3 (Fig. S1B). There was an approximate 6-fold increase in the fraction of Rasgrf1 DNA associated with the H3K27me3 modification compared to the fraction of GAPDH DNA (Fig. S1C), demonstrating that the assay was specific for H3K27me3. We also did not detect any significant changes in the levels of H3K27me3 on the cellular controls at the three time points tested.
We then examined the kinetics of association of H3 and H3K27me3 with different regions of the viral genome, including the LAT promoter and 5′ exon, the immediate early ICP4 and ICP0 promoters, the early ICP8 and TK promoters, and the late UL48 promoter. ChIP with the histone H3-specific antibody immunoprecipitated significant amounts of all 5 viral promoters by 7 dpi (Fig. 1B), compared to amounts immunoprecipitated with control antibody (Fig. 1A), indicating that histone H3 associated with all 5 viral promoters by 7 dpi. In contrast, the H3K27me3 modification was not observed until 14 dpi on the viral lytic gene promoters. The ICP8, TK, and UL48 promoters showed the highest levels of H3K27Me3 (Fig. 1C). The differences in the association of H3K27me3 with the ICP8, TK, and UL48 promoters from that of the ICP0 promoter were 6.4-fold, 9.6-fold, and 3.4-fold, respectively.
FIG 1.

Association of histone H3K27me3 with the wild-type (WT) herpes simplex virus 1 (HSV-1) genome during establishment of latent infection. Mice were infected with WT HSV-1, and at 7, 10, or 14 dpi, the mice were sacrificed. Trigeminal ganglia were collected, and chromatin immunoprecipitation (ChIP) was carried out. Shown are percentages of viral DNA immunoprecipitated with IgG control antibody (A), with an antibody against total histone H3 (B), or with an antibody against H3K27me3 (C). Pr, promoter.
Because it appeared that total H3 was present on the viral genomes at a time preceding the H3K27me3 modification, we analyzed the levels of H3 and H3K27me3 compared to their levels with the IgG control at 7 dpi. Statistical analyses carried out on the ChIPs of 7-dpi TG revealed that the association of lytic genes with total histone H3 was increased significantly compared to their association with the IgG control on all the promoters tested (Fig. 2). In contrast, the association of viral promoters with the H3K27me3 modification was not increased significantly compared to that with the IgG control. Therefore, histone H3 association with viral lytic promoters was detectable earlier than H3K27me3 modification (18).
FIG 2.
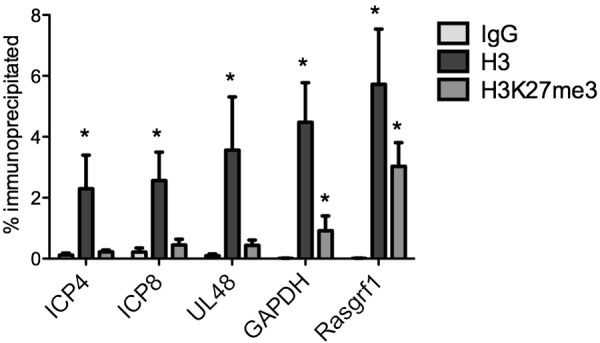
Association of HSV-1 lytic promoters with total chromatin and H3K27me3 at 7 dpi. Mice were infected with WT HSV-1, and at 7 dpi, the mice were sacrificed and trigeminal ganglia were collected. ChIP was carried out on trigeminal ganglia. The percentages of viral lytic gene promoters (ICP4, ICP8, and UL48) and cellular genes (GAPDH and Rasgrf1) immunoprecipitated with nonspecific IgG, total histone H3, and H3K27me3 were determined. Values that were statistically increased compared to the value for the nonspecific control (P ≤ 0.05, one-tailed paired t test) are indicated (*).
The association of H3K27me3 with viral lytic promoters increases between 10 and 14 dpi in the absence of viral DNA replication.
HSV-1 undergoes a period of acute replication starting at 2 dpi following ocular infection of CD-1 mice, and the infection is then cleared (18). Therefore, it was possible that the increase in H3K27me3 levels seen after 10 dpi was due to clearance of viral genomes not associated with H3K27me3 following the resolution of the acute infection and not de novo formation of H3K27me3 on viral genomes. We therefore infected mice with the dlsptk thymidine kinase (TK)-null mutant virus, which does not undergo viral DNA replication in neurons (23, 24). Because viral DNA replication results in a decrease in the fraction of viral DNA associated with histone H3 (25), the use of a viral mutant that does not undergo DNA replication in neurons allowed us to enrich for viral genomes associated with H3K27me3 during an acute infection that might otherwise be masked by ongoing viral DNA replication.
The specificity of the ChIP carried out on dlsptk virus-infected TG was confirmed by analyzing the amount of cellular DNA immunoprecipitated with the H3- and H3K27me3-specific antibodies. Both GAPDH and Rasgrf1 DNAs were enriched following immunoprecipitation with the histone H3 antibody compared to the nonspecific antibody control (see Fig. S2A and S2B in the supplemental material). There was an approximate 12-fold-increased association of Rasgfr1 DNA with the H3K27me3 modification compared to GAPDH DNA (Fig. S2C). Although it appeared that the association of Rasgfr1 with H3K27me3 increased between 10 and 14 dpi, the proportions of histone H3 with the K27me3 modification were similar for all three time points (Fig. S2D). Consistent with our observations for wild-type virus, histone H3 associated with the viral gene promoters by 7 dpi (Fig. 3B), whereas little viral DNA was immunoprecipitated with the IgG control (Fig. 3A). The levels of H3K27me3 on the dlsptk genome (Fig. 3C) appeared to be increased compared to those on the WT viral genome (compare Fig. 1C to 3C) by 7 dpi; however, the ChIP assays on WT- and dlsptk-infected TG (Fig. 1 and 3) were not carried out side by side. To determine if H3K27me3 levels were increased on the dlsptk genome, we infected mice with either WT or dlsptk virus, and the ChIP assays were carried out in parallel. We detected an approximately 2-fold increase in the proportion of H3 with the K27me3 modification following infection with dlsptk virus compared to that following infection with WT virus (results not shown). Therefore, the TK-null mutant virus showed an earlier although still limited association of H3K27me3 with its genome. Furthermore, the association of H3K27me3 with the dlsptk lytic promoters increased between 10 and 14 dpi (Fig. 3C). Because we saw some variation in the total H3 levels on the dlsptk promoters, we also normalized the proportion of histone H3 with the K27me3 modification (Fig. 3D). Following normalization to total H3, a dramatic increase in the K27me3 levels was evident between 10 and 14 dpi especially on the ICP4, ICP0, ICP8, and TK promoters. There was no increase in H3K27me3 levels on the LAT promoter or 5′ exon during the same time period. Thus, even in the absence of viral replication during the acute stage of infection, H3K27me3 accumulated on the viral genome at a time later than the time of clearance of the wild-type virus acute infection, indicating that the increase in H3K27me3 association was not a result of the clearance of virus that had undergone DNA replication.
FIG 3.

Association of H3K27me3 with the HSV-1 genome following infection with a TK-null mutant virus. Mice were infected with HSV-1 dlsptk virus, and at 7, 10, or 14 dpi the mice were sacrificed and trigeminal ganglia were collected. ChIP was carried out on trigeminal ganglia. Shown are the percentages of viral DNA sequences immunoprecipitated with IgG control antibody (A), with an antibody against total histone H3 (B), and with an antibody against H3K27me3 (C). (D) The percentages of viral DNA immunoprecipitated with an antibody against H3K27me3 were normalized to the percentages of DNA precipitated with an antibody against total histone H3.
ICP0 gene transcript levels are higher than those of the ICP8 and UL48 genes at 14 dpi.
Because we observed low levels of H3K27me3 on the ICP4 and ICP0 promoters at 14 days following infection with the wild-type virus, we examined whether there were differences in gene expression that correlated with H3K27me3 levels on lytic promoters. To this end, we carried out real-time–quantitative PCR (RT-qPCR) using primers specific for the ICP0, ICP8, and UL48 gene transcripts. Following infection with WT virus, we observed an approximate 2-fold increase in ICP0 RNA compared to ICP8 RNA (see Fig. S3 in the supplemental material). We also observed higher ICP8 RNA levels than UL48 levels (approximately 3-fold more) (Fig. S3). Therefore, in TG isolated from infected mice, an increase in ICP0 RNA levels compared to ICP8 and UL48 RNA levels correlated with reduced H3K27me3 association with the ICP0 (italicized) promoter. However, the correlation was not complete, because we observed increased levels of H3K27me3 on the ICP8 promoter compared to levels on the UL48 promoter at 14 dpi but higher levels of ICP8 RNA than UL48 RNA.
Suz12 is recruited to the viral genome during the establishment of latency.
Polycomb repressive complex 2 (PRC2) is responsible for the addition of H3K27me3 to cellular chromatin (26). We therefore tested whether we could detect PRC2 on viral promoters during the establishment of latent infection, at the time at which we first detected H3K27me3 on the viral genome. We first examined whether we could detect PRC2 on the WT viral genome at 14 dpi, because this was the earliest time at which the lytic gene promoters were found to be enriched for H3K27me3. To this end, we carried out ChIP using an antibody specific for Suz12, a component of PRC2. We first validated the antibody by analyzing the enrichment of Suz12 at cellular DNA. Following immunoprecipitations with Suz12, there was no enrichment of GAPDH DNA compared to levels with the nonspecific antibody control, whereas there was an approximate 5-fold increase in Rasgrf1 DNA immunoprecipitated with the Suz12 antibody compared to that with the nonspecific antibody control (Fig. S4A). Because the Suz12 ChIP assay detected the association of cellular DNA correlating with H3K27me3, we analyzed the enrichment of Suz12 at viral promoters in the same samples (Fig. 4A). We detected an enrichment of Suz12 on the ICP8, UL48, and LAT gene promoters, compared to its level on the Rasgfr1 sequences. Although detectable over levels with the nonspecific antibody control, the levels of Suz12 on the ICP0 and ICP4 promoters were less than in the cellular positive control. We could not detect an enrichment of Suz12 on the LAT 5′ exon over levels with the nonspecific antibody control.
FIG 4.

Association of the Polycomb group protein Suz12 with the HSV-1 genome. Mice were infected with either WT or dlsptk virus, and at 7, 10, or 14 dpi, the mice were sacrificed and trigeminal ganglia were collected. ChIP was carried out on trigeminal ganglia using an antibody specific for Suz12. (A) The percentages of viral DNA sequences immunoprecipitated from WT infected ganglia with an antibody against Suz12 were normalized to the percentages of Rasgrf1 DNA immunoprecipitated in the same reaction. (B) The percentages of viral DNA sequences immunoprecipitated from dlsptk-infected ganglia with an antibody against Suz12 were normalized to the percentages of Rasgrf1 DNA immunoprecipitated in the same reaction.
Having shown that Suz12 was present on the WT viral genome at 14 dpi, we examined whether the kinetics of Suz12 recruitment mirrored the increase in H3K27me3 levels seen on the dlsptk genomes between 7 and 14 dpi. The levels of Suz12 on the Rasgrf1 region of cellular DNA remained constant between 7 and 14 dpi and were enriched by approximately 10-fold over levels on GAPDH sequences (see Fig. S4B in the supplemental material). We observed an increase in Suz12 levels on all four lytic gene promoters tested between 10 and 14 dpi (ICP0, 6-fold; ICP4, 3-fold; ICP8, 3-fold; UL48, 3.8-fold) (Fig. 4B). Consistently with the H3K27me3 results, the levels of Suz12 were highest on the ICP8 and UL48 gene promoters at all of the times tested.
Suz12 recruitment occurs independently of LAT expression.
The increase in H3K27me3 levels between 10 and 14 dpi argued for a potential trigger of Suz12 recruitment during this time period. Noncoding RNAs have been found to function as cofactors for the recruitment of PRC2 to regions of cellular chromatin (27–33). Using two independent LAT-null viruses, we previously identified a role for the LAT in promoting wild-type levels of H3K27me3 on the lytic gene promoters during latent infection (12).
Proença et al. (20) found that the activity of the LAT promoter increased in ganglia between 5 and 15 dpi. We therefore examined whether levels of LAT intron and exon RNAs increased in our system during the same time frame, in which we observed increases in H3K27me3 levels. We carried out RT-qPCR analysis with RNA extracted from mice infected with WT virus at 7, 10, and 14 dpi (Fig. 5A). Primers specific for the LAT intron and 5′ and 3′ exons were used, and the RNA copy numbers were normalized to the viral DNA copy numbers in the samples extracted from the same mice. As expected, the LAT intron was more abundant than the 5′ and 3′ exons. We observed a slight increase (2.7-fold) in LAT intron expression following infection with WT virus between 10 and 14 dpi (Fig. 5A).
FIG 5.

Role of latency-associated transcript (LAT) in the recruitment of Suz12 during the establishment of latent infection. (A) Kinetics of LAT expression by WT HSV-1. The absolute copy numbers of the LAT 5′ exon, intron, and 3′ exon transcripts were normalized to the copy numbers of viral genomes isolated from the same mouse following infection with wild-type HSV-1. (B) Mice were infected with either a KdlLAT LAT promoter deletion mutant virus or the KFSLAT rescued virus, and at 14 dpi, the mice were sacrificed and trigeminal ganglia were collected. ChIP was carried out on trigeminal ganglia using an antibody specific for Suz12. The percentages of viral DNA sequences immunoprecipitated from KdlLAT- or KFSLAT-infected ganglia with an antibody against Suz12 were normalized to the percentages of Rasgrf1 DNA immunoprecipitated in the same reaction.
We further investigated whether Suz12 association with viral lytic gene promoters was affected by the presence or absence of the LAT. We infected mice with the KdlLAT LAT deletion mutant virus in parallel with the KFSLAT rescued virus. All experiments and ChIP assays for the two viruses were carried out side by side. The enrichment of Suz12 on the cellular Rasgfr1 sequence was similar to that on the GAPDH sequence following infection with both viruses (see Fig. S5B in the supplemental material). The levels of Suz12 on the ICP4, ICP8, TK, and UL48 gene promoters were comparable following infection with either KdlLAT or KFSLAT virus (Fig. 5B). The slight increase in the levels of Suz12 on the ICP0 gene promoter following infection with the LAT deletion virus was not statistically significant (P = 0.137, one-tailed paired t test). Importantly, we did not detect any change in Suz12 recruitment to the lytic gene promoters in the absence of the LAT.
Bmi1 is not detectable on lytic promoters previously found to be associated with H3K27me3 during latent infection.
Because it appeared that the LAT did not play a role in the initial recruitment of the PRC2 complex during the establishment of latent infection, we hypothesized that it might instead play a role in the recruitment of the PRC1 complex for maintenance of the H3K27me3 modification. However, in the course of our experiments investigating Bmi1 recruitment to viral lytic gene promoters following infection with KdlLAT and KFSLAT, we noticed that the levels of occupancy by Bmi1 were much lower than the levels of occupancy on the cellular positive control (results not shown). Therefore, we decided to investigate whether Bmi1 was indeed recruited to WT genomes during the maintenance of latent infection. To this end, we infected mice with WT HSV and prepared chromatin from latently infected TG at least 28 dpi. A 3.2-fold increase in the enrichment of Rasgfr1 DNA compared to GAPDH DNA following immunoprecipitation with the Bmi1 antibody confirmed the specificity of the ChIP assay (see Fig. S6 in the supplemental material). The relative enrichment of viral genes following ChIP with Bmi1-specific antibody was normalized to the Bmi1 occupancy of the negative-control GAPDH gene. Of the regions of the viral genome tested, the only regions enriched at least 2-fold above their level in the cellular negative control were the LAT promoter, LAT intron, and TK promoter (Fig. 6). However, in a number of experiments, these regions were not enriched even compared to levels with the IgG control. Therefore, we carried out a direct comparison with the amount of viral DNA immunoprecipitated with the Bmi1 antibody and the nonspecific antibody control (Table 1). The only region of DNA analyzed that was significantly enriched for Bmi1, compared to IgG, was the cellular positive control, Rasgrf1. Thus, we did not detect significant association of the lytic promoters with Bmi1.
FIG 6.

Association of the Polycomb group protein Bmi1 with the HSV-1 genome. Mice were infected with wild-type HSV-1, and at 28 dpi or later, TG were collected. Chromatin immunoprecipitation was carried out on the trigeminal ganglia using an antibody against Bmi1. The fractions of viral DNA sequences immunoprecipitated with the Bmi1-specific antibody are shown normalized to the fraction of cellular GAPDH DNA. Results from individual experiments are represented.
TABLE 1.
Immunoprecipitation of viral and cellular DNA with anti-Bmi1 and control antibodies
| Primer | Mean fraction immunoprecipitated with: |
Mean fold enrichment relative to IgG control |
P value | |
|---|---|---|---|---|
| IgG | Bmi1 | |||
| LAT promoter | 0.0748 | 0.416 | 5.6 | 0.3125 |
| 5′ exon | 0.0616 | 0.105 | 1.7 | 0.2188 |
| ICP4 | 0.0808 | 0.0620 | 0.77 | 0.9375 |
| ICP0 | 0.138 | 0.135 | 0.98 | 0.3750 |
| ICP8 | 0.122 | 0.0529 | 0.43 | 0.6875 |
| TK | 0.0391 | 0.0806 | 2.1 | 0.1250 |
| UL48 | 0.158 | 0.0551 | 0.35 | 0.2188 |
| Rasgrf1 | 0.0113 | 0.128 | 11 | 0.0156 |
| GAPDH | 0.0244 | 0.0387 | 1.6 | 0.1094 |
DISCUSSION
Lytic gene promoters on the HSV genome are associated with heterochromatin during latent infection of murine sensory ganglia (11–13). We and others observed that one of the major heterochromatin marks associated with HSV latent chromatin is H3K27me3 (12, 13), a form of heterochromatin associated with developmentally regulated genes called facultative heterochromatin (16). The observation in this study that the PRC2 complex is recruited to the viral genome during the establishment of latent infection argues further that the virus utilizes this repressive mark, at least in part, to silence or maintain silencing of its genome during latent infection. The association of lytic promoters with fHC, which is thought to be dynamic and capable of being converted to euchromatin, would be advantageous for reactivation of HSV from latent infection. However, unlike with some forms of cellular facultative heterochromatin, we were unable to detect the PRC1 complex on viral lytic promoters during the maintenance of latent infection.
Little was known about the kinetics of association of various forms of chromatin with the HSV lytic genes beyond our original observations that the HSV-1 genome is associated with histone H3 by 7 dpi and that the heterochromatin marker H3K9me2 is associated with the viral genome by 15 dpi (11). In this study, we confirmed that during establishment of latent infection by HSV-1 in murine trigeminal ganglia, the viral lytic gene promoters are associated with histone H3 by 7 dpi. Histone H3K27me3 modification was detected on WT HSV lytic gene chromatin by 14 dpi, arguing that nucleosomes are deposited prior to K27 methylation. Hence, we hypothesize that there are multiple steps in the silencing and the maintenance of that silencing during latent infection.
Mechanisms of HSV gene silencing following initial infection of neurons.
In this infection model, acute viral replication in the trigeminal ganglia largely declined by 7 dpi (18). Therefore, the bulk of H3K27me3 addition onto viral genomes appears to occur after resolution of the infection. This argues that other earlier events are responsible for the initial silencing of the viral genome. The initial mechanisms of silencing may involve the association of unmodified histones, other heterochromatin markers on HSV DNA, or other undefined mechanisms.
Other factors that have been hypothesized to explain the lack of lytic gene expression in sensory neurons include the following. (i) The transactivator complex is unable to localize in the nucleus of an infected neuron or to stimulate IE gene expression (7). The transactivator complex includes VP16, the cellular HCF-1, Oct1, histone K9 demethylases, and histone K4 methyltransferases (7, 34). In addition to recruiting the transactivator complex, VP16 recruits ATP-dependent chromatin remodeling complexes to IE promoters (35). Hence, if VP16 is unable to recruit cellular factors to IE promoters in neurons, the result could be an increase in total chromatin, increased heterochromatin, and decreased euchromatin association with IE promoters. (ii) Initial silencing may be due to cellular repressive mechanisms. The cellular repressor REST-CoRest complex has been hypothesized to contribute to lytic gene silencing. Inactivation of this complex in infected cells resulted in increased viral replication in vivo by 7 dpi (36).
In this study, we did not detect a role for LATs in the initial recruitment of Suz12. However, silencing of lytic gene expression during the acute stage of infection may involve RNAs expressed from the LAT locus acting in a distinct manner, independently of Suz12 recruitment. Infection with a LAT deletion mutant virus resulted in increased numbers of neurons expressing lytic mRNAs by 3 to 5 dpi (9). Whether this was due to a direct effect of LAT RNAs down-regulating RNA expression, a result of deletion of cis-acting sequences in the LAT region, such as insulators, or an indirect effect due to viral miRNAs down-regulating lytic protein expression is not known. Clearly, more-detailed studies utilizing recombinant viruses lacking specific elements within the LAT region are required.
Thus, it is conceivable that other mechanisms restrict IE gene transcription as the initial acute infection is cleared and that facultative heterochromatin modifications are incorporated onto the histones associated with lytic promoters to maintain gene silencing. Consistently with this, a recent study found that dense chromatin increases H3K27 methylation (37), raising the possibilities that the initial silencing of the viral genome involves association of dense chromatin with the viral genome and, then, that H3K27me3 modification serves to maintain or further silence gene expression. More-detailed studies of the first days of infection could yield more information on these possibilities.
H3K27me3 association with lytic promoters during the establishment of latent infection.
We hypothesize that H3K27 methylation plays a role in the maintenance of latent infection by silencing lytic gene expression. Although a limited number of genes have been surveyed thus far, it is noteworthy that the early ICP8 and TK genes and the late UL48 gene had higher levels of H3K27me3 than the IE ICP0 and ICP4 genes. We also detected higher levels of ICP0 RNA at 14 dpi than of UL48 and ICP8 RNAs. Thus, this heterochromatic mark may have a greater silencing effect on E and L genes than on IE genes, or H3K27me3 levels may be higher on E and L genes because they undergo lower levels of transcription. Noteworthy was the level of expression of ICP0 gene transcripts, which was higher than those of ICP8 and UL48 RNAs. One model of RNA interference (RNAi)-mediated transcriptional silencing involves complementary transcripts (38); therefore, it is conceivable that the LAT and ICP0 gene transcripts form complementary RNAs that silence the rest of the genome. The presence of ICP0 mRNA does not necessarily indicate the presence of ICP0 protein, especially since an HSV-1 miRNA, miR-H2, which is expressed during both acute and latent infection of the TG (15), targets ICP0 mRNA and prevents the accumulation of ICP0 protein levels but not mRNA when tested in transfected cells (39).
The LAT and the mechanism of H3K27me3 modification of viral chromatin.
Previous studies in our system have shown that viruses that lack LAT expression have reduced levels of the H3K27me3 modification on histones associated with HSV lytic gene promoters (12), although one other study comparing a mutant HSV-1 strain 17 with a wild-type strain 17 virus observed that this mutant showed increased H3K27me3 associated with viral promoters (13). In the current study, we observed that recruitment of Suz12, a component of PRC2 that serves as the methyltransferase complex, was not increased by LAT expression; instead, Suz12 was associated with the viral lytic gene promoters at equal levels in the presence and absence of LAT transcription. This argues that the LAT may promote the accumulation of H3K27me3 by a mechanism(s) other than recruitment of Suz12 to the viral lytic promoters. Most models for long noncoding RNA silencing show the PRC2 complex of Ezh2, Suz12, and Eed being recruited to chromatin as a complex involving the RNA (38). Based on our results, it is conceivable that a complex that does not involve Suz12 may be regulated by LAT or that components of the complex are recruited onto chromatin separately and that LAT promotes the association of a component other than Suz12. Alternatively, the LAT may regulate another step and shift the equilibrium by stimulating the activity of the PRC2 complex or inhibiting a demethylase enzyme.
Role of PRC1 in HSV lytic gene silencing.
In this study, we were unable to detect significant enrichment of HSV lytic promoters with a component of the PRC1 complex, Bmi1. At first glance, this seems contradictory to the findings of a previous study (13). However, our results are similar to the results obtained in the study by Kwiatkowski et al. (13), which also found that the enrichment of lytic promoters following ChIP for Bmi1 was less than 2-fold more than that of the cellular negative control (APRT). The two different conclusions reached in our study and the previous study highlight the challenges in using ChIP assays to determine the presence or absence of a protein on regions of DNA. It is possible that the PRC1 complex is present on the viral genome during latency at other sites not analyzed in this study. Global analysis of ChIP assays using antibodies to PRC1 proteins would be required to determine if PRC1 binds specific regions of the HSV genome during latent infection. It is also possible that Bmi1 is recruited at a time point later than 28 dpi.
Our results raise the possibility that the form of facultative heterochromatin on the viral lytic promoters is not bound by PRC1. A study by Ku et al. found that a proportion of bivalent genes enriched for H3K27me3 and H3K4me3 in embryonic stem cells were not enriched for PRC1 (40). Hence, H3K27me3 is not always bound by PRC1. The presence of the H3K27me3 modification in the absence of PRC1 binding is still predicted to maintain gene silencing because methylation of the lysine 27 residue of histone H3 has been found to prevent its acetylation (41). Lytic gene promoters that are associated with H3K27me3 in the absence of Bmi1 may also be more readily converted into euchromatin to allow reactivation to occur. Clearly, additional functional studies are required to investigate the contribution of both the PRC2 and PRC1 complexes to the initiation and maintenance of latent infection.
MATERIALS AND METHODS
Cells and viruses.
Vero cells were maintained as described previously (25). The WT strain of HSV-1 (KOS) used in this study was grown and titrated as described previously (9). The HSV-1 KdlLAT LAT promoter deletion mutant virus, the KFSLAT repaired virus, and their propagation and titration have been described elsewhere (9). The HSV-1 KOS dlsptk TK-null mutant virus has been described previously (23).
Mouse infections.
Six-week-old male CD-1 mice (Charles River Laboratories) were anesthetized by intraperitoneal injection of ketamine hydrochloride (3.4 mg) and xylazine hydrochloride (0.5 mg). For the majority of experiments, the scarified corneas of mice were inoculated with 2 × 106 PFU/eye of virus (in a 5-µl volume), as described previously (42). For dlsptk infections, mice were inoculated with 5 × 106 PFU of virus/eye. Mice were housed in accordance with institutional and National Institutes of Health guidelines on the care and use of animals in research, and all procedures were approved by the Institutional Animal Care and Use Committee of Harvard Medical School.
Chromatin immunoprecipitations.
The following antibodies were used for chromatin immunoprecipitations (ChIP): anti-H3K27me3 (Millipore; catalog number 07-449), anti-histone H3 (Abcam; catalog number AB1791), anti-Bmi1 (Abcam; catalog number AB14389), anti-Suz12 (AB12093), and normal rabbit IgG (Millipore; catalog number 12-370).
See materials and methods in the supplemental material for detailed descriptions of chromatin immunoprecipitation and quantification of viral gene expression.
SUPPLEMENTAL MATERIAL
Supplemental materials and methods. Download
Cellular controls for ChIP on TG isolated from WT-virus-infected mice. Mice were infected with WT HSV-1, and at 7, 10, and 14 dpi, mice were sacrificed and the TG isolated. ChIP was carried out as described in the materials and methods. Shown are the percentages of cellular DNA immunoprecipitated with IgG control antibody (A), with a histone H3 antibody (B), or with an H3K27me3 antibody (C). Download
Cellular controls for ChIP on TG isolated from dlsptk-infected mice. Mice were infected with a dlsptk virus and at 7, 10, and 14 dpi, mice were sacrificed and the TG isolated. ChIP was carried out as described in the materials and methods. Shown are the percentages of cellular DNA immunoprecipitated with IgG control antibody (A), with a histone H3 antibody (B), and with an H3K27me3 antibody (C). (D) The fraction of cellular DNA immunoprecipitated with the H3K27me3 antibody was normalized to the fraction immunoprecipitated with the total H3 antibody. Download
Viral lytic gene expression at 14 days following infection with WT virus. Mice were infected with WT HSV-1, and at 14 dpi, the mice were sacrificed and trigeminal ganglia were collected. The absolute copy numbers of lytic gene transcripts for ICP0, ICP4, and UL48 are shown. Download
Cellular controls for the Polycomb group protein Suz12. Mice were infected with either WT or dlsptk virus, and at 7, 10, or 14 dpi, the mice were sacrificed and trigeminal ganglia were collected. ChIP was carried out on trigeminal ganglia using an antibody specific for Suz12. Shown are the percentages of cellular GAPDH and Rasgrf1 DNA sequences immunoprecipitated from WT-virus-infected ganglia with either rabbit IgG or Suz12-specific antibody (A) and from dlsptk virus-infected ganglia (B) at 7, 10, and 14 dpi. Download
Cellular controls for the Suz12 ChIP carried out on TG isolated from mice infected with KdlLAT or KFSLAT. Download
Cellular controls for the Bmi1 ChIP carried out on TG isolated from mice infected with WT HSV-1. Download
ACKNOWLEDGMENTS
This research was supported by grants NS35133 and AI099081.
Footnotes
Citation Cliffe AR, Coen DM, Knipe DM. 2013. Kinetics of facultative heterochromatin and Polycomb group protein association with the herpes simplex viral genome during establishment of latent infection. mBio 4(1):00590-12. doi:10.1128/mBio.00590-12.
REFERENCES
- 1. Rock DL, Fraser NW. 1983. Detection of HSV-1 genome in central nervous system of latently infected mice. Nature 302:523–525 [DOI] [PubMed] [Google Scholar]
- 2. Deshmane SL, Fraser NW. 1989. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J. Virol. 63:943–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. 1987. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 235:1056–1059 [DOI] [PubMed] [Google Scholar]
- 4. Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses. In Knipe DM, Howley PM, Fields virology, 6th ed, in press Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 5. Sears AE, Hukkanen V, Labow MA, Levine AJ, Roizman B. 1991. Expression of the herpes simplex virus 1 alpha transinducing factor (VP16) does not induce reactivation of latent virus or prevent the establishment of latency in mice. J. Virol. 65:2929–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kristie TM, Vogel JL, Sears AE. 1999. Nuclear localization of the C1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. Proc. Natl. Acad. Sci. U. S. A. 96:1229–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Knipe DM, Lieberman PM, Jung JU, McBride AA, Morris KV, Ott M, Margolis D, Nieto A, Nevels MJPR, Kristie TM. 2013. Snapshots: chromatin control of viral infection. Virology 435:141–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Du T, Zhou G, Roizman B. 2011. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc. Natl. Acad. Sci. U. S. A. 108:18820–18824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garber DA, Schaffer PA, Knipe DM. 1997. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J. Virol. 71:5885–5893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen SH, Kramer MF, Schaffer PA, Coen DM. 1997. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 71:5878–5884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang OY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. 2005. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. U. S. A. 102:16055–16059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol. 83:8182–8190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kwiatkowski DL, Thompson HW, Bloom DC. 2009. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J. Virol. 83:8173–8181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454:780–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kramer MF, Jurak I, Pesola JM, Boissel S, Knipe DM, Coen DM. 2011. Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology 417:239–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trojer P, Reinberg D. 2007. Facultative heterochromatin: is there a distinctive molecular signature? Mol. Cell 28:1–13 [DOI] [PubMed] [Google Scholar]
- 17. Simon JA, Kingston RE. 2009. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat. Rev. Mol. Cell Biol. 10:697–708 [DOI] [PubMed] [Google Scholar]
- 18. Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA. 1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J. Virol. 63:2893–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Speck PG, Simmons A. 1991. Divergent molecular pathways of productive and latent infection with a virulent strain of herpes simplex virus type 1. J. Virol. 65:4001–4005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Proença JT, Coleman HM, Connor V, Winton DJ, Efstathiou S. 2008. A historical analysis of herpes simplex virus promoter activation in vivo reveals distinct populations of latently infected neurones. J. Gen. Virol. 89:2965–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Margolis TP, Sedarati F, Dobson AT, Feldman LT, Stevens JG. 1992. Pathways of viral gene expression during acute neuronal infection with HSV-1. Virology 189:150–160 [DOI] [PubMed] [Google Scholar]
- 22. Lindroth AM, Park YJ, McLean CM, Dokshin GA, Persson JM, Herman H, Pasini D, Miró X, Donohoe ME, Lee JT, Helin K, Soloway PD. 2008. Antagonism between DNA and H3K27 methylation at the imprinted Rasgrf1 locus. PLoS Genet. 4:e1000145 http://dx.doi.org/10.1371/journal.pgen.1000145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Coen DM, Kosz-Vnenchak M, Jacobson JG, Leib DA, Bogard CL, Schaffer PA, Tyler KL, Knipe DM. 1989. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc. Natl. Acad. Sci. U. S. A. 86:4736–4740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kramer MF, Chen SH, Knipe DM, Coen DM. 1998. Accumulation of viral transcripts and DNA during establishment of latency by herpes simplex virus. J. Virol. 72:1177–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cliffe AR, Knipe DM. 2008. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 82:12030–12038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298:1039–1043 [DOI] [PubMed] [Google Scholar]
- 27. Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. 2008. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322:750–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guil S, Soler M, Portela A, Carrère J, Fonalleras E, Gómez A, Villanueva A, Esteller M. 2012. Intronic RNAs mediate EZH2 regulation of epigenetic targets. Nat. Struct. Mol. Biol. 19:664–670 [DOI] [PubMed] [Google Scholar]
- 29. Ng SY, Johnson R, Stanton LW. 2012. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 31:522–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT. 2010. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 40:939–953 doi: 10.1016/j.molcel.2010.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. 2010. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329:689–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL. 2009. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U. S. A. 106:11667–11672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY. 2007. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129:1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. 2009. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med. 15:1312–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Herrera FJ, Triezenberg SJ. 2004. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J. Virol. 78:9689–9696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Du T, Zhou G, Khan S, Gu H, Roizman B. 2010. Disruption of HDAC/CoREST/REST repressor by dnREST reduces genome silencing and increases virulence of herpes simplex virus. Proc. Natl. Acad. Sci. U. S. A. 107:15904–15909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yuan W, Wu T, Fu H, Dai C, Wu H, Liu N, Li X, Xu M, Zhang Z, Niu T, Han Z, Chai J, Zhou XJ, Gao S, Zhu B. 2012. Dense chromatin activates polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science 337:971–975 [DOI] [PubMed] [Google Scholar]
- 38. Morris KV. 2011. The emerging role of RNA in the regulation of gene transcription in human cells. Semin. Cell Dev. Biol. 22:3512–3518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Umbach JL, Nagel MA, Cohrs RJ, Gilden DH, Cullen BR. 2009. Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J. Virol. 83:10677–10683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, Adli M, Kasif S, Ptaszek LM, Cowan CA, Lander ES, Koseki H, Bernstein BE. 2008. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 4:e1000242 http:/dx.doi.org/.10.1371/journal.pgen.1000242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pasini D, Malatesta M, Jung HR, Walfridsson J, Willer A, Olsson L, Skotte J, Wutz A, Porse B, Jensen ON, Helin K. 2010. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 38:4958–4969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tenser RB, Dunstan ME. 1979. Herpes simplex virus thymidine kinase expression in infection of the trigeminal ganglion. Virology 99:417–422 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental materials and methods. Download
Cellular controls for ChIP on TG isolated from WT-virus-infected mice. Mice were infected with WT HSV-1, and at 7, 10, and 14 dpi, mice were sacrificed and the TG isolated. ChIP was carried out as described in the materials and methods. Shown are the percentages of cellular DNA immunoprecipitated with IgG control antibody (A), with a histone H3 antibody (B), or with an H3K27me3 antibody (C). Download
Cellular controls for ChIP on TG isolated from dlsptk-infected mice. Mice were infected with a dlsptk virus and at 7, 10, and 14 dpi, mice were sacrificed and the TG isolated. ChIP was carried out as described in the materials and methods. Shown are the percentages of cellular DNA immunoprecipitated with IgG control antibody (A), with a histone H3 antibody (B), and with an H3K27me3 antibody (C). (D) The fraction of cellular DNA immunoprecipitated with the H3K27me3 antibody was normalized to the fraction immunoprecipitated with the total H3 antibody. Download
Viral lytic gene expression at 14 days following infection with WT virus. Mice were infected with WT HSV-1, and at 14 dpi, the mice were sacrificed and trigeminal ganglia were collected. The absolute copy numbers of lytic gene transcripts for ICP0, ICP4, and UL48 are shown. Download
Cellular controls for the Polycomb group protein Suz12. Mice were infected with either WT or dlsptk virus, and at 7, 10, or 14 dpi, the mice were sacrificed and trigeminal ganglia were collected. ChIP was carried out on trigeminal ganglia using an antibody specific for Suz12. Shown are the percentages of cellular GAPDH and Rasgrf1 DNA sequences immunoprecipitated from WT-virus-infected ganglia with either rabbit IgG or Suz12-specific antibody (A) and from dlsptk virus-infected ganglia (B) at 7, 10, and 14 dpi. Download
Cellular controls for the Suz12 ChIP carried out on TG isolated from mice infected with KdlLAT or KFSLAT. Download
Cellular controls for the Bmi1 ChIP carried out on TG isolated from mice infected with WT HSV-1. Download