

ABSTRACT
Clostridium perfringens enterotoxin (CPE) contributes to several important human gastrointestinal (GI) diseases. This toxin and its derivatives are also being explored for translational applications, i.e., cancer therapy or drug delivery. Some, but not all, members of the 24-member claudin (Cldn) family of mammalian tight junction proteins can serve as CPE receptors. Among the human Cldns (hCldns), hCldn-3 and -4 are known to convey CPE sensitivity when expressed by fibroblast transfectants. However, other Cldns are also reportedly expressed in the intestines, where they might contribute to natural CPE-mediated GI disease, and in other organs, where they might react with CPE-based therapeutics. Therefore, the current study assessed whether two additional hCldns beside hCldn-3 and -4 are also functional CPE receptors. Using Cldn-expressing transfectants, hCldn-8 and -14 were shown to convey CPE-mediated cytotoxicity at pathophysiologically relevant concentrations of this toxin, although ~2-to-10-fold less efficiently than hCldn-4. Site-directed mutagenesis then demonstrated that the N146 residue in hCldn-14 and the S151 residue in hCldn-8 are largely responsible for modulating the weaker CPE binding properties of hCldn-8 and -14 versus hCldn-4, which broadens understanding of Cldn:CPE binding interactions. Since Cldn-8 and -14 are reportedly expressed in mammalian intestines, the current results support the possibility that these two hCldns contribute to natural CPE-mediated gastrointestinal disease and could be CPE-based therapeutic targets for cancers overexpressing those claudins. However, these results also suggest caution during therapeutic use of CPE, which might trigger toxic side effects in normal human tissues producing hCldn-8 or -14, as well as in those producing hCldn-3 or -4.
IMPORTANCE Clostridium perfringens enterotoxin (CPE) is responsible for the gastrointestinal symptoms of the second-most-common bacterial food-borne illness and is also being explored for use as a cancer therapeutic or for increasing drug delivery. Until now, the only known human CPE receptors were claudin-3 and -4. This work shows that human claudin-8 and -14 can also bind CPE and convey cytotoxicity, although slightly less efficiently than claudin-3 and -4. The claudin-8 and -14 residues responsible for this weaker CPE binding were identified, shedding new light on CPE:claudin interactions.
IMPORTANCE
Clostridium perfringens enterotoxin (CPE) is responsible for the gastrointestinal symptoms of the second-most-common bacterial food-borne illness and is also being explored for use as a cancer therapeutic or for increasing drug delivery. Until now, the only known human CPE receptors were claudin-3 and -4. This work shows that human claudin-8 and -14 can also bind CPE and convey cytotoxicity, although slightly less efficiently than claudin-3 and -4. The claudin-8 and -14 residues responsible for this weaker CPE binding were identified, shedding new light on CPE:claudin interactions.
Introduction
Clostridium perfringens enterotoxin (CPE) causes the gastrointestinal (GI) symptoms of C. perfringens type A food poisoning (1, 2), which currently ranks as the second-most-common bacterial food-borne illness in the United States, where one million cases occur annually (3). This toxin is also involved in many cases of antibiotic-associated diarrhea (4, 5) and several other human and veterinary GI diseases (4–6). In addition, there is current translational interest in using CPE, or binding-capable derivatives of this toxin, for (i) chemotherapy against tumors overexpressing CPE receptors, which thus exhibit exquisite sensitivity to the toxin (7–9), and (ii) increasing drug delivery (10–16).
CPE, a 319-amino-acid single polypeptide (17), belongs structurally to the aerolysin toxin family (18, 19). The enterotoxin has a unique action that begins with its binding to protein receptors described below. This binding results in formation of a small (~90-kDa) CPE-containing complex in host cell plasma membranes (20, 21) that is requisite, but insufficient, for cytotoxicity (21, 22). The toxin in the small complex then rapidly oligomerizes to form an ~450-kDa complex named CPE hexamer-1 (CH-1), which migrates anomalously on sodium dodecyl sulfate (SDS)-PAGE with an apparent size of ~155 kDa (23, 24). CH-1 formation initially occurs on the enterocyte membrane surface, but this prepore then quickly inserts into the plasma membrane to form an active pore (20, 25, 26). Plasma membrane permeability alterations (27–30) caused by the CPE pore include a Ca+2 influx that triggers apoptotic or oncotic cell death pathways (31, 32). Morphological damage then develops in these CPE-treated cells, which allows formation of a second, ~600-kDa CPE complex named CH-2 that migrates on SDS-PAGE with an apparent size of ~200 kDa (23, 24, 33). CH-2 formation apparently induces internalization of occludin into host cells (24). During CPE-associated intestinal disease, the cytotoxic effects of this enterotoxin lead to histopathologic damage, which is thought to trigger the development of diarrhea and abdominal cramps (34, 35).
Tight junctions, the most apically located junctions in epithelial and endothelial cells, help to regulate paracellular permeability and mediate cell-to-cell adhesion (36, 37). Among the most important structural and functional components of tight junctions are claudins (Cldns), which are a 24 member family of ~20-to-25-kDa proteins (38, 39). Cldn-1 to -10, -14, -15, -17, and -19 belong to the classical claudin family due to their high sequence similarity (40). The classical Cldns are thought to consist of a short cytoplasmic N-terminal domain, four transmembrane domains, two extracellular loops (ECLs), and a cytoplasmic C-terminal tail (38, 39). Posttranslational glycosylation of Cldns has not yet been demonstrated, to our knowledge.
Substantial evidence now implicates certain Cldns as functional receptors mediating CPE binding to host cells. For example, while mouse fibroblasts naturally do not produce claudins or respond to this toxin (41–44), these cells bind and become sensitive to CPE when transfected to express Cldn-3 or -4 from several mammalian species, including humans (42–47). The Ka for CPE binding to human Cldn-3 (hCldn-3) or -4 is reportedly on the order of 107 M−1 (43). Further supporting the importance of those two hCldns as CPE receptors, coimmunoprecipitation studies demonstrated that the enterotoxin physically interacts with hCldn-3 and -4 upon binding to naturally CPE-sensitive, human enterocyte-like Caco-2 cells (23). Similarly, fibroblast transfectants expressing Cldn-6, -7, -8, or -14 (from unspecified species) were also shown to acquire CPE binding ability, with a Ka for CPE binding that is approximately an order of magnitude lower than that of hCldn-4 (45). Importantly, the CPE sensitivity of transfectants expressing hCldn-6, -7, -8, or -14 has not yet been reported, to our knowledge. However, it is also clear that not all Cldns bind CPE, since fibroblast transfectants expressing hCldn-1, -2, -5, or -10 bind little or no CPE (44, 48).
Using transfectants expressing Cldn-1 or Cldn-3 chimeras, it was determined that amino acids present in the C-terminal half of Cldn-3 or -4 confer CPE binding ability and sensitivity upon fibroblast transfectants (45). Follow-up studies using a similar Cldn chimera-expressing transfectant approach then specifically mapped CPE binding activity to the proposed ECL-2 region of hCldn-4 receptors (44). Furthermore, by using various approaches, several laboratories determined that an Asn residue located in the middle of ECL-2 (e.g., N149 of hCldn-4) in mouse Cldn-3 and human or mouse Cldn-4 is critical for CPE binding (44, 47, 48).
Binding studies using Cldn-expressing fibroblast transfectants also revealed that, even among the CPE binding Cldns possessing the N149 residue or its equivalent, considerable variations exist in their binding affinity for this toxin. For example, both Cldn-8 and -14 (of unspecified species origin) have the N149 residue or its equivalent and yet exhibit a Ka for CPE binding that is about an order of magnitude lower than that of hCldn-3 or -4 (43, 45). Of potential pathophysiologic relevance, expression of both Cldn-8 and -14 has been detected in the mammalian intestines and other organs, i.e., Cldn-8 is expressed in kidney, prostate, and both the small and large intestine, while there are reports of Cldn-14 expression in the liver, inner ear, kidney, and large intestine (49–51). The reported presence of Cldn-8 and -14 in the intestines raises a question: might other hCldns beside hCldn-3 and -4 contribute as receptors during CPE-mediated GI disease? To begin addressing this issue, the current study assessed whether hCldn-8 and -14 bind CPE and, if so, whether they can convey cytotoxicity at pathophysiologically relevant CPE concentrations.
RESULTS
Soluble recombinant hCldn-14 or -8 exhibits weaker CPE binding properties than recombinant hCldn-4.
The current study first compared the CPE binding properties of hCldn-8 and -14 versus hCldn-4, as this issue has apparently not yet been directly addressed in the literature. For this purpose, a fixed amount (1 µg) of CPE was preincubated at 37°C for 20 min with various concentrations of a soluble recombinant hCldn (rhCldn) before that preincubation mixture was added to CPE-sensitive Caco-2 cells and the development of CPE-induced cytotoxicity was monitored. If, for example, hCldn-8 or -14 binds CPE less strongly than hCldn-4, it should be necessary to preincubate CPE with more rhCldn-8 or -14 versus rhCldn-4 to obtain equivalent protection levels for Caco-2 cells in this assay.
Control experiments first confirmed previous reports (44) that Caco-2 cells develop extensive (~70%) cytotoxicity when challenged with 1 µg/ml of CPE that had been preincubated in the absence of any rhCldn. That CPE-induced cytotoxicity was inhibited, in a dose-dependent manner (Fig. 1A), when Caco-2 cells were challenged with CPE that had been preincubated with soluble rhCldn-4. This protective effect of rhCldn-4 was specific, since preincubating the same amount of CPE with up to a 280-fold molar excess of soluble rhCldn-1, which binds CPE very poorly or not at all (44), did not prevent the development of CPE-induced cytotoxicity (Fig. 1A).
FIG 1 .

Cytotoxic effects of treating Caco-2 cells with CPE (Clostridium perfringens enterotoxin) preincubated with soluble rhCldns. (A) Cytotoxicity detected in Caco-2 cell cultures after treatment for 60 min at 37°C with the indicated preincubation mix containing 1 µg of CPE/ml and specified molar excesses of each soluble rhCldn. Cytotoxicity was determined using the Roche LDH assay. % of maximal CPE cytotoxicity = percent CPE-induced cytotoxicity in the presence of the specified rhCldn/percent CPE-induced cytotoxicity in the absence of any rhCldn. Results shown represent the means of three experimental repetitions. Error bars represent standard deviations (SD). (B) Western immunoblot analysis of Caco-2 cells after treatment for 60 min at 37°C with mixes containing 1 µg of CPE/ml that had been preincubated with a 70- to 280-fold molar excess of soluble rhCldn-1, -4, -8, or -14. After collection of cell lysates, Western immunoblotting was performed with the appropriate Cldn antibody. Results shown are representative of three experimental repetitions.
Preincubating CPE with soluble recombinant forms of hCldn-8 or -14 also interfered, in a dose-dependent manner, with the development of CPE-induced cytotoxicity. However, consistent with slightly weaker CPE binding by hCldn-8 or -14 versus hCldn-4, ~3-fold more hCldn-8 or -14 than rhCldn-4 was needed during the preincubation to obtain equivalent protection against CPE-induced cytotoxicity (Fig. 1A).
To confirm that the protective effects of rhCldn-4, -8, and -14 shown in Fig. 1A were due to interference with toxin binding to Caco-2 cells, and hence downstream inhibition of CPE large-complex formation, CPE Western blotting was performed using Caco-2 cells that had been challenged with preincubation mixtures containing CPE and various concentrations of each soluble rhCldn. This immunoblotting analysis demonstrated that preincubating CPE with a 70-fold molar excess of rhCldn-4 sharply reduced subsequent CPE binding to, and large-complex formation in, Caco-2 cells (Fig. 1B). In contrast, a similar preincubation with up to even a 280-fold molar excess of rhCldn-1 did not affect the amount of CPE binding or CH-1/CH-2 complex formation in this assay (Fig. 1B). Consistent with the cytotoxicity results shown in Fig. 1A, preincubating CPE with increasing concentrations of soluble rhCldn-8 or -14 reduced CPE binding to, and large-complex formation in, Caco-2 cells; however, this inhibition was less efficient than that observed using rhCldn-4 (Fig. 1B).
CPE effects on stable transfectants expressing hCldn-8 or -14.
Having demonstrated during the studies represented in Fig. 1 that rhCldn-8 and -14 both exhibit slightly weaker CPE binding properties than rhCln-4, the current study next investigated whether hCldn-8 and -14 can still convey CPE action on host cells. To address this question, fibroblast transfectants were prepared that stably express hCldn-8 or -14.
Western blotting studies first confirmed that the parental fibroblasts do not naturally express any of the studied claudins (Fig. 2), consistent with previous results (42–45). Western blotting also confirmed (Fig. 2) the identity of previously constructed (44) stable transfectants expressing either (i) hCldn-4, as these cells would be used as a positive control for an hCldn-expressing transfectant known to exhibit strong CPE sensitivity (44), or (ii) hCldn-1, as these cells would be used as a negative control for an hCldn-expressing transfectant that was shown previously to be insensitive to even relatively high (12 µg/ml) CPE doses (44).
FIG 2 .

Expression of specified hCldn or hCldn variants by mouse fibroblast transfectants. Stable transfectants were lysed and subjected to Western blot analysis for Cldn expression as described in Materials and Methods. Note that Cldn antibodies are directed against the cytoplasmic C-terminal tail, not the ECL-2 region. The table in the figure shows rhCldn expression levels as determined by Western blotting during three experimental repetitions.
Similar Western blot analysis then demonstrated production of the expected hCldn by the newly constructed stable transfectants expressing hCldn-8 or -14 (Fig. 2). When quantitative Western blotting of rhCldn production was performed using lysates of each transfectant run against a standard curve of the corresponding purified rhCldn, these transfectants were each shown to produce approximately similar amounts of their respective rhCldns (Fig. 2).
Control cytotoxicity studies (Fig. 3A) then confirmed previous reports (44) that the viability of parental mouse fibroblasts is unaffected by CPE treatment, even at high toxin doses. Also consistent with previous studies (44), fibroblast transfectants stably producing hCldn-4 exhibited a strong cytotoxic response within 30 min of treatment with 1 µg/ml of CPE (Fig. 3A), while no cytotoxicity was observed when fibroblast transfectants stably expressing rhCldn-1 were treated with high CPE doses (Fig. 3A).
FIG 3 .
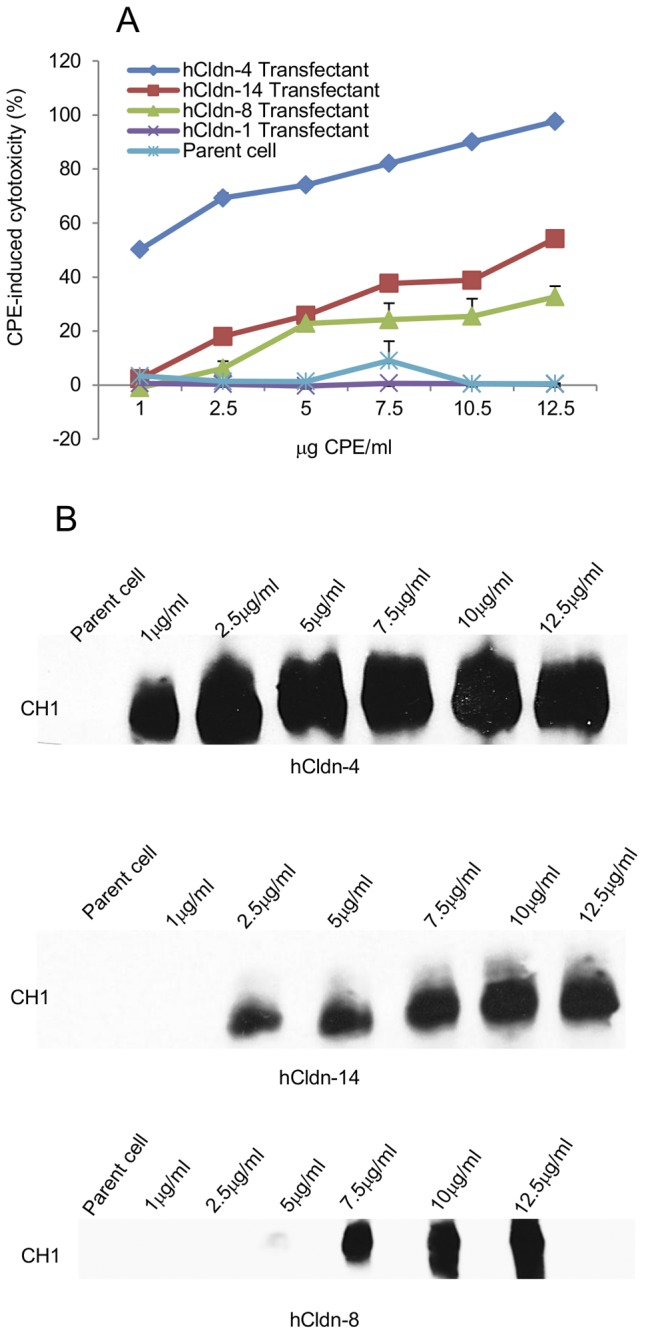
Cytotoxic effects of CPE treatment on mouse fibroblast transfectants expressing specified hCldns. (A) Cytotoxicity developing in mouse fibroblast transfectants expressing the specified hCldn after a 60-min treatment at 37°C with 1 to 12.5 µg of CPE/ml. CPE-induced cell cytotoxicity was expressed as the percentage of dead cells caused specifically by CPE treatment, i.e., after subtraction of any background nonviable cells. Results shown represent the means of three experimental repetitions. Error bars represent the SD. (B) CPE Western blot analysis of transfected fibroblasts expressing hCldn-4, -8, or -14 after a 60-min treatment at 37°C with 1 to 12.5 µg of CPE/ml. CPE binding to transfectant expressing hCldn-1 was not detected (data not shown). CPE immunoreactivity was detected by Western blot analysis using rabbit polyclonal anti-CPE serum. Results shown are representative of three experimental repetitions.
When the newly prepared fibroblast transfectants stably expressing rhCldn-14 or -8 were similarly CPE challenged, those cells showed sensitivity to CPE (Fig. 3A). However, relative to that seen with the hCldn-4 transfectants, CPE-induced cytotoxicity was reduced using transfectants producing hCldn-14 or -8. Western immunoblotting detected CPE binding and CH-1 formation after CPE treatment of the transfectants stably expressing hCldn-4, -8, or -14, although this detection required use of higher CPE doses for assaying the hCldn-8- or -14-producing transfectants (Fig. 3B).
C-terminal sequence comparisons of hCldn-1, -4, -8, and -14.
The experiments represented in Fig. 1, 2, and 3 showed that rhCldn-8 and -14 bind CPE less strongly than rhCldn-4 and yet still convey CPE-induced cytotoxicity at pathophysiologically relevant toxin concentrations (1). Therefore, a C-terminal sequence alignment was performed for the hCldns used in this study. As reported previously (44), this sequence comparison indicated (Fig. 4) that each of the three CPE binding hCldns (i.e., hCldn-4, -8, and -14) used in this study possesses an ECL-2 Asn residue equivalent to the N149 of hCldn-4. However, while hCldn-4 has a P150 residue adjacent to its N149 (44, 47), hCldn-8 instead has an S151 adjacent to its equivalent N150 residue. Similarly, while hCldn-4 has a D146 located 3 residues from the ECL-2 N149 that is critical for CPE binding, hCldn-14 instead contains an N146 at this ECL-2 residue (Fig. 4).
FIG 4 .
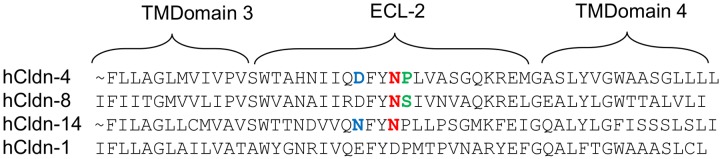
Comparative sequence alignment of the C-terminal half of hCldn-1, -4, -8, and -14. Sequences shown were obtained from GenBank. The ECL-2 N residue (N149 of hCldn-4, or the N149 or N150 equivalents in hCldn-14 or -8, respectively) important for CPE binding (48) is highlighted in red for hCldn-4, -8, and -14. Highlighted in blue are the N146 of hCld-14 and the corresponding D146 of hCldn-4. Highlighted in green are the S151 of hCldn-8 and the corresponding P150 of hCldn-4.
These sequence comparisons suggested that (i) the P150 and D146 residues in the ECL-2 of hCldn-4 may be important for strong CPE binding and that (ii) the S151 and N146 variations at those ECL-2 residues could help to explain why the experiments represented in Fig. 1, 2, and 3 detected weaker CPE binding properties, respectively, for hCldn-8 and -14 relative to hCldn-4.
Experimental identification of ECL-2 residues modulating the weaker CPE binding of soluble rhCldn-14 and -8.
Soluble rhCldn variants were prepared to begin testing our hypothesis that the weaker binding of rhCldn-8 and -14 versus rhCldn-4, as detected in experiments shown in Fig. 1, 2, and 3, involved (respectively) the S151 residue in ECL-2 of hCldn-8 and the N146 ECL-2 residue in hCldn-14. For this purpose, ECL-2 residue D146 in hCldn-4 was changed by site-directed mutagenesis to N, as found in the ECL-2 of hCldn-14 (creating rhCldn-4D146N), while ECL-2 residue N146 of hCldn-14 was mutated to D (creating rhCldn-14N146D), as found in the hCldn-4 ECL-2. Similarly, the P150 present in the ECL-2 of hCldn-4 was changed to S (creating rhCldn-4P150S), while the corresponding S151 in the ECL-2 of hCldn-8 was mutated to the P found in the hCldn-4 ECL-2 (creating rhCldn-8S151P). All mutations were confirmed by DNA sequencing (results not shown).
Before constructing stable transfectants expressing each of these rhCldn variants, the CPE binding properties of a soluble version of each variant were first assessed with the preincubation challenge assay used for the experiments represented in Fig. 1. For this purpose, each recombinant site-directed rhCldn variant, and the corresponding soluble rhCldn with a wild-type sequence, was expressed in Escherichia coli and then affinity purified. Using those samples, preincubating CPE with a 70-fold molar excess of rhCldn-4 was sufficient to cause an ~70% inhibition of CPE-induced cytotoxicity; however, it was necessary to preincubate the same amount of CPE with higher concentrations of either rhCldn-4D146N (Fig. 5A) or rhCldn-4P150S (Fig. 5B) to provide Caco-2 cells with an equivalent level of protection. In contrast, compared to the results obtained using a preincubation mixture containing rhCldn-14 and CPE, lower concentrations of rhCldn-14N146D were needed during the preincubation to obtain 70% interference with the development of CPE-induced cytotoxicity in Caco-2 cells (Fig. 5A). Similarly, compared to the preincubation results obtained using rhCldn-8, lower concentrations of rhCldn-8S151P were needed for a similar block in the development of CPE-induced cytotoxicity in Caco-2 cells (Fig. 5B)
FIG 5 .

Cytotoxic effects of preincubating CPE with soluble rhCldns or rhCldn variants. (A) Cytotoxicity in Caco-2 cell cultures treated for 60 min at 37°C with the indicated preincubation mix containing 1 µg/ml of CPE/ml and the specified molar excesses of (i) soluble rhCldn-4 or -14 or (ii) rhCldn variant rhCldn-4D146N or rhCldn-14N146D. Cytotoxicity was determined using the Roche LDH assay. % of maximal CPE cytotoxicity = percent CPE-induced cytotoxicity in the presence of the specified rhCldn or rhCldn variant/percent CPE-induced cytotoxicity in the absence of any rhCldn or rhCldn variant. Results shown represent the means of three experimental repetitions. Error bars shown represent the SD. (B) Cytotoxicity in Caco-2 cell cultures treated for 60 min at 37°C with the indicated preincubation mix containing 1 µg/ml of CPE/ml and the specified molar excesses of (i) soluble rhCldn-4 or -8 or (ii) the rhCldn-4P150S or rhCldn-8S151P variant. Cytotoxicity was calculated using the LDH assay. % of maximal CPE cytotoxicity = percent CPE-induced cytotoxicity in the presence of the specified rhCldn or rhCldn variant/percent CPE-induced cytotoxicity in the absence of any rhCldn or rhCldn variant. Results shown represent the means of three experimental repetitions. Error bars shown represent the SD. (C and D) CPE Western immunoblot analyses of Caco-2 cells after a 60-min treatment at 37°C with preincubation mixes containing the specified molar excess of soluble rhCldn and 1 µg of CPE/ml. Locations of the CH-1 (CPE hexamer-1) and CH-2 complexes are noted on the left side of the figure. Results shown are representative of three experimental repetitions.
Western immunoblotting demonstrated that the stronger protective effects noted in Fig. 5A and B using rhCldn-14N146D or rhCldn-8S151P versus rhCldn-8 or -14 were due to those rhCldn variants more efficiently inhibiting CPE binding and large-complex formation in Caco-2 cells (compare Fig. 1B results to those shown in Fig. 5C and D). Similarly, these Western blot studies also determined (compare Fig. 1B results to those shown in Fig. 5C and D) that the weaker protective effects of rhCldn-4D146N and rhCldn-4P150S, relative to equivalent amounts of rhCldn-4 determined during the experiments represented in Fig. 5A and B, were due to the less efficient inhibition of CPE binding and large-complex formation in Caco-2 cells.
CPE sensitivity of mouse fibroblast transfectants expressing hCldn-4D146N, hCldn-4P150S, hCldn-14N146D, or hCldn-8S151P variants.
The results presented in Fig. 5 showed that changing (i) the ECL-2 D146 residue in rhCldn-4 to N, (ii) the corresponding ECL-2 N146 residue in rhCldn-14 to D, (iii) the ECL-2 P150 residue in rhCldn-4 to S, or (iv) the corresponding ECL-2 S151 residue in rhCldn-8 to P significantly affected the ability (relative to that of the parent rhCldn) of those rhCldn variants, in soluble form, to protect Caco-2 cells against CPE in the preincubation challenge model. Therefore, experiments were performed to evaluate directly whether mouse fibroblast transfectants stably expressing these hCldn variants show altered CPE binding ability and sensitivity relative to stable transfectants expressing the parental hCldns.
For this study, mouse fibroblast transfectants stably expressing the hCldn-4D146N, hCldn-14N146D, hCldn-4P150S, or hCldn-8S151P variants were prepared. Western blotting (Fig. 2) confirmed that each transfectant expressed the appropriate hCldn. These analyses also demonstrated that production levels of each hCldn variant were approximately similar to the rhCldn levels made by stable transfectants expressing hCldn-4, -8, or -14.
Once constructed, the stable transfectants producing the hCldn variants were tested for their CPE sensitivity. These experiments showed (Fig. 6A and B) that transfectants expressing hCldn-4D146N or hCldn-4P150S were less CPE sensitive than transfectants producing hCldn-4. However, the transfectants expressing those hCldn-4 variants developed CPE-induced cytotoxicity similar to that seen with transfectants producing hCldn-14 or hCldn-8, respectively.
FIG 6 .

Cytotoxic effects of CPE treatment on mouse fibroblast transfectants expressing specified hCldn variants. (A and B) Cytotoxicity developing in mouse fibroblast transfectants expressing the specified hCldn or hCldn variants after a 60-min treatment at 37°C with 1 to 12.5 μg of CPE/ml. CPE-induced cell cytotoxicity was expressed as the percentage of dead cells caused specifically by CPE treatment, i.e., after subtraction of any background nonviable cells. Results shown represent the means of three experimental repetitions. Error bars shown represent the SD. (C and D) CPE Western blot analysis of transfected fibroblasts expressing the specified hCldn or hCldn variant after a 60-min treatment at 37°C with 1 to 12.5 of CPE/ml. CPE immunoreactivity was detected by Western blot analysis using rabbit polyclonal anti-CPE serum. Results shown are representative of three experimental repetitions.
Conversely, transfectants stably expressing hCldn-14N146D or hCldn-8S151P were more CPE sensitive than transfectants stably producing hCldn-8 or -14. At equivalent CPE doses, those transfectants expressing the hCldn-8 or -14 variants developed CPE-induced cytotoxicity similar to that detected using transfectants stably producing hCldn-4 (Fig. 6A and B).
Western blot analyses (Fig. 6C and D) demonstrated that, using the same CPE concentration, mouse fibroblast transfectants stably expressing hCldn-14N146D or hCldn-8S151P showed CPE binding and CH-1 formation characteristics similar to those of transfectants producing hCldn-4. However, the transfectants expressing either of those two hCldn-8 or -14 variants bound more CPE and formed more CH-1 than transfectants producing either hCldn-8 or -14. Similar Western blot experiments also showed that, with the same CPE dose, transfectants stably expressing hCldn-4D146N or hCldn-4P150S exhibited less CPE binding and CH-1 formation than did transfectants stably producing hCldn-4. The CPE binding and CH-1 formation characteristics of transfectants stably producing those two hCldn-4 variants were similar to those of transfectants stably expressing hCldn-8 or -14, respectively.
DISCUSSION
There is considerable interest in understanding the interactions between CPE and Cldn receptors due to the biomedical importance of natural CPE-mediated GI diseases, as well as in the translational exploitation of CPE or CPE derivatives for cancer therapy or to increase drug absorption. However, it had not yet been established which of the ~24 hCldns can act as functional CPE receptors, except for (i) hCldn-3 and -4, which can convey CPE-induced cytotoxicity, and (ii) hCldn-1, -2, -5, and -10, which are not CPE receptors (44, 48)
Results from previous studies were not always in agreement regarding differences in CPE binding properties among various Cldns. For example, an early study using transfectants expressing Cldns of unspecified species origin reported that Cldn-8 and -14 possess binding affinity for CPE that is an order of magnitude lower than the strong CPE binding properties of Cldn-4 (45). However, since that initial study, the CPE binding properties of Cldn-8 and -14 have become less clear, with reports that mouse Cldn-8 or -14 binding to CPE ranges from weak to strong, relative to the binding of this toxin by mouse or human Cldn-4 (48). In addition, while CPE binding to hCldn-8 or -14 apparently had not been experimentally assessed, it was proposed (52) that hCldn-8 is unlikely to bind CPE because it lacks a motif purportedly essential for Cldn binding of this toxin (further discussion later).
Therefore, to our knowledge, results from the current study have established for the first time that hCldn-8 and -14 bind CPE, although slightly less well than hCldn-4. Furthermore, since fibroblasts do not naturally produce Cldns (41), the demonstrated CPE sensitivity of fibroblast transfectants expressing only hCldn-8 or -14 revealed that hCldns with weaker CPE binding properties than hCldn-4 can still convey CPE-induced cytotoxicity, although less efficiently. Last, these findings indicated that hCldns other than hCldn-3 or -4 can serve as functional CPE receptors capable of mediating cytotoxicity at CPE concentrations present during natural GI disease (1). These three conclusions were supported by the observation that higher CPE concentrations are necessary to obtain equivalent CPE binding/cytotoxicity levels in transfectants expressing hCldn-8 or -14 versus hCldn-4, which strongly binds CPE.
Results from several experiments performed in the current work indicated that these transfectant sensitivity differences were due to variations in the CPE binding properties of these hCldns, rather than to differences in Cldn expression levels. First, Western blot analyses revealed that the transfectants under study all express approximately similar amounts of their respective Cldns. Furthermore, Western blotting established a correlation between a transfectant’s levels of CPE binding and CH-1 formation and its CPE sensitivity. Finally, preincubation experiments using identical amounts of purified soluble rhCldns directly demonstrated slightly weaker binding properties for rhCldn-8 or -14 versus rhCldn-4, since ~3-fold more rhCldn-8 or -14 than rhCldn-4 was needed to obtain an equivalent inhibition of CPE binding to Caco-2 cells.
Establishing that both hCldn-8 and -14 can mediate cytotoxicity at pathophysiologically relevant CPE doses (1) has potential biological relevance. Since expression of those two Cldns has reportedly been detected in the mammalian intestines (49–51), the current findings open the possibility that hCldn-8 and -14 could contribute to natural intestinal diseases caused by CPE, including C. perfringens type A food poisoning. However, Cldn-3 and -4 are also expressed in the mammalian intestines (49–51), so those two Cldns also likely contribute to CPE action during disease. The small differences in CPE binding properties among hCldn-4, -8, and -14, as detected during the current study, may be of limited importance during in vivo disease. However, the specific contributions of different Cldns during CPE-mediated disease would be influenced by their expression levels in the intestines. To our knowledge, information on the relative abundances of various Cldns in the human intestines is limited.
There is now considerable interest in translational applications of CPE and CPE derivatives for cancer therapy or for increasing drug absorption (see the introduction). For those purposes, it has often been assumed that a CPE-based therapeutic would affect only human cells expressing Cldn-3 or -4 receptors. Establishing that other rhCldns can also convey CPE sensitivity suggests both positive and negative implications for applied uses of CPE or CPE-based derivatives. First, it suggests that CPE or CPE derivatives could be effective (i) against human tumors overexpressing rhCldn-8 or -14 or (ii) for increasing drug absorption in normal human tissues expressing rhCldn-8 or -14. However, these results also open the possibility that toxic side effects from CPE therapeutics could occur in normal human tissues expressing hCldn-8 or -14, as well as in those producing hCldn-3 or -4.
It is unclear why reports differ regarding the relative CPE binding properties of different Cldns. Study-to-study variations regarding the CPE binding properties of some Cldns likely involve such factors as (i) the CPE species used, with some studies employing native CPE, while others used CPE fragments, CPE-based synthetic peptides, or CPE fusion proteins, and (ii) the Cldn used, with studies utilizing Cldns from different mammalian species and also differing in their use of full-length Cldns, Cldn-based peptides, and Cldn peptide fusion proteins (48, 52). In this respect, our studies sought to maximize biomedical relevance by employing native CPE and full-length hCldns.
The current results now clearly indicate that rhCldn-8 can bind CPE, although slightly less strongly than hCldn-4. This finding is interesting since, as mentioned earlier, it has been proposed (52) that hCldn-8 would not bind CPE because it lacks the ECL-2 motif NP(L/M) (V/L/T) (P/A/D) suggested to be necessary for CPE binding (48, 52). The N and L/M residues in this motif were thought to be particularly important for CPE binding (48, 52), so our results are notable because the hCldn-8 ECL-2 sequence corresponding to the NP (L/M) (V/L) (P/A/D) motif is NSIVN; i.e., the putative ECL-2 CPE binding motif of hCldn-8 has an I rather than an L or M present at residue 3. Therefore, the current study has revealed more flexibility in the proposed Cldn ECL-2 CPE binding motif than had been previously appreciated. However, our results are consistent with the proposal that residue 3 of the ECL-2 motif requires a hydrophobic amino acid for CPE binding (48). Combining all results (44, 47, 48, 53–55), residue N149 of Cldn-4 (or its equivalent in other CPE binding Cldns) appears to be the only amino acid in the putative ECL-2 motif that is invariantly necessary for a Cldn to possess intermediate or strong CPE binding properties.
It has also been proposed (47, 48, 55) that the ECL-2 of some or all CPE binding Cldns forms a helix-turn-helix, with this ECL-2 turn region containing the putative CPE binding motif discussed above. It was further suggested that, during toxin binding, the helix-turn-helix of ECL-2 docks with a cavity present in the C-terminal binding domain of CPE (47, 48). The proposed ECL-2 turn region of CPE binding Cldn receptors often contains two prolines, which were postulated to help stabilize the ECL2 turn region and thus contribute to CPE binding. For example, the putative ECL-2 turn region/CPE binding motif sequence for mouse Cldn-3, which strongly binds CPE (48), is NPLVP. However, the strong CPE binder hCldn-4 (references 43 and 44 and this study) naturally lacks the second P in its putative ECL-2 turn region sequence of NPLVA, so the model has been modified (48) to indicate that an A or D (52) at this position is also permissive for CPE binding, with sequences located C-terminal to the fifth residue in the CPE binding motif presumably helping to maintain a favorable ECL-2 structure for CPE binding.
Consistent with this model, our results now indicate that CPE binding characteristics similar to those of hCldn-4 can be imparted to hCldn-8 by substituting P for the natural S151 residue present in the putative NSIVN turn sequence of the hCldn-8 ECL-2. This finding supports the idea of the importance of the first P residue in the putative Cldn turn region for strong CPE binding. However, our hCldn-8S151P variant with strong CPE binding properties still lacked the second P found in the putative ECL-2 turn region of mouse Cldn-4, indicating that an N (as well as a P, A, or D) can suffice at the fifth residue in the putative ECL-2 turn region/binding motif to support strong CPE binding.
The toxin binding contributions of amino acids located N-terminal to the N149 residue of hCldn-4 (or its equivalent residue in other CPE receptor claudins) have received limited research attention, with the exception of a previous report indicating that alanine substitutions introduced at the ECL-2 F147 and Y148 of hCldn-4 did not affect CPE binding properties (54). The current report now establishes that the residue located immediately N-terminal to those FY residues in receptor Cldns modulates CPE binding affinity. Specifically, an N-to-D change at residue 146 in hCldn-14 produced an hCldn-14N146D variant with CPE binding properties stronger than those of wild-type hCldn-14, while the converse change from D to N in hCldn-4 produced an hCldn-4D146N variant with CPE binding weaker than that of wild-type hCldn-4. These mutations induced a charge difference in the hCldn variants, which is interesting since the contribution of electrostatic interactions to CPE binding by Cldns is controversial. It was proposed (54) that electrostatic interactions are important for CPE binding to Cldns, but later data (55) indicated that, except for the N149 residue of hCldn-4 (or its equivalent in other CPE receptor Cldns), electrostatic interactions are not very influential for CPE:Cldn binding interactions. Solving the structure of CPE bound to Cldns is under way and will be immensely helpful to better explain the nature of CPE:Cldn binding interactions, but our hCldn-14 data nevertheless already broaden perspectives on which ECL-2 residues modulate CPE binding affinity by showing that an amino acid located N-terminal to the N149 ECL-2 residue of hCldn receptors also influences CPE binding properties.
In summary, the current report establishes for the first time that hCldns other than hCldn-3 or -4 can bind and convey CPE-induced cytotoxicity at pathophysiologically relevant toxin concentrations. Furthermore, findings obtained using hCldn variants suggest implications for understanding natural CPE-mediated gastrointestinal disease and for translational use of CPE. They also indicate that previously proposed models for CPE:Cldn interactions should be broadened. Further analyses of CPE:hCldn interactions are under way.
MATERIALS AND METHODS
CPE purification.
CPE was purified to homogeneity from C. perfringens type A strain NCTC8239, as described previously (56).
Plasmid cDNA constructs.
Full-length hCldn-4 and -8 cDNA (Invitrogen) was PCR amplified using (i) primers Cldn14XhoIFwd (5′-GGCAGCCTCGAGGCCTAGGCAGGCAGCCGCACCATGG-3′) and Cldn14HindIIIRev (5′-CCGTCGAAGCTTTCACACCTAGTCGTTCAGCCTGTACCCGCTG-3′) to amplify hCldn-14 cDNA and (ii) primers Cldn8XhoIFwd (5′-GGCCGGCTCGAGATGGCAACCCATGCCTTAGAAATC-3′) and Cldn8HindIIIRev (5′-CCGTCGAAGCTTCTACACATACTGACTTCTGGAGTAGAC-3′) to amplify hCldn-8 cDNA. The PCR for this amplification used Taq polymerase (New England Biolabs) and the following conditions: 94°C for 5 min; 35 cycles of 94°C for 30 s, 68°C for 45 s, and 72°C for 2 min; and 72°C for 7 min. Full-length cDNA hCldn-1 and -4 cDNAs were amplified using (i) primers Cldn1BamHI (5′-GGCCGGATCCAACTCTCCGCCTTCTGCAC-3′) and Cldn1EcoRI (5′-GGCCGAATTCTTGAGTATGATTACTCAA-3′) for hCldn-1 cDNA and (ii) primers Cldn4XhoIFwd (5′-GGCAGCCTCGAGGCCATTATGGCCTCCATGGGGCTAC-3′) and Cldn4HindIII rev (5′-CCGTCGAAGCTTGAATTCGCCCTTTTACACGTAGTTGC-3′) for hCldn-4 cDNA. PCR conditions used were 94°C for 5 min; 35 cycles of 94°C for 30 s, 68°C for 45 s, and 72°C for 2 min; and 72°C for 7 min. Each amplicon was then cloned into TOPO-TA (Invitrogen) and subcloned using XhoI and HindIII into pTrcHisA (Invitrogen) for expression as a soluble rhCldn.
Site-directed mutations were introduced into the cloned hCldn-4, -8, and -14 cDNAs using a QuikChange Lightning site-directed mutagenesis kit (Stratagene). PCR products encoding the hCldn variants were then subcloned into pCEP4 for transfection. The primers used for preparing the hCldn-14N146D variant were SDM14-4 A436G Rev (5′-CAGCGGGTTGTAGAAGTCCTGCACCA-3′) and SDM14-4 A436G Fwd (5′-CCAACGACGTGGTGCAGGACTTCTAC-3′); those used for preparing the hCldn-4D146N variant were SDM4-14 G436A Rev (5′-CACCAGCGGATTGTAGAAGTTTTGGA-3′) and SMD4-14G436AFwd (5′-CGGCCCACAACATCATCCAAAACTTC-3′); those used for preparing the hCldn-8S151P variant were SDM8-4SerProFwd (5′-ATCATCAGAGATTTCTATAACCCGATA-3′) and SDM8-4SerProRev (5′-TTGGGCAACATTCACTATCGGGTTATA-3′); and those used for preparing the hCldn-4P150S mutant were SDM4-8ProSerFwd (5′-ATCATCCAAGACTTCTACAATTCACTG-3′) and SDM4-8ProSerRev (5′-CTGCCCGGAGGCCACCAGTGAATTGTA-3′). The PCR conditions used for construction of site-directed rhCldn variants were 95°C for 2 min; 18 cycles of 95°C for 20 s, 60°C for 10 s, and 68°C for 30 s; and 68°C for 5 min.
DNA sequencing.
All wild-type Cldn and Cldn variant sequences were verified by DNA sequencing at the University of Pittsburgh Genomics and Proteomics Core Laboratories.
Antibodies.
Rabbit polyclonal antibodies against Cldn-1, -4, -8, or -14 antibody were purchased from Invitrogen. These antibodies were raised against synthetic peptides corresponding to unique epitopes present in the cytoplasmic C-terminal tail of each Cldn. Rabbit polyclonal CPE antiserum had been prepared previously (23). Goat polyclonal anti-rabbit horseradish peroxidase conjugate was purchased from Sigma.
Cell culture.
Caco-2 human colorectal adenocarcinoma cells were maintained in minimum essential Eagle’s medium (MEM) supplemented with 10% fetal bovine serum, 1% nonessential amino acids, 100 units/ml penicillin, 100 mg/ml streptomycin, and 1% glutamine. Mouse parental NT6 fibroblasts were cultured in MEM, 10% fetal bovine serum, 100 units/ml penicillin, and 100 mg/ml streptomycin. Mouse fibroblast transfectants expressing either rhCldn-8 or -14 were maintained in MEM, 10% fetal bovine serum, 100 units/ml penicillin, 100 mg/ml streptomycin, and 50 mg/ml hygromycin. All cells were maintained at 37°C in 5% atmospheric CO2 and grown to confluence in 75-cm2.
Preparation of mouse fibroblast transfectants stably expressing rhCldn or rhCldn variants.
To prepare mouse transfectants stably expressing hCldn-8, -14, -4D146N, -14N146D, -4P150S, or -8S151P, volumes of pCEP4 vector carrying a cDNA encoding one of these hCldns or hCldn variants were separately transfected into mouse fibroblasts. The stable transfectants were prepared as follows. On day 1, the mouse fibroblast cells were grown to 60% confluence. On day 2, 0.75 µg of DNA was mixed with 12 µl of Lipofectamine (Invitrogen) and 200 µl of Opti-MEM (Gibco); that DNA/Lipofectamine mix was then added to cells in a total volume of 2 ml of MEM and the cultures were incubated for 4 to 7 h at 37°C. A 2-ml volume of media was added, and the culture was incubated overnight at 37°C. On day 3, the cells were fed with fresh MEM. On day 4, the cells were trypsinized, plated at 1:50, 1:30, and 1:10 dilutions, put under selective pressure with 200 µg/ml of hygromycin, and then grown for 10 to 12 days with a change of medium every 4 days. When colonies were easily discernible by the naked eye (when they reached several millimeters in diameter), they were cloned. On day 12, colonies with sufficient expression of the desired rhCldn, as determined by Western blotting, were selected and grown for 4 to 5 days under conditions of hygromycin selection and stable cell lines were screened by immunoblot analysis. At least five stably expressing clonal cell lines were generated for each construct. All stable transfectants were cultured at 37°C in 5% atmospheric CO2 in MEM–10% fetal bovine serum–100 U of penicillin/ml–100 mg of streptomycin/ml–50 mg of hygromycin/ml.
Affinity enrichment of rhCldn species.
rhCldn or rhCldn variants were expressed as His6-tagged proteins from pTrcHisA and then affinity enriched using Ni2+-nitrilotriacetic acid (NTA) affinity chromatography (Qiagen), as described in the manufacturer’s protocol. Fractions containing the rhCldn species were identified by Western immunoblotting, using (as appropriate) rabbit polyclonal antibodies against Cldn-1, -4, -8, or -14. Protein was then quantified by a Lowry protein assay (57). In addition, Coomassie blue staining was performed, which showed the presence of a single band of ~25 kDa for each rhCldn preparation.
Analysis of CH-1 or CH-2 formation in CPE-treated cells.
Confluent Caco-2 cells or fibroblast transfectants were treated with 1 µg/ml of CPE in Hanks’ balanced salt solution without calcium and magnesium (HBSS; Mediatech) for 60 min, scraped, washed once, and lysed with 2× sodium dodecyl sulfate (SDS) buffer. Samples were then electrophoresed on SDS-containing gels containing 4% polyacrylamide and transferred onto a nitrocellulose membrane (Bio-Rad). Western immunoblotting was performed on this membrane, as described previously (23), to detect the presence of SDS-resistant CH-1 and CH-2 CPE complexes.
Receptor preincubation studies.
Specified molar excesses of each affinity-enriched, soluble rhCldn species were preincubated with 1 µg/ml of CPE in 10 ml of HBSS, with gentle rocking, for 20 min at 37°C. Confluent Caco-2 cells were then treated with those preincubation mixes for 60 min at 37°C and processed for analysis of CPE large-complex formation, morphological damage, and cytotoxicity.
Evaluation of CPE cytotoxic effects.
A Live/Dead viability/cytotoxicity assay kit (Roche) for mammalian cells was used to evaluate CPE-induced cytotoxicity (44). The assay, performed as described by the supplier, measures lactate dehydrogenase (LDH) release. It was conducted after (i) treatment of mouse transfectants with 1, 2.5, 5, 7.5, 10, or 12.5 µg/ml of CPE for 60 min at 37°C or (ii) treatment of Caco-2 monolayers for 60 min at 37°C with 1 µg/ml of CPE. For the Caco-2 cell experiments, CPE was preincubated for 20 min at 37°C with a 100-fold molar excess of the appropriate affinity-enriched rhCldn species prior to addition to this preincubation mix to the monolayer, as described earlier. Cytotoxicity was expressed as a percentage of the total cells that were determined to be nonviable.
Western blot analysis of rhCldn expression by transfectants.
Western blot analysis of rhCldn expression by transfectants was carried out as previously described (44) using, as primary antibody, rabbit polyclonal antibodies against Cldn-1, -4, -8, or -14. Goat polyclonal anti-rabbit horseradish peroxidase conjugate was used as the secondary antibody.
ACKNOWLEDGMENT
This research was generously supported by grant R37-AI19844-30 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Citation Shrestha A, McClane BA. 2013. Human claudin-8 and -14 are receptors capable of conveying the cytotoxic effects of Clostridium perfringens enterotoxin. mBio 4(1):e00594-12. doi:10.1128/mBio.00594-12.
REFERENCES
- 1. McClane BA, Robertson SL, Li J. 2013. Clostridium perfringens, p 465–489 In Doyle MP, Buchanan RL, Food microbiology: fundamentals and frontiers, 4th ed. ASM Press, Washington, DC [Google Scholar]
- 2. Sarker MR, Carman RJ, McClane BA. 1999. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol. Microbiol. 33:946–958 [DOI] [PubMed] [Google Scholar]
- 3. Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson M-A, Roy SL, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States—major pathogens. Emerg. Infect. Dis. 17:7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carman RJ. 1997. Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea of man and other animals. Rev. Med. Microbiol. 8(Suppl. 1):S43–S45 [Google Scholar]
- 5. McClane BA, Uzal FA, Miyakawa MF, Lyerly D, Wilkins TD. 2006. The enterotoxic Clostridia, p 688–752 In Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E, The prokaryotes, 3rd ed. Springer Press, New York, NY. [Google Scholar]
- 6. Songer JG. 1996. Clostridial enteric diseases of domestic animals. Clin. Microbiol. Rev. 9:216–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao Z, McClane BA. 15 September 2012. Use of Clostridium perfringens enterotoxin and the enterotoxin receptor-binding domain (C-CPE) for cancer treatment: opportunities and challenges. J. Toxicol. http://dx.doi.org/10.1155/2012/981626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kojima T, Kyuno D, Sawada N. 2012. Targeting claudin-4 in human pancreatic cancer. Expert Opin. Ther. Targets 16:881–887 [DOI] [PubMed] [Google Scholar]
- 9. Neesse A, Griesmann H, Gress TM, Michl P. 2012. Claudin-4 as therapeutic target in cancer. Arch. Biochem. Biophys. 524:64–70 [DOI] [PubMed] [Google Scholar]
- 10. Deli MA. 2009. Potential use of tight junction modulators to reversibly open membranous barriers and improve drug delivery. Biochim. Biophys. Acta 1788:892–910 [DOI] [PubMed] [Google Scholar]
- 11. Harada M, Kondoh M, Ebihara C, Takahashi A, Komiya E, Fujii M, Mizuguchi H, Tsunoda S, Horiguchi Y, Yagi K. 2007. Role of tyrosine residues in modulation of claudin-4 by the C-terminal fragment of Clostridium perfringens enterotoxin. Biochem. Pharmacol. 73:206–214 [DOI] [PubMed] [Google Scholar]
- 12. Kondoh M, Takahashi A, Fujii M, Yagi K, Watanabe Y. 2006. A novel strategy for a drug delivery system using a claudin modulator. Biol. Pharm. Bull. 29:1783–1789 [DOI] [PubMed] [Google Scholar]
- 13. Matsuhisa K, Kondoh M, Suzuki H, Yagi K. 2012. Comparison of mucosal absorption-enhancing activity between a claudin-3/-4 binder and a broadly specific claudin binder. Biochem. Biophys. Res. Commun. 423:229–233 [DOI] [PubMed] [Google Scholar]
- 14. Rosenthal R, Heydt MS, Amasheh M, Stein C, Fromm M, Amasheh S. 2012. Analysis of absorption enhancers in epithelial cell models. Ann. N. Y. Acad. Sci. 1258:86–92 [DOI] [PubMed] [Google Scholar]
- 15. Suzuki H, Kondoh M, Takahashi A, Yagi K. 2012. Proof of concept for claudin-targeted drug development. Ann. N. Y. Acad. Sci. 1258:65–70 [DOI] [PubMed] [Google Scholar]
- 16. Takahashi A, Komiya E, Kakutani H, Yoshida T, Fujii M, Horiguchi Y, Mizuguchi H, Tsutsumi Y, Tsunoda S, Koizumi N, Isoda K, Yagi K, Watanabe Y, Kondoh M. 2008. Domain mapping of a claudin-4 modulator, the C-terminal region of C-terminal fragment of Clostridium perfringens enterotoxin, by site-directed mutagenesis. Biochem. Pharmacol. 75:1639–1648 [DOI] [PubMed] [Google Scholar]
- 17. Czeczulin JR, Hanna PC, McClane BA. 1993. Cloning, nucleotide sequencing, and expression of the Clostridium perfringens enterotoxin gene in Escherichia coli. Infect. Immun. 61:3429–3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Briggs DC, Naylor CE, Smedley JG, III, Lukoyanova N, Robertson S, Moss DS, McClane BA, Basak AK. 2011. Structure of the food-poisoning Clostridium perfringens enterotoxin reveals similarity to the aerolysin-like pore-forming toxins. J. Mol. Biol. 413:138–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kitadokoro K, Nishimura K, Kamitani S, Fukui-Miyazaki A, Toshima H, Abe H, Kamata Y, Sugita-Konishi Y, Yamamoto S, Karatani H, Horiguchi Y. 2011. Crystal structure of Clostridium perfringens enterotoxin displays features of beta-pore-forming toxins. J. Biol. Chem. 286:19549–19555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wieckowski EU, Kokai-Kun JF, McClane BA. 1998. Characterization of membrane-associated Clostridium perfringens enterotoxin following pronase treatment. Infect. Immun. 66:5897–5905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wieckowski EU, Wnek AP, McClane BA. 1994. Evidence that an ~50kDa mammalian plasma membrane protein with receptor-like properties mediates the amphiphilicity of specifically-bound Clostridium perfringens enterotoxin. J. Biol. Chem. 269:10838–10848 [PubMed] [Google Scholar]
- 22. McClane BA, Wnek AP. 1990. Studies of Clostridium perfringens enterotoxin action at different temperatures demonstrate a correlation between complex formation and cytotoxicity. Infect. Immun. 58:3109–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robertson SL, Smedley JG, III, Singh U, Chakrabarti G, Van Itallie CM, Anderson JM, McClane BA. 2007. Compositional and stoichiometric analysis of Clostridium perfringens enterotoxin complexes in Caco-2 cells and claudin 4 fibroblast transfectants. Cell. Microbiol. 9:2734–2755 [DOI] [PubMed] [Google Scholar]
- 24. Singh U, Van Itallie CM, Mitic LL, Anderson JM, McClane BA. 2000. Caco-2 cells treated with Clostridium perfringens enterotoxin form multiple large complex species, one of which contains the tight junction protein occludin. J. Biol. Chem. 275:18407–18417 [DOI] [PubMed] [Google Scholar]
- 25. Chen J, Theoret JR, Shrestha A, Smedley JG, III, McClane BA. 2012. Cysteine scanning mutagenesis supports the importance of Clostridium perfringens enterotoxin amino acids 80–106 for membrane insertion and pore formation. Infect. Immun. 80:4078–4088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smedley JG, III, Uzal FA, McClane BA. 2007. Identification of a prepore large-complex stage in the mechanism of action of Clostridium perfringens enterotoxin. Infect. Immun. 75:2381–2390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matsuda M, Ozutsumi K, Iwahashi H, Sugimoto N. 1986. Primary action of Clostridium perfringens type A enterotoxin on HeLa and Vero cells in the absence of extracellular calcium: rapid and characteristic changes in membrane permeability. Biochem. Biophys. Res. Commun. 141:704–710 [DOI] [PubMed] [Google Scholar]
- 28. McClane BA. 1994. Clostridium perfringens enterotoxin acts by producing small molecule permeability alterations in plasma membranes. Toxicology 87:43–67 [DOI] [PubMed] [Google Scholar]
- 29. McClane BA, Hanna PC, Wnek AP. 1988. Clostridium perfringens type A enterotoxin. Microb. Pathog. 4:317–323 [DOI] [PubMed] [Google Scholar]
- 30. McClane BA, McDonel JL. 1980. Characterization of membrane permeability alterations induced in Vero cells by Clostridium perfringens enterotoxin. Biochim. Biophys. Acta 600:974–985 [DOI] [PubMed] [Google Scholar]
- 31. Chakrabarti G, McClane BA. 2005. The importance of calcium influx, calpain, and calmodulin for the activation of Caco-2 cell death pathways by Clostridium perfringens enterotoxin. Cell. Microbiol. 7:129–146 [DOI] [PubMed] [Google Scholar]
- 32. Chakrabarti G, Zhou X, McClane BA. 2003. Death pathways activated in Caco-2 cells by Clostridium perfringens enterotoxin. Infect. Immun. 71:4260–4270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Singh U, Mitic LL, Wieckowski EU, Anderson JM, McClane BA. 2001. Comparative biochemical and immunochemical studies reveal differences in the effects of Clostridium perfringens enterotoxin on polarized Caco-2 cells versus Vero cells. J. Biol. Chem. 276:33402–33412 [DOI] [PubMed] [Google Scholar]
- 34. Sherman S, Klein E, McClane BA. 1994. Clostridium perfringens type A enterotoxin induces concurrent development of tissue damage and fluid accumulation in the rabbit ileum. J. Diarrhoeal Dis. Res 12:200–207 [PubMed] [Google Scholar]
- 35. Smedley JG, III, Saputo J, Parker JC, Fernandez-Miyakawa ME, Robertson SL, McClane BA, Uzal FA. 2008. Noncytotoxic Clostridium perfringens enterotoxin (CPE) variants localize CPE intestinal binding and demonstrate a relationship between CPE-induced cytotoxicity and enterotoxicity. Infect. Immun. 76:3793–3800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Anderson JM, Van Itallie CM. 2009. Physiology and function of the tight junction. Cold Spring Harb. Perspect. Biol. 1:a002584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Farquhar MG, Palade GE. 1963. Junctional complexes in various epithelia. J. Cell Biol. 17:375–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Piontek J, Winkler L, Wolburg H, Müller SL, Zuleger N, Piehl C, Wiesner B, Krause G, Blasig IE. 2008. Formation of tight junction: determinants of homophilic interaction between classic claudins. FASEB J. 22:46–58 [DOI] [PubMed] [Google Scholar]
- 39. Van Itallie CM, Anderson JM. 2006. Claudins and epithelial paracellular transport. Annu. Rev. Physiol. 68:403–429 [DOI] [PubMed] [Google Scholar]
- 40. Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J, Blasig IE. 2008. Structure and function of claudins. Biochim. Biophys. Acta 1778:631–645 [DOI] [PubMed] [Google Scholar]
- 41. Anderson JM, Van Itallie CM. 1999. Tight junctions: closing in on the seal. Curr. Biol. 9:R922–R924 [DOI] [PubMed] [Google Scholar]
- 42. Katahira J, Inoue N, Horiguchi Y, Matsuda M, Sugimoto N. 1997. Molecular cloning and functional characterization of the receptor for Clostridium perfringens enterotoxin. J. Cell Biol. 136:1239–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Katahira J, Sugiyama H, Inoue N, Horiguchi Y, Matsuda M, Sugimoto N. 1997. Clostridium perfringens enterotoxin utilizes two structurally related membrane proteins as functional receptors in vivo. J. Biol. Chem. 272:26652–26658 [DOI] [PubMed] [Google Scholar]
- 44. Robertson SL, Smedley JG, III, McClane BA. 2010. Identification of a claudin-4 residue important for mediating the host cell binding and action of Clostridium perfringens enterotoxin. Infect. Immun. 78:505–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fujita K, Katahira J, Horiguchi Y, Sonoda N, Furuse M, Tsukita S. 2000. Clostridium perfringens enterotoxin binds to the second extracellular loop of claudin-3, a tight junction membrane protein. FEBS Lett. 476:258–261 [DOI] [PubMed] [Google Scholar]
- 46. Sonoda N, Furuse M, Sasaki H, Yonemura S, Katahira J, Horiguchi Y, Tsukita S. 1999. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: evidence for direct involvement of claudins in tight junction barrier. J. Cell Biol. 147:195–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Veshnyakova A, Piontek J, Protze J, Waziri N, Heise I, Krause G. 2012. Mechanism of Clostridium perfringens enterotoxin interaction with claudin-3/-4 protein suggests structural modifications of the toxin to target specific claudins. J. Biol. Chem. 287:1698–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Veshnyakova A, Protze J, Rossa J, Blasig IE, Krause G, Piontek J. 2010. On the interaction of Clostridium perfringens enterotoxin with claudins. Toxins (Basel) 2:1336–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Charoenphandhu N, Wongdee K, Tudpor K, Pandaranandaka J, Krishnamra N. 2007. Chronic metabolic acidosis upregulated claudin mRNA expression in the duodenal enterocytes of female rats. Life Sci. 19:1729–1737 [DOI] [PubMed] [Google Scholar]
- 50. Fujita H, Chiba H, Yokozaki H, Sakai N, Sugimoto K, Wada T, Kojima T, Yamashita T, Sawada N. 2006. Differential expression and subcellular localization of claudin-7, -8, -12, -13, and -15 along the mouse intestine. J. Histochem. Cytochem. 54:933–944 [DOI] [PubMed] [Google Scholar]
- 51. Holmes JL, Van Itallie CM, Rasmussen JE, Anderson JM. 2006. Claudin profiling in the mouse during postnatal intestinal development and along the gastrointestinal tract reveals complex expression patterns. Gene Expr. Patterns 6:581–588 [DOI] [PubMed] [Google Scholar]
- 52. Mitchell LA, Koval M. 2010. Specificity of interaction between Clostridium perfringens enterotoxin and claudin-family tight junction proteins. Toxins (Basel) 2:1595–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ling J, Liao H, Clark R, Wong MS, Lo DD. 2008. Structural constraints for the binding of short peptides to claudin-4 revealed by surface plasmon resonance. J. Biol. Chem. 283:30585–30595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kimura J, Abe H, Kamitani S, Toshima H, Fukui A, Miyake M, Kamata Y, Sugita-Konishi Y, Yamamoto S, Horiguchi Y. 2010. Clostridium perfringens enterotoxin interacts with claudins via electrostatic attraction. J. Biol. Chem. 285:401–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Winkler L, Gehring C, Wenzel A, Müller SL, Piehl C, Krause G, Blasig IE, Piontek J. 2009. Molecular determinants of the interaction between Clostridium perfringens enterotoxin fragments and claudin-3. J. Biol. Chem. 284:18863–18872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McDonel JL, McClane BA. 1988. Production, purification and assay of Clostridium perfringens enterotoxin. Methods Enzymol. 165:94–103 [DOI] [PubMed] [Google Scholar]
- 57. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275 [PubMed] [Google Scholar]