

Abstract
The gut microbiota, the intestinal mucosa and the host immune system are among the large biological networks involved in the development of inflammatory bowel disease (IBD), which includes Crohn disease (CD) and ulcerative colitis (UC). Host genetics and environmental factors can significantly modulate the interactive relationships among these biological systems and influence predilection toward IBD. High monozygotic twin discordance rates and the rapid rise in the prevalence of IBD indicate that environmental influences may be as important or even more important in their pathogenesis than genetic susceptibility. However, the nature and timing of environmental factors critical for inducing IBD remain largely unknown. The molecular mechanisms and the key biological component(s) that may be affected by such factors are also in question. Epigenetic changes, such as DNA methylation (the methylation of cytosines followed by a guanine in CpG dinucleotides) can be modified by environmental influences during finite developmental periods and have been implicated in the pathogenesis of IBD. Mucosal DNA methylation can also react to changes in the commensal microbiota, underscoring the intercalating relationships among the large biological systems involved in gastrointestinal disorders. Therefore, transient environmental influences during specific periods of development may induce critical change(s) in an isolated or concomitant fashion within the intestinal biomic networks and lead to increased susceptibility to IBD. The present review focuses on the emerging paradigm shift considering IBD to originate from critical environmental effects during pre- and postnatal development.
Keywords: Diet, DNA methylation, Epigenetics, Inflammatory bowel disease, Microbiome
Abstract
Le microbiote intestinal, la muqueuse intestinale et le système immunitaire de l’hôte font partie des grands réseaux biologiques qui participent à l’apparition des maladies inflammatoires de l’intestin (MII), lesquelles incluent la maladie de Crohn (MC) et la colite ulcéreuse (CU). La génétique de l’hôte et les facteurs environnementaux peuvent moduler de manière significative les relations interactives entre ces systèmes biologiques et influer sur la prédilection vers les MII. D’après les taux de discordance élevés chez des jumeaux monozygotes et l’augmentation rapide de la prévalence des MII, la pathogenèse des influences environnementales serait aussi importante, sinon plus, que la susceptibilité génétique. Cependant, on ne sait pas grand-chose de la nature et du moment d’exposition aux facteurs environnementaux nécessaires pour induire une MII. Les mécanismes moléculaires et les principaux éléments biologiques susceptibles d’être touchés par ces facteurs sont également remis en question. Les changements épigénétiques, tels que la méthylation de l’ADN (méthylation des cytosines suivie par une guanine dans les dinucléotides CpG), peuvent être modifiés par des influences environnementales pendant des périodes précises du développement et contribuer à la pathogenèse des MII. La méthylation de l’ADN muqueux peut également réagir aux changements du microbiote commensal, ce qui fait ressortir les relations intercalantes entre les grands systèmes biologiques participant aux troubles gastro-intestinaux. Ainsi, des influences environnementales transitoires pourraient induire des changements capitaux de manière isolée ou concomitante dans les réseaux biomiques intestinaux, suscitant une plus grande susceptibilité aux MII. La présente analyse porte sur le changement de paradigme émergent selon lequel les MII proviendraient d’effets capitaux de l’environnement pendant le développement prénatal et postnatal.
Genetic predisposition to inflammatory bowel disease (IBD) is an intensive area of research, which was demonstrated by a recent request for application by the National Institutes of Health of the United States (RFA-DK-11-032). However, the emergence of the disorders over the past five to six decades with the spread of industrialization around the world (1,2), and the high monozygotic twin discordance rates (3) in both Crohn disease (CD) and ulcerative colitis (UC), argue for nongenetic factors being as or even more important for IBD pathogenesis than genetic susceptibility. Epigenetic changes are environmentally responsive molecular mechanisms that can modify gene expression independently of the genetic code. They were proposed to play an important role in IBD pathogenesis in 2000 (4). Yet, a recent PubMed search with “genetics” and “IBD” as keywords yielded 7218 publications, while a search using “epigenetics” and “IBD” yielded only 15 articles (>480 fold difference in favour of genetics). Therefore, the present review intends to highlight the potential importance of epigenetic mechanisms with regard to IBD and support the developmental origins paradigm shift in the basic scientific and clinical approaches toward the disease group.
NONGENETIC FACTORS ARE IMPORTANT IN IBD PATHOGENESIS
Twin studies (3,5,6) indicate that the relative genetic contribution to CD and UC is low when compared with celiac disease (CeD) (7), and is comparable with irritable bowel syndrome (IBS) (8,9) in the case of UC (Figure 1). These observations raise the importance of nongenetic processes in the etiology of IBD. Compared with CeD, in which a single human leukocyte antigen (HLA) locus can explain 40% of heritability (10), more than 71 loci have been associated with CD (11) and more than 47 such genomic regions have been described for UC (12). Most IBD susceptibility loci contribute to disease development with low ORs (1 to 1.5), revealing the complexity of even the relatively low genetic attribution to these disorders. Naturally, the unreliability of the clinical subphenotypes of IBD may also add to the difficulties in determining phenotype-genotype associations, as has similarly been described for investigations into the etiology of IBS (13).
Figure 1).
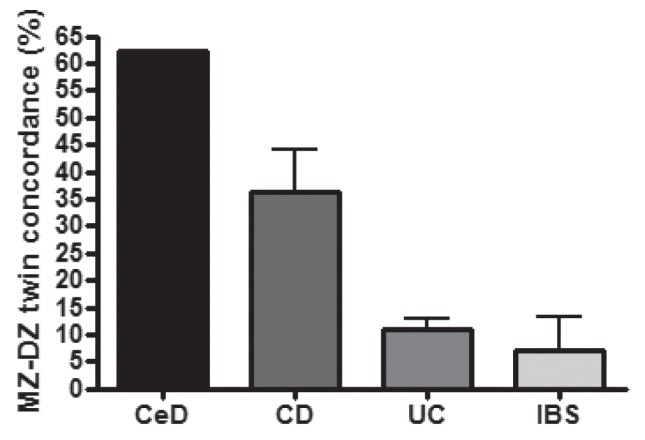
Estimation of relative genetic contribution to common gastrointestinal diseases based on monozygotic (MZ) and dizygotic (DZ) twin concordance rates. CeD Celiac disease; CD Crohn disease; IBS Irritable bowel syndrome; UC Ulcerative colitis
There is also diverse genetic susceptibility to IBD in different ethnic backgrounds in spite of similar disease phenotypes (14–17). This genetic diversity suggests that common changes of industrialization inducing nongenetic modification in genetically vulnerable hosts may be more important than the genetic predisposition itself toward inducing disease. The increased incidence of IBD in populations migrating from low incidence to high incidence areas of the world (18) also supports this latter conclusion.
EPIGENETICS AND IBD
The term ‘epigenetics’ was introduced by Conrad Hal Waddington (19) to describe all physiological processes that bridge genetics to individual phenotypes. Over time, the field has evolved to study biological mechanisms, which are mitotically (during the lifetime of one individual) heritable and can modulate pretranslational gene expression independently of the genetic code (20). In spite of the explosive expansion of epigenetic research, it remains debatable whether epigenetic mechanisms control transcription or if transcriptional velocity coordinates the establishment of epigenetic modifications, which then can firmly set the level of gene expression at select genomic loci (21).
The most stable epigenetic alteration is the methylation of the fifth cytosine carbon (5-mC) at CpG dinucleotides (Figure 2A). This molecular modification at gene promoters generally correlates with transcriptional downregulation. However, there are multiple exceptions to this rule (21,22), and gene body methylation has a variable association with transcriptional activity. Intragenic DNA methylation most commonly positively correlates with the transcription of intermediately expressed genes (23). DNA methylation, which is critical for development and differentiation (24), is catalyzed by DNA methyltransferases dependent on dietary substrates and cofactors (25). More recently, it has been recognized that methylated cytosines can be hydroxymethylated (5-hmC) (26), leading to an added complexity of epigenetic regulation through cytosine modifications. Similar to methylation, hydroxymethylation of cytosines also appears to play an important role in mammalian development and differentiation (27).
Figure 2).
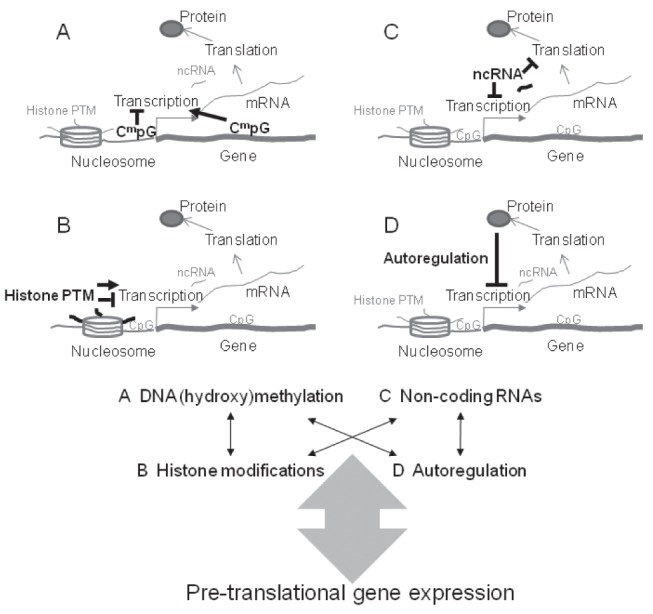
Upper panel Epigenetic mechanisms. A DNA methylation (and hydroxymethylation) can occur in cytosine-guanine dinucleotides (Cm pG). It usually negatively correlates ( ⊤ ) with transcription when present in gene promoter regions, but most commonly positively associates (→) with transcription when present within the gene. B Histone modification or post-translational modification (PTM) of histones correlates with gene transcription in a variable manner. C Noncoding RNAs (ncRNAs) can inhibit gene transcription and messenger RNA (mRNA) translation. They can also increase mRNA decay, thereby inducing pretranslational inhibition of gene expression. D Autoregulation provides a feedback loop for proteins to inhibit their own transcription. Lower panel Dynamic interaction between epigenetic mechanisms can provide a complex modulation of pretranslational gene expression
Another well studied epigenetic mechanism is the post-translational modification of histones (Figure 2B). Eight histone molecules comprise the nucleosomes around which DNA (147 base pairs) is coiled in the chromatin of eukaryotes (28). The aminoterminal tails of histones extend from the surface of the nucleosomes (29) and contribute to the higher organization of chromatin. Histone variants, nucleosome spacing and nuclear spatial positioning also contribute to the functional structure of chromatin (30). Post-translational modification of histones usually occurs at lysine, arginine, serine and threonine residues of the tails. There are currently 67 different types of histone alterations recognized (31), highlighting the intriguing complexity of this epigenetic mechanism. Perhaps the most clearly described positive correlation between histone modifications and gene expression is the acetylation of lysines in histone 3 and histone 4 tails. In the meantime, the exact mechanisms by which this transcriptional regulation is mediated remain unclear (32), but it frequently appears to occur in concert with decreased DNA methylation in less dense chromatin structures (euchromatin) where gene expression is usually more intense (as opposed to the case of densely packaged chromatin or heterochromatin).
Another field of epigenetics that has generated significant findings is the study of noncoding RNAs (ncRNAs, Figure 2C), which can be discriminated by their length (ie, long [lncRNA >200 bases], medium [mncRNA 50 to 200 bases] and short/small [sncRNA <50 bases; including microRNAs [miRNAs]) (33). These molecules can interfere with messenger RNA (mRNA) translation, induce mRNA degradation (mostly the sncRNAs) (34) or modulate gene expression by DNA-ncRNA, or ncRNA-protein (with protein complexes involved in gene expression regulation) interactions (33).
A less frequently discussed epigenetic mechanism is autoregulation, in which proteins (such as transcription factors) can bind their promoter region thereby leading to an autoregulatory feedback loop of their own transcription (Figure 2D) (35).
Importantly, all of the above epigenetic processes can interact with one another in a dynamic fashion, thereby providing an intricate complexity for nongenetically mediated molecular processes to modulate transcription (Figure 2, lower panel). One descriptive example for this intriguing intercalating epigenetic network is the expression of NF-E2-related factor 2 (NRF2), a master regulator of oxidative and xenobiotic stress responses (36). Both DNA methylation (37) and histone modification (38) modulate its transcription, whereas more than 30 different transcription factors regulated by NRF2 affect more than 60 miRNAs that can inhibit NRF2 expression (39).
The concept of epigenetic disease associations is erupting in all areas of medicine from neurology (40) to cardiology (41), even through anesthesiology (42). The potential importance for epigenetics in IBD was raised in 2000 (4); however, consecutive reviews in this area are just recently appearing (43). This delay in advancement is likely related to the inherent difficulties of studying the epigenetics of IBD:
IBD is specific to humans (because similar disorders are still markedly different even in nonhuman primates [44]) leading to significant compromise when examining the diseases in animal models.
Multiple biological networks, tissues and cell types are involved in IBD pathogenesis including the commensal microbiota, the gut mucosa and the host immune system (45,46). Secondary to the critical involvement of epigenetic changes in differentiation, a large number of these are cell-type specific, making it difficult to determine which tissue to study to delineate epigenetic associations of IBD. Furthermore, the isolation and manipulation of tissues may induce nonspecific epigenetic changes and mask those that are vital.
CD, and even UC, are likely not single diseases but common manifestations of distinct subtypes. Even the similar subtypes may arise from critical changes in different biological systems (47). Nevertheless, phenotypic characterization and appropriate separation of disease subtypes should ideally precede epigenetic studies in IBD.
Epigenetic changes are influenced by race, age, sex and location within tissues (such as within the colon) (48), and are likely to be modulated by drugs (48,49), and by the prolonged presence of disease and comorbidities. Therefore, the examination of treatment-naive patients at diagnosis with stringently selected controls is a feasible approach toward epigenetics in IBD.
Epigenetic changes can associate with genetic modification. Therefore, the most ideal population to study epigenetic disease associations is monozygotic (MZ) twins discordant for the disorder. However, healthy twin observations (50) predicted that a twin with a disease in discordant MZ pairs may be epigenetically less different from the healthy twin sibling at critical pathogenic loci, compared with unrelated healthy individuals. Therefore, pathogenic epigenetic changes may be more difficult to detect (depending on the sensitivity of the detection method) in MZ twins than in the general population.
Even if epigenetic differences between patients and controls are identified, cause or effect relationships must be determined, which is extremely difficult to perform in humans (see below).
One can imagine that it is exceedingly difficult to incorporate all of the above considerations into studies on IBD and epigenetics. In spite of such hurdles, increasing attempts have been made to delineate epigenetic associations with the disease group. Both targeted gene methylation (51) and DNA methylation microarray (52) assessments have detected numerous colonic mucosal associates of inflammation in IBD. As for peripheral blood, a recent study indicated epigenetic derrangement in peripheral blood leukocyte (PBL) DNA at genes linked to the T-helper (Th) cell 17 pathway in association with adult ileal CD (53). However, a large number of the putative associations coincided with T cell subset-specific DNA methylation variation, indicating that the results may have been confounded by disease-specific white blood cell composition variation (Kellermayer R, Inflamm Bowel Dis, In press). Isolation of specific cell subsets of PBLs may lead to further advancement in the future because interferon-gamma methylation in isolated peripheral T cells correlated with IBD subgroups, for example (54). It is likely secondary to the lack of select PBL subset isolation that we have not been able to determine DNA methylation correlates of IBD, either from PBLs, or from peripheral blood mononuclear cell DNA, when studying treatment-naive pediatric cases and adults (55).
There are some indications that histone modifications (43,56) and miRNAs (57–59) may contribute to the development, progression and/or maintenance of IBD. However, only DNA methylation has been shown to be stably transmitted through repetitive cell divisions (30,60), thereby having the capacity to permanently convey epigenetic information during the lifetime of an individual. Additionally, only DNA methylation has been described to directly communicate environmental exposures to phenotypic outcome in mammals (61). Therefore, we will further focus on DNA methylation with regard to the epigenetic aspects of the developmental origins of IBD.
ENVIRONMENTAL ORIGINS OF IBD
The developmental origins of disease hypothesis postulates that transient environmental exposures can induce critical changes in biological structures during finite periods of development, thereby modifying susceptibility to disorders later in life. The hypothesis has different implications for disease origins in the developing (62) versus the developed world (63).
Multiple characteristics of the tremendously changing industrialized environment have been targeted as potential causes for the rising incidence of IBD over the past five to six decades. Among these are pollution (64), refrigeration (65), increased hygiene (66), decreased infection with pathogenic organisms (67) and increased consumption of total fats, omega-6 fatty acids and meat, with a decreased intake of fruits, vegetables and fibre (68). The single prospective nutritional study linked omega-6 fatty acid consumption to the development of UC (69). In the meantime, clear results from human epidemiological studies (even if prospective and well controlled) are very difficult to obtain for obvious ethical and technical reasons (70). Therefore, the timing and the nature of environmental factors critical to IBD development have remained largely unknown, as have the molecular mechanisms that may communicate the environmental effects toward disease induction.
Observations of human migration would indicate that the peri-conceptual, prenatal and early postnatal developmental periods may be more important with regard to the developmental origins of IBD. This conclusion is based on studies showing that it is the second generation of immigrants from low-incidence countries to high-incidence countries of IBD who develop a similar (71) – or even augmented (72,73) – disease distribution compared with the endemic population. However, it has also been observed that transient exposures to higher levels of industrialization even in young adulthood (after 18 years of age) may increase one’s chances of developing IBD or, at least, UC (74).
According to our current knowledge, biological (including epigenetic) plasticity is higher during periconceptual and prenatal development than postnatally secondary to the increased number of molecular processes in flux (25). Therefore, it is likely that prenatal environmental exposures are more important in the etiology of common diseases, such as IBD, although these present later in life (Figure 3A). However, environmental influences can hypothetically trigger critical pathogenic changes at any time within an individual before the onset of disease, which can occur during a broad age range from young childhood to adulthood in the case of IBD. To further complicate the question of ‘nature (ie, genetic origins) versus nurture (ie, nongenetic origins)’ in the disease group, very-early onset IBD appears to present a subgroup in which genetic changes may play a more important role than in adults (75). Nevertheless, the epidemiology of IBD (discussed above) strongly supports the importance of ‘nurture’ in the etiology of most cases. It should also be noted that nongenetic factors can be important modulators even in clearly monogenic gastrointestinal disorders (such as Alagille syndrome [76,77]), the importance of which increase through genetically less clearly defined diseases (such as CeD [10]) to common multietiological illnesses (such as IBD and IBS) (Figure 3B).
Figure 3).
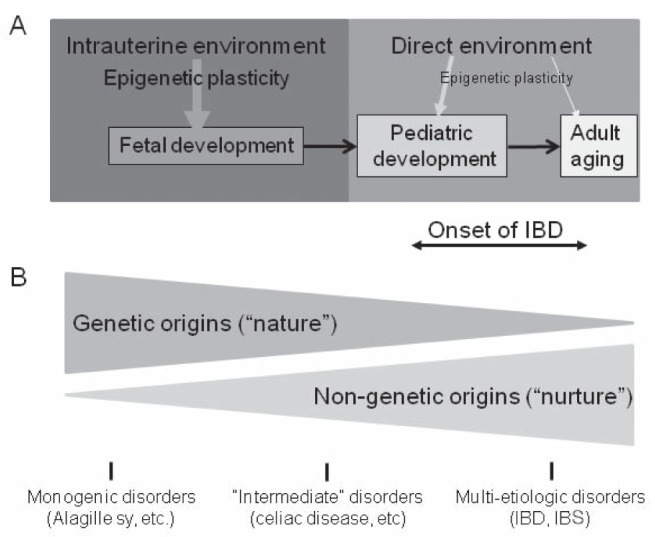
Developmental plasticity, epigenetics and inflammatory bowel diseases (IBDs). Upper panel Developmental plasticity is higher during fetal development secondary to the massive molecular processes (intense cell division and differentiation) in flux. Therefore, environmental influences can significantly impact the fetus (thicker arrow) through the involvement of epigenetic mechanisms. Developmental plasticity progressively decreases postnatally (indicated by thinner arrows for environmental influences affecting pediatric development and adult aging). IBD presents in children (25%) and in young adults. Nevertheless, environmentally induced fetal changes are likely to contribute more to the onset of the disorders than postnatal modifications secondary to this decline in developmental (including epigenetic) plasticity. Lower panel Nongenetic mechanisms, which can be environmentally more labile than the genetic code, contribute more to common gastrointestinal disorders such as IBD and irritable bowel syndrome (IBS) than celiac disease or monogenic gastrointestinal diseases
DNA METHYLATION AND THE ENVIRONMENTAL ORIGINS OF IBD
As detailed earlier, DNA methylation is the only molecular process that has been clearly shown to communicate transient nutritional influences to definite phenotype modification in mammals. The first example of this nutritional imprinting to occur was described in the viable yellow Agouti (Avy) mouse (78). The effect of supplementing a dam diet with methyl-donor micronutrients on offspring Agouti gene methylation was studied. Methyl-donor micronutrients (MDs) feed into the mammalian one-carbon pool, thereby increasing methyl-group abundance for DNA methyl-transferases to catalyze DNA methylation. Maternal MD supplementation increased offspring methylation at Agouti, which correlated with decreased gene expression and, consequently, more brown-coloured offspring than yellow. A handful of similarly behaving mammalian genomic loci have been identified (61), and some observations indicate that such nutritionally sensitive epigenetic modifications are also present in humans (79,80). Other examples of human and nonhuman mammalian epigenomic plasticity have been clearly demonstrated (61,63); however, the mechanism for the establishment remains unknown.
Clear conclusions with regard to epigenetic disease etiology must be made prospectively with the use of transient environmental exposures, and the critical tissues must be examined in the exposed subjects. The execution of such experiments in humans to unravel the environmental epigenetic origins of IBD is exceedingly difficult. Not surprisingly, only limited animal model examples currently exist to support this paradigm.
The difficulty of determining the critically affected tissue by environmental exposures presents a challenge even in animal models of IBD because the diseases are believed to arise from an uncontrolled immune response against intestinal microbes or microbial components that is transmitted by the intestinal mucosa (45,46). Therefore, a systems biological approach to disease pathogenesis is feasible. Indeed, concomitant and potentially interactive epigenetic and microbiota changes can occur in the background of genetic predisposition to colitis in mammals, thereby modulating predilection toward intestinal inflammation (81). Consistent with the systems biological approach, one must consider environmental influences to induce epigenetic changes relevant for IBD differently during various stages of mammalian development. Transuteral, transplacental and/or amniotic transmission communicates maternal exposures toward the fetus in lack of a commensal microbiota during prenatal development (Figure 4A). There is a dramatic change during delivery with respect to these interactions because direct environmental communication is established and progressive generation of the gut microbiota ensues (Figure 4B). Therefore, the interactive communication among the environment, the intestinal microbiota and the epigenome of the various host systems involved in IBD must be considered postnatally.
Figure 4).
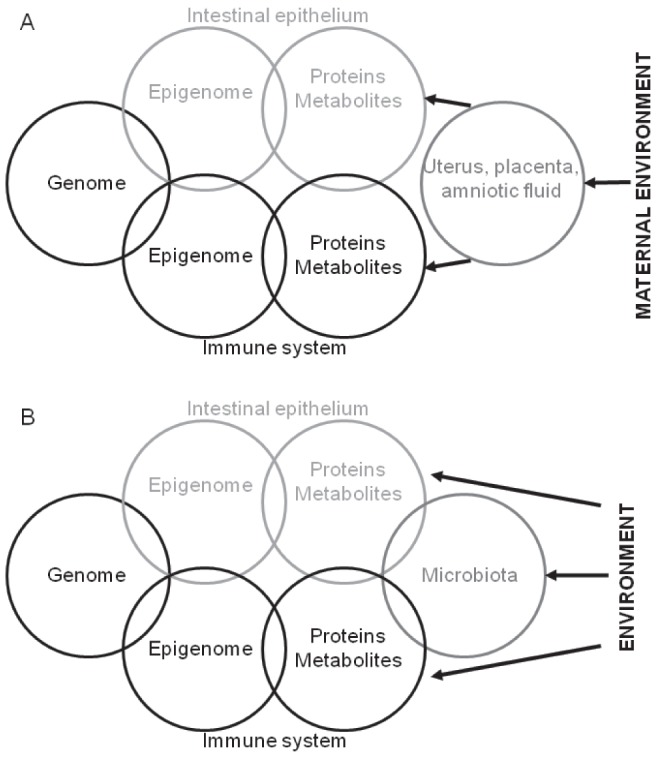
Prenatal (A) and postnatal (B) environmental influences can impact the interactive biological networks (genome and epigenome of the intestines and the associated immune system) involved in inflammatory bowel disease. The uterus, placenta, and/or amniotic fluid communicate maternal exposures toward the fetus in lack of a commensal microbiota during prenatal development. However, postnatally, environmental exposures directly affect the interactive communication between the intestinal microbiota and the epigenome of the various host systems involved in inflammatory bowel disease
Epigenetic plasticity extends beyond infancy in the colonic mucosa of mice and it may be relevant for modulating the propensity toward colitis (82). Metabolite profiling and drug exposures have certainly indicated the role of DNA methylation in mucosal inflammation (83). Nevertheless, only prenatal and infantile exposure to the same methyl-donor supplementation – as used in the Avy mouse – augmented murine colitis susceptibility, but pediatric supplementation did not (84). This maternally transmitted dietary exposure induced colonic mucosal DNA methylation and microbiota changes supporting the dynamic interaction between developmental nutritional epigenetics, microbes, and mammalian colitis. The micronutrient exposure-induced microbiota variation appeared to be independent of the maternally inherited microbiota. This observation suggests that prenatal developmental programming of colonic mucosal and immunological maturation imprints/programs postnatal microbiota shifts in the intestinal mucosa (85). Observations in sheep sustain the importance of maternally transmitted nutritional exposures (maternal obesity) modulating colitis susceptibility in lambs (86) and in young adult rats (maternal diet with varied fatty acid composition) (87); however, the epigenetic aspects of these interventions were not examined.
While the above-mentioned findings highlight the potential importance of prenatal biological/epigenetic plasticity in the etiology of human IBD, other results suggest that early postnatal exposures are similarly important. Neonatal stress has been observed to increase susceptibility toward colitis in adult mice in association with hypothalamic gene expression changes; however, epigenetic modifications were not studied (88). The importance of microbial composition changes during infancy has been observed in mammalian models with respect to mucosal immune responses (89) and colitis susceptibility (90). The oral and gut mucosa can react to microbial changes by epigenetic modification at select genes relevant for intestinal immunomodulation such as TLR4 (91), hBD2 (the gene of the antimicrobial peptide human β-defensin 2), CC chemokine ligand 20, and others (92). Such epigenetic modification may be dynamic and dependent on the presence of specific microbes (93). However, very recently, infantile exposure to specific pathogen-free microbiota in germ-free mice has been shown to decrease the methylation, hydroxymethylation and gene expression of Cxcl16 (the ligand of chemokine receptor Cxcr6) in the colon and lung, whereas later (at five weeks of age [ie, late pediatric development in mice]) exposure to the microbiota did not (94). Cxcl16 was critical for the accumulation of invariant natural killer cells in the tissues and for sensitization to oxazolone-induced colitis. Infantile pharmacological interventions with inhibitors and activators of DNA methylation successfully modulated the methylation and expression of Cxcl16. Unfortunately, the mechanism by which DNA methylation was communicated between the microbiota and Cxcl16 could not be determined. Nevertheless, this work provides the first evidence for microbial influences to induce epigenetic changes relevant for mammalian colitis during a specific period of postnatal development.
CONCLUSIONS.
The epidemiology of IBD and increasing number of animal model observations support the relevance of the environmental origins hypothesis in the pathogenesis of the disorders. Epigenetic mechanisms, such as DNA methylation, are likely to be key molecular processes for communicating environmental exposures toward IBD susceptibility in humans. A paradigm shift that includes these perspectives could aid the establishment of novel preventative (ie, elimination of critical epigenetically active environmental exposures), diagnostic (ie, IBD subset specific epigenetic changes) and therapeutic (ie, locus specific inhibition or activation of epigenetic changes) measures for IBD.
REFERENCES
- 1.Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54. doi: 10.1053/j.gastro.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140:1785–94. doi: 10.1053/j.gastro.2011.01.055. [DOI] [PubMed] [Google Scholar]
- 3.Halfvarson J. Genetics in twins with Crohn’s disease: Less pronounced than previously believed? Inflamm Bowel Dis. 2011;17:6–12. doi: 10.1002/ibd.21295. [DOI] [PubMed] [Google Scholar]
- 4.Petronis A, Petroniene R. Epigenetics of inflammatory bowel disease. Gut. 2000;47:302–6. doi: 10.1136/gut.47.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spehlmann ME, Begun AZ, Burghardt J, Lepage P, Raedler A, Schreiber S. Epidemiology of inflammatory bowel disease in a German twin cohort: Results of a nationwide study. Inflamm Bowel Dis. 2008;14:968–76. doi: 10.1002/ibd.20380. [DOI] [PubMed] [Google Scholar]
- 6.Jess T, Riis L, Jespersgaard C, et al. Disease concordance, zygosity, and NOD2/CARD15 status: Follow-up of a population-based cohort of Danish twins with inflammatory bowel disease. Am J Gastroenterol. 2005;100:2486–92. doi: 10.1111/j.1572-0241.2005.00224.x. [DOI] [PubMed] [Google Scholar]
- 7.Nistico L, Fagnani C, Coto I, et al. Concordance, disease progression, and heritability of coeliac disease in Italian twins. Gut. 2006;55:803–8. doi: 10.1136/gut.2005.083964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bengtson MB, Ronning T, Vatn MH, Harris JR. Irritable bowel syndrome in twins: Genes and environment. Gut. 2006;55:1754–9. doi: 10.1136/gut.2006.097287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mohammed I, Cherkas LF, Riley SA, Spector TD, Trudgill NJ. Genetic influences in irritable bowel syndrome: A twin study. Am J Gastroenterol. 2005;100:1340–4. doi: 10.1111/j.1572-0241.2005.41700.x. [DOI] [PubMed] [Google Scholar]
- 10.Plaza-Izurieta L, Castellanos-Rubio A, Irastorza I, et al. Revisiting genome wide association studies (GWAS) in coeliac disease: Replication study in Spanish population and expression analysis of candidate genes. J Med Genet. 2011;48:493–6. doi: 10.1136/jmg.2011.089714. [DOI] [PubMed] [Google Scholar]
- 11.Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–25. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson CA, Boucher G, Lees CW, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43:246–52. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saito YA, Mitra N, Mayer EA. Genetic approaches to functional gastrointestinal disorders. Gastroenterology. 2010;138:1276–85. doi: 10.1053/j.gastro.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo QS, Xia B, Jiang Y, Qu Y, Li J. NOD2 3020insC frameshift mutation is not associated with inflammatory bowel disease in Chinese patients of Han nationality. World J Gastroenterol. 2004;10:1069–71. doi: 10.3748/wjg.v10.i7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inoue N, Tamura K, Kinouchi Y, et al. Lack of common NOD2 variants in Japanese patients with Crohn’s disease. Gastroenterology. 2002;123:86–91. doi: 10.1053/gast.2002.34155. [DOI] [PubMed] [Google Scholar]
- 16.Ozen SC, Dagli U, Kilic MY, et al. NOD2/CARD15, NOD1/CARD4, and ICAM-1 gene polymorphisms in Turkish patients with inflammatory bowel disease. J Gastroenterol. 2006;41:304–10. doi: 10.1007/s00535-005-1780-z. [DOI] [PubMed] [Google Scholar]
- 17.Juyal G, Prasad P, Senapati S, et al. An investigation of genome-wide studies reported susceptibility loci for ulcerative colitis shows limited replication in north Indians. PLoS ONE. 2011;6:e16565. doi: 10.1371/journal.pone.0016565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams CN. Does the incidence of IBD increase when persons move from a low- to a high-risk area? Inflamm Bowel Dis. 2008;14(Suppl 2):S41–2. doi: 10.1002/ibd.20562. [DOI] [PubMed] [Google Scholar]
- 19.Van Speybroeck L. From epigenesis to epigenetics: The case of C. H. Waddington. Ann N Y Acad Sci. 2002;981:61–81. [PubMed] [Google Scholar]
- 20.Jaenisch R, Bird A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 21.Waterland RA, Kellermayer R, Rached MT, et al. Epigenomic profiling indicates a role for DNA methylation in early postnatal liver development. Hum Mol Genet. 2009;18:3026–38. doi: 10.1093/hmg/ddp241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rishi V, Bhattacharya P, Chatterjee R, et al. CpG methylation of half-CRE sequences creates C/EBPalpha binding sites that activate some tissue-specific genes. Proc Natl Acad Sci USA. 2010;107:20311–6. doi: 10.1073/pnas.1008688107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3:462–74. doi: 10.18632/oncotarget.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–26. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 25.Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr. 2007;27:363–88. doi: 10.1146/annurev.nutr.27.061406.093705. [DOI] [PubMed] [Google Scholar]
- 26.Ficz G, Branco MR, Seisenberger S, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 27.Orr BA, Haffner MC, Nelson WG, Yegnasubramanian S, Eberhart CG. Decreased 5-hydroxymethylcytosine is associated with neural progenitor phenotype in normal brain and shorter survival in malignant glioma. PLoS ONE. 2012;7:e41036. doi: 10.1371/journal.pone.0041036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnstone RW. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287–99. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 29.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 30.Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet. 2010;11:285–96. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan M, Luo H, Lee S, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–28. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gardner KE, Allis CD, Strahl BD. Operating on chromatin, a colorful language where context matters. J Mol Biol. 2011;409:36–46. doi: 10.1016/j.jmb.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nie L, Wu HJ, Hsu JM, et al. Long non-coding RNAs: Versatile master regulators of gene expression and crucial players in cancer. Am J Transl Res. 2012;4:127–50. [PMC free article] [PubMed] [Google Scholar]
- 34.Aalto AP, Pasquinelli AE. Small non-coding RNAs mount a silent revolution in gene expression. Curr Opin Cell Biol. 2012;24:333–40. doi: 10.1016/j.ceb.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murugan R, Kreiman G. On the minimization of fluctuations in the response times of autoregulatory gene networks. Biophys J. 2011;101:1297–306. doi: 10.1016/j.bpj.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci USA. 1994;91:9926–30. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khor TO, Huang Y, Wu TY, Shu L, Lee J, Kong AN. Pharmacodynamics of curcumin as DNA hypomethylation agent in restoring the expression of Nrf2 via promoter CpGs demethylation. Biochem Pharmacol. 2011;82:1073–8. doi: 10.1016/j.bcp.2011.07.065. [DOI] [PubMed] [Google Scholar]
- 38.Wang B, Zhu X, Kim Y, et al. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic Biol Med. 2012;52:928–36. doi: 10.1016/j.freeradbiomed.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papp D, Lenti K, Modos D, et al. The NRF2-related interactome and regulome contain multifunctional proteins and fine-tuned autoregulatory loops. FEBS Lett. 2012;586:1795–802. doi: 10.1016/j.febslet.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dauncey MJ. Recent advances in nutrition, genes and brain health. Proc Nutr Soc. 2012:1–11. doi: 10.1017/S0029665112000237. [DOI] [PubMed] [Google Scholar]
- 41.Schnabel RB, Baccarelli A, Lin H, Ellinor PT, Benjamin EJ. Next steps in cardiovascular disease genomic research – sequencing, epigenetics, and transcriptomics. Clin Chem. 2012;58:113–26. doi: 10.1373/clinchem.2011.170423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Naguib M, Bie B, Ting AH. Fundamental concepts of epigenetics for consideration in anesthesiology. Curr Opin Anaesthesiol. 2012;25:434–43. doi: 10.1097/ACO.0b013e3283556211. [DOI] [PubMed] [Google Scholar]
- 43.Scarpa M, Stylianou E. Epigenetics: Concepts and relevance to IBD pathogenesis. Inflamm Bowel Dis. 2012;18:1982–96. doi: 10.1002/ibd.22934. [DOI] [PubMed] [Google Scholar]
- 44.Sonnenberg A, Melton SD, Genta RM, Lewis AD. Absence of focally enhanced gastritis in macaques with idiopathic colitis. Inflamm Bowel Dis. 2011;12:2456–61. doi: 10.1002/ibd.21696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sartor RB. Key questions to guide a better understanding of host-commensal microbiota interactions in intestinal inflammation. Mucosal Immunol. 2011;4:127–32. doi: 10.1038/mi.2010.87. [DOI] [PubMed] [Google Scholar]
- 46.Kellermayer R. Genetic drift. “Omics” as the filtering gateway between environment and phenotype: The inflammatory bowel diseases example. Am J Med Genet A. 2010;152A:3022–5. doi: 10.1002/ajmg.a.33726. [DOI] [PubMed] [Google Scholar]
- 47.Nagy-Szakal D, Kellermayer R. The remarkable capacity for gut microbial and host interactions. Gut Microbes. 2011;2:178–82. doi: 10.4161/gmic.2.3.16107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wallace K, Grau MV, Levine AJ, et al. Association between folate levels and CpG Island hypermethylation in normal colorectal mucosa. Cancer Prev Res (Phila) 2010;3:1552–64. doi: 10.1158/1940-6207.CAPR-10-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan B, O’Connor TR, Wang Y. 6-Thioguanine and S-methylthioguanine are mutagenic in human cells. ACS Chem Biol. 2010;5:1021–7. doi: 10.1021/cb100214b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaminsky ZA, Tang T, Wang SC, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41:240–5. doi: 10.1038/ng.286. [DOI] [PubMed] [Google Scholar]
- 51.Saito S, Kato J, Hiraoka S, et al. DNA methylation of colon mucosa in ulcerative colitis patients: Correlation with inflammatory status. Inflamm Bowel Dis. 2011;9:1955–65. doi: 10.1002/ibd.21573. [DOI] [PubMed] [Google Scholar]
- 52.Lin Z, Hegarty JP, Cappel JA, et al. Identification of disease-associated DNA methylation in intestinal tissues from patients with inflammatory bowel disease. Clin Genet. 2011;80:59–67. doi: 10.1111/j.1399-0004.2010.01546.x. [DOI] [PubMed] [Google Scholar]
- 53.Nimmo ER, Prendergast JG, Aldhous MC, et al. Genome-wide methylation profiling in Crohn’s disease identifies altered epigenetic regulation of key host defense mechanisms including the Th17 pathway. Inflamm Bowel Dis. 2012;18:889–99. doi: 10.1002/ibd.21912. [DOI] [PubMed] [Google Scholar]
- 54.Gonsky R, Deem RL, Landers CJ, et al. Distinct IFNG methylation in a subset of ulcerative colitis patients based on reactivity to microbial antigens. Inflamm Bowel Dis. 2011;17:171–8. doi: 10.1002/ibd.21352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harris RA, Nagy-Szakal D, Pedersen N, et al. Genome-wide peripheral blood leukocyte DNA methylation microarrays identified a single association with inflammatory bowel diseases. Inflamm Bowel Dis. 2012 Mar 29; doi: 10.1002/ibd.22956. (E-pub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosen MJ, Frey MR, Washington MK, et al. STAT6 activation in ulcerative colitis: A new target for prevention of IL-13-induced colon epithelial cell dysfunction. Inflamm Bowel Dis. 2011;17:2224–34. doi: 10.1002/ibd.21628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu F, Guo NJ, Tian H, et al. Peripheral blood microRNAs distinguish active ulcerative colitis and Crohn’s disease. Inflamm Bowel Dis. 2011;17:241–50. doi: 10.1002/ibd.21450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen Y, Ge W, Xu L, et al. miR-200b is involved in intestinal fibrosis of Crohn’s disease. Int J Mol Med. 2012;29:601–6. doi: 10.3892/ijmm.2012.894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zahm AM, Thayu M, Hand NJ, Horner A, Leonard MB, Friedman JR. Circulating MicroRNA is a biomarker of pediatric Crohn disease. J Pediatr Gastroenterol Nutr. 2011;53:26–33. doi: 10.1097/MPG.0b013e31822200cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wigler M, Levy D, Perucho M. The somatic replication of DNA methylation. Cell. 1981;24:33–40. doi: 10.1016/0092-8674(81)90498-0. [DOI] [PubMed] [Google Scholar]
- 61.Faulk C, Dolinoy DC. Timing is everything: The when and how of environmentally induced changes in the epigenome of animals. Epigenetics. 2011;6:791–7. doi: 10.4161/epi.6.7.16209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uauy R, Kain J, Corvalan C. How can the Developmental Origins of Health and Disease (DOHaD) hypothesis contribute to improving health in developing countries? Am J Clin Nutr. 2011;94:1759S–64S. doi: 10.3945/ajcn.110.000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wadhwa PD, Buss C, Entringer S, Swanson JM. Developmental origins of health and disease: Brief history of the approach and current focus on epigenetic mechanisms. Semin Reprod Med. 2009;27:358–68. doi: 10.1055/s-0029-1237424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beamish LA, Osornio-Vargas AR, Wine E. Air pollution: An environmental factor contributing to intestinal disease. J Crohns Colitis. 2011;5:279–86. doi: 10.1016/j.crohns.2011.02.017. [DOI] [PubMed] [Google Scholar]
- 65.Malekzadeh F, Alberti C, Nouraei M, et al. Crohn’s disease and early exposure to domestic refrigeration. PLoS One. 2009;4:e4288. doi: 10.1371/journal.pone.0004288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gent AE, Hellier MD, Grace RH, Swarbrick ET, Coggon D. Inflammatory bowel disease and domestic hygiene in infancy. Lancet. 1994;343:766–7. doi: 10.1016/s0140-6736(94)91841-4. [DOI] [PubMed] [Google Scholar]
- 67.Elliott DE, Weinstock JV. Helminth-host immunological interactions: Prevention and control of immune-mediated diseases. Ann N Y Acad Sci. 2012;1247:83–96. doi: 10.1111/j.1749-6632.2011.06292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hou JK, Abraham B, El-Serag H. Dietary intake and risk of developing inflammatory bowel disease: A systematic review of the literature. Am J Gastroenterol. 2011;106:563–73. doi: 10.1038/ajg.2011.44. [DOI] [PubMed] [Google Scholar]
- 69.Tjonneland A, Overvad K, Bergmann MM, et al. Linoleic acid, a dietary n-6 polyunsaturated fatty acid, and the aetiology of ulcerative colitis: A nested case-control study within a European prospective cohort study. Gut. 2009;58:1606–11. doi: 10.1136/gut.2008.169078. [DOI] [PubMed] [Google Scholar]
- 70.Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–7. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- 71.Carr I, Mayberry JF. The effects of migration on ulcerative colitis: A three-year prospective study among Europeans and first- and second- generation South Asians in Leicester (1991–1994) Am J Gastroenterol. 1999;94:2918–22. doi: 10.1111/j.1572-0241.1999.01438.x. [DOI] [PubMed] [Google Scholar]
- 72.Li X, Sundquist J, Hemminki K, Sundquist K. Risk of inflammatory bowel disease in first- and second-generation immigrants in Sweden: A nationwide follow-up study. Inflamm Bowel Dis. 2011;17:1784–91. doi: 10.1002/ibd.21535. [DOI] [PubMed] [Google Scholar]
- 73.Pinsk V, Lemberg DA, Grewal K, Barker CC, Schreiber RA, Jacobson K. Inflammatory bowel disease in the South Asian pediatric population of British Columbia. Am J Gastroenterol. 2007;102:1077–83. doi: 10.1111/j.1572-0241.2007.01124.x. [DOI] [PubMed] [Google Scholar]
- 74.Barreiro-de Acosta M, Alvarez Castro A, Souto R, Iglesias M, Lorenzo A, Dominguez-Munoz JE. Emigration to western industrialized countries: A risk factor for developing inflammatory bowel disease. J Crohns Colitis. 2011;5:566–9. doi: 10.1016/j.crohns.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 75.Muise AM, Snapper SB, Kugathasan S. The age of gene discovery in very early onset inflammatory bowel disease. Gastroenterology. 2012;143:285–8. doi: 10.1053/j.gastro.2012.06.025. [DOI] [PubMed] [Google Scholar]
- 76.Kamath BM, Krantz ID, Spinner NB, Heubi JE, Piccoli DA. Monozygotic twins with a severe form of Alagille syndrome and phenotypic discordance. Am J Med Genet. 2002;112:194–7. doi: 10.1002/ajmg.10610. [DOI] [PubMed] [Google Scholar]
- 77.Kellermayer R. Genetic drift. Physiologic noise obscures genotype-phenotype correlations. Am J Med Genet A. 2007;143A:1306–7. doi: 10.1002/ajmg.a.31825. [DOI] [PubMed] [Google Scholar]
- 78.Cooney CA, Dave AA, Wolff GL. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J Nutr. 2002;132:2393S–400S. doi: 10.1093/jn/132.8.2393S. [DOI] [PubMed] [Google Scholar]
- 79.Waterland RA, Kellermayer R, Laritsky E, et al. Season of conception in rural Gambia affects DNA methylation at putative human metastable epialleles. PLoS Genet. 2010;6:e1001252. doi: 10.1371/journal.pgen.1001252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Khulan B, Cooper WN, Skinner BM, et al. Periconceptional maternal micronutrient supplementation is associated with widespread gender related changes in the epigenome: A study of a unique resource in the Gambia. Hum Mol Genet. 2012;21:2086–101. doi: 10.1093/hmg/dds026. [DOI] [PubMed] [Google Scholar]
- 81.Nagy-Szakal D, Kellermayer R. The remarkable capacity for gut microbial and host interactions. Gut Microbes. 2011;2:178–82. doi: 10.4161/gmic.2.3.16107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kellermayer R, Balasa A, Zhang W, et al. Epigenetic maturation in colonic mucosa continues beyond infancy in mice. Hum Mol Genet. 2010;19:2168–76. doi: 10.1093/hmg/ddq095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kominsky DJ, Keely S, MacManus CF, et al. An endogenously anti-inflammatory role for methylation in mucosal inflammation identified through metabolite profiling. J Immunol. 2011;186:6505–14. doi: 10.4049/jimmunol.1002805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schaible TD, Harris RA, Dowd SE, Smith CW, Kellermayer R. Maternal methyl-donor supplementation induces prolonged murine offspring colitis susceptibility in association with mucosal epigenetic and microbiomic changes. Hum Mol Genet. 2011;20:1687–96. doi: 10.1093/hmg/ddr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nagy-Szakal D, Ross MC, Dowd SE, et al. Maternal micronutrients can modify colonic mucosal microbiota maturation in murine offspring. Gut Microbes. 2012;3:426–33. doi: 10.4161/gmic.20697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yan X, Huang Y, Wang H, et al. Maternal obesity induces sustained inflammation in both fetal and offspring large intestine of sheep. Inflamm Bowel Dis. 2011;17:1513–22. doi: 10.1002/ibd.21539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Innis SM, Dai C, Wu X, Buchan AM, Jacobson K. Perinatal lipid nutrition alters early intestinal development and programs the response to experimental colitis in young adult rats. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1376–85. doi: 10.1152/ajpgi.00258.2010. [DOI] [PubMed] [Google Scholar]
- 88.Veenema AH, Reber SO, Selch S, Obermeier F, Neumann ID. Early life stress enhances the vulnerability to chronic psychosocial stress and experimental colitis in adult mice. Endocrinology. 2008;149:2727–36. doi: 10.1210/en.2007-1469. [DOI] [PubMed] [Google Scholar]
- 89.Mulder IE, Schmidt B, Stokes CR, et al. Environmentally-acquired bacteria influence microbial diversity and natural innate immune responses at gut surfaces. BMC Biol. 2009;7:79. doi: 10.1186/1741-7007-7-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Atarashi K, Tanoue T, Shima T, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–41. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Takahashi K, Sugi Y, Nakano K, et al. Epigenetic control of the host gene by commensal bacteria in large intestinal epithelial cells. J Biol Chem. 2011;286:35755–62. doi: 10.1074/jbc.M111.271007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yin L, Chung WO. Epigenetic regulation of human beta-defensin 2 and CC chemokine ligand 20 expression in gingival epithelial cells in response to oral bacteria. Mucosal Immunol. 2011;4:409–19. doi: 10.1038/mi.2010.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sepulveda AR, Yao Y, Yan W, et al. CpG methylation and reduced expression of O6-methylguanine DNA methyltransferase is associated with Helicobacter pylori infection. Gastroenterology. 2010;138:1836–44. doi: 10.1053/j.gastro.2009.12.042. [DOI] [PubMed] [Google Scholar]
- 94.Olszak T, An D, Zeissig S, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–93. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]