

Abstract
Progesterone receptor (PR) isoforms (PRA and PRB) are implicated in the progression of breast cancers frequently associated with imbalanced PRA/PRB expression ratio. Antiprogestins represent potential antitumorigenic agents for such hormone-dependent cancers. To investigate the mechanism(s) controlling PR isoforms degradation/stability in the context of agonist and antagonist ligands, we used endometrial and mammary cancer cells stably expressing PRA and/or PRB. We found that the antiprogestin RU486 inhibited the agonist-induced turnover of PR isoforms through active mechanism(s) involving distinct MAPK-dependent phosphorylations. p42/44 MAPK activity inhibited proteasome-mediated degradation of RU486-bound PRB but not PRA in both cell lines. Ligand-induced PRB turnover required neosynthesis of a mandatory down-regulating partner whose interaction/function is negatively controlled by p42/44 MAPK. Such regulation strongly influenced expression of various endogenous PRB target genes in a selective manner, supporting functional relevance of the mechanism. Interestingly, in contrast to PRB, PRA stability was specifically increased by MAPK kinase kinase 1-induced p38 MAPK activation. Selective inhibition of p42/p44 or p38 activity resulted in opposite variations of the PRA/PRB expression ratio. Moreover, MAPK-dependent PR isoforms stability was independent of PR serine-294 phosphorylation previously proposed as a major sensor of PR down-regulation. In sum, we demonstrate that MAPK-mediated cell signaling differentially controls PRA/PRB expression ratio at posttranslational level through ligand-sensitive processes. Imbalance in PRA/PRB ratio frequently associated with carcinogenesis might be a direct consequence of disorders in MAPK signaling that might switch cellular responses to hormonal stimuli and contribute towards pathogenesis.
Progesterone receptor (PR), a steroid-activated transcription factor, is an important pharmacological target for contraception, female reproductive disorders, as well as for hormone-dependent breast and uterine cancers. Alternative transcription of PR gene results in equal expression of two major isoforms PRA and PRB (1, 2). PRA lacks the 164 N-terminal amino acids, also called the B-upstream segment (BUS) present in PRB (3). Each isoform having distinct genomic targets (4) and exerting tissue-specific effects (5), PRA/PRB expression ratio is a key biological determinant selecting tissue responsiveness to hormone and growth factors stimuli. Neosynthesized PR is stabilized by interacting with heat shock protein 90-containing complexes (6). Upon ligand binding, PR dissociates from these chaperones and undergoes conformational changes leading to its homo- and heterodimerization and sequential interactions with transcriptional coregulators (coactivators and corepressors). Ligand also induces posttranslational modifications, notably phosphorylations, ubiquitination, and sumoylation, and regulates PR functions at multiple levels as well as its down-regulation via proteasomes (7–11). Beside alternative transcription of PR isoforms, only few studies reported the preferential regulation of one isoform at the posttranscriptional level (12). However, aberrant PRA/PRB expression is frequently observed in breast and endometrial cancers (2, 13), suggesting potential alterations in down-regulation mechanisms affecting PR isoforms stabilities via posttranslational modifications.
In PR, at least 14 phosphorylation sites are targeted by multiple kinases, mostly within serine-proline motifs in N-terminal domain affecting PR transcriptional activity and turnover (7, 14–17). Among these phosphorylation events, PRB serine-294 phosphorylation (pS294) (pS294-PRB) has been shown to act as an important sensor for growth factor inputs that affects PR function and plays a critical role in cross talk with growth factor signaling pathways (17, 18). Blocking of progestin-induced receptor turnover by proteasome inhibitors blocks PR transcriptional activities (9). The underlying mechanisms of this paradoxical link between PR stabilization and transcriptional inactivation are yet to be fully understood but likely involve direct coupling of proteasomes with transcriptional machinery as already demonstrated for estrogen receptor (19). RU486 (Mifepristone), a widely used PR antagonist, has been proposed for hormone-dependent breast cancer treatment (20). Although RU486 blocks PR transcriptional activity by favoring corepressors recruitment, it was found that PR turnover was highly reduced after RU486 treatment (8, 21, 22). Like progesterone, RU486 stimulates similar early cascade of events, including chaperone dissociation, dimerization, and posttranslational modifications, such as sumoylation (10) and phosphorylation (8, 22). Mutation of breast cancer 1, a PR-interacting protein, leads to deregulated PRA/PRB ratio, resulting in mammary tumorigenesis that was prevented by RU486 (23). It thus becomes of major importance to explore the mechanisms regulating posttranslational modifications of PR isoforms and their respective turnover.
In this study, we investigated the effects of RU486 on PR isoforms turnover in endometrial and mammary cancer cells stably expressing PRA or PRB or both. We report that, in contrast to other antagonists and progestin R5020, RU486 strongly inhibits PRB and PRA degradation. Further investigations revealed that down-regulations of PRB and PRA are negatively controlled by key phosphorylation events involving distinct MAPK, resulting in selective PR isoform stabilization. Furthermore, these phosphorylation events are differentially controlled by ligands and antagonize PRB degradation via proteasome. Our data support the existence of a switching mechanism differentially regulating PR isoform expression ratio via MAPK-dependent phosphorylations, which might have important consequences in progression of hormone-dependent cancers.
Results
Antagonist RU486 inhibits agonist-induced down-regulation of PRA and PRB
Both PR isoforms when coexpressed undergo agonist-induced degradation to similar extent (22). However, PRB is degraded much more rapidly as compared with PRA in cells expressing either of PR isoforms (24). Given that PR transcriptional activity is coupled to its proteasome-mediated down-regulation, we wondered whether antagonist RU486 that inhibits PR target gene transcription could impair agonist-induced PR protein degradation. To investigate the mechanisms controlling differential PR isoforms protein stability/degradation independently of transcriptional contributions from endogenous PR promoters, we used endometrial (Ishikawa) and mammary cancer cells (MDA-MB-231) stably expressing recombinant PRA or PRB under the control of same promoter (25, 26). In these models, PR isoform expression was comparable with that of endogenous expression levels detected in wild-type breast cancer cells T47D (Supplemental Fig. 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). As expected, in both cell types, agonist R5020 (10−8 m)-induced PRA- or PRB-mediated up-regulation of FK506 binding protein 5 (FKBP5) gene expression was abrogated by 100-fold excess of RU486 (10−6 m) (Fig. 1, lower panels). Under similar hormonal conditions, RU486 was found to abolish agonist-induced PRA or PRB turnover and led to both PR isoforms accumulation with an electrophoretic upshift, characteristic of phosphorylated PR species (Fig. 1, upper panels). Therefore, in both endometrial and mammary cancer cells, silencing of agonist-induced PR isoforms-mediated target gene transcription by RU486 is accompanied with PR isoforms accumulation through unknown mechanisms.
Fig. 1.

RU486 abrogates agonist-dependent PRA and PRB down-regulation and transcriptional activity. Ishikawa or MDA-MB-231 cells stably expressing PRA or PRB were treated either by vehicle or R5020 (10−8 m) alone or in combination with RU486 (10−6 m) during either 6 h for the analysis of FKBP5 gene transcripts by real time quantitative PCR (lower panel) or 24 h for immunoblot detection of PR isoforms using anti-PR antibody (upper panel). Statistical significance is shown for agonist-induced transactivation as compared with vehicle-treated cells (asterisks) or for comparison between agonist alone or with antagonist-treated cells (XX).
RU486 stabilizes pS294-PRB
To understand the mechanisms by which RU486 stabilizes PR isoforms, we first hypothesized that RU486 might inhibit agonist-induced PRB pS294, which has been described as a major signal for PRB turnover and hypertranscriptional activity (14). To test this possibility, Ishikawa PRB or Ishikawa PRA cells were treated by R5020 (10−8 m) alone or in combination with equal concentration of RU486 for 6 h or 14 h. pS294-PRB and total PRB levels (Fig. 2A) or pS130-PRA (equivalent residue of PRB S294) and total PRA levels (Supplemental Fig. 2) were analyzed by Western blotting. We found that RU486 was unable to inhibit the agonist-induced pS294-PRB (Fig. 2A) or pS130-PRA (Supplemental Fig. 2) after 6 h, whereas down-regulation of pS294-PRB in RU486-treated cells occurred to a much lesser extent than with agonist alone after 14 h (Fig. 2A). Moreover, RU486 markedly slowed down the agonist-induced degradation of PRB (Fig. 2A), indicating that RU486 did not inhibit agonist-induced pS294-PRB but instead strongly stabilized it. We next examined whether RU486 and ZK98299, another PR antagonist, could induce pS294-PRB and impact PRB turnover. After 6 h, pS294-PRB levels were higher accompanied by lack of PRB turnover in RU486-treated cells, as compared with R5020 (Fig. 2B, inset). In contrast, ZK98299, as expected (8, 22), resulted in a weak overall PRB phosphorylation (lack of electrophoretic upshift), including pS294-PRB, and still provoked intermediary PRB degradation (Fig. 2B, inset). This suggested that pS294-PRB turnover might be interrupted by RU486 binding. Therefore, we next compared R5020- or RU486-induced pS294-PRB kinetics in Ishikawa PRB cells under similar ligand concentration (10−8 m) (Fig. 2C). Quantification of electrophoretic bands (Fig. 2C, graphs) allowed analyzing the time course of ligand-induced pS294-PRB and PRB degradation. R5020 induced a robust early pS294-PRB (Fig. 2C, left graph) reaching a peak at 1 h and then decreased concomitantly to PRB degradation (Fig. 2C, left and middle graphs). RU486 also induced pS294-PRB but with slower kinetics reaching a plateau at 12–14 h, which remained stable thereafter (Fig. 2C, right graph), parallel to PRB accumulation profile (Fig. 2C, middle graph). As expected (22), analysis of PRA phosphorylation on S130 in Ishikawa PRA cells showed that PRA also undergoes agonist-induced pS130-PRA but with much slower kinetics and to a lesser extent as compared with PRB (Supplemental Fig. 3). Although agonist R5020 or antagonist RU486 induced pS294-PRB as early as 15 min, ligand-induced pS130-PRA is detectable only after 1 h of hormonal treatment (Supplemental Fig. 3). Although both R5020 and RU486 induced pS294-PRB (and pS130-PRA), only the agonist-bound PR isoform is signaled toward degradation, whereas antagonist-bound PR failed to undergo expected pS294- or pS130-driven PR isoform down-regulation. Therefore, we asked whether RU486-bound PRB might be insensitive to ubiquitination. Parental Ishikawa cells were transiently transfected with HA-tagged ubiquitin and PRB expression vectors, pretreated with proteasome inhibitor MG132, and incubated or not by ligands during 4 h. Immunoprecipitated PRB was analyzed by Western blotting using anti-HA antibody. RU486 markedly reduced basal PRB ubiquitination (Fig. 2D). Taken together, our results indicate that RU486, despite inducing pS294, stabilizes PRB in part by inhibiting ubiquitination processes. Thus, turnover of R5020- or RU486-bound PR isoform is inversely correlated, irrespective of the pS294 status.
Fig. 2.
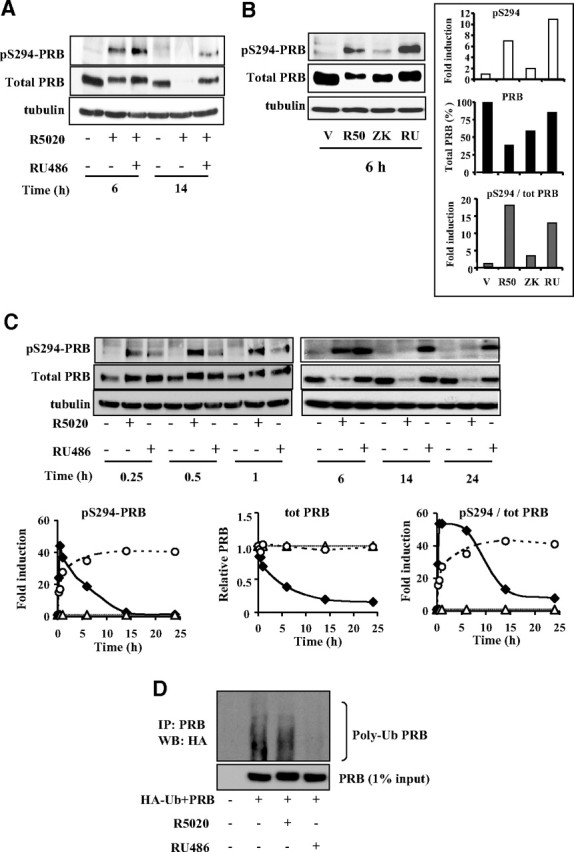
Unlike R5020 and ZK98299, RU486 induces stable PRB- pS294. A, Ishikawa PRB cells were treated by ligands as in Fig. 1 during 6 or 14 h, and whole cells extracts were immunoblotted using PRB serine 294 phospho-specific antibody, anti-PRB, or antitubulin antibody. B, Ishikawa PRB cells were treated with vehicle (V) or R5020 (R50) (10−8 m) or ZK98299 (ZK) (10−6 m) or RU486 (RU) (10−8 m) during 6 h, and whole cell extracts were immunoblotted as in A. Numerized band densities corresponding to pS294-PRB (upper inset) or total PRB (middle inset) are normalized to vehicle or tubulin controls and plotted as fold induction or percentage of total PRB in ligand-free condition. Ligand-induced pS294/PRB (lower inset) is presented as fold induction of vehicle-treated cells. C, Ishikawa PRB cells were treated without or with R5020 (10−8 m) or RU486 (10−8 m) during the indicated time periods, and whole cell extracts were immunoblotted as in A. pS294-PRB and PRB band densities were normalized to vehicle or tubulin controls and plotted as fold induction of ligand-free species for each time point (left and middle panels), and the corresponding ratio is shown in the right panel (white triangle, vehicle; black diamond, R5020; and white circle, RU486). D, Parental Ishikawa cells lacking PRB expression were transiently transfected with HA-ubiquitin and PRB expression vectors during 48 h, pretreated with MG132 (5 μm) during 30 min, and then incubated without or with R5020 (10−8 m) or RU486 (10−8 m) during 4 h. After PRB immunoprecipitation (IP) using monoclonal anti-PR antibody, ubiquitinated-PRB was analyzed by Western blotting (WB) using anti-HA antibody (upper panel). PRB levels corresponding to 1% input were detected by anti-PR antibody (lower panel).
Phosphorylated p42/p44 are pivotal for PRB but not PRA stability
MAPK were reported to enhance PRB transcriptional activity and turnover through PRB phosphorylation at S294 in the presence of agonist ligands (27). Therefore, we wondered whether PRB or PRA stabilization by RU486 could be related to alterations in p42/44 MAPK-dependent phosphorylation events. Ishikawa cells stably expressing either PRB or PRA were incubated with vehicle or R5020 or RU486 for 1, 6, or 24 h in the absence or presence of U0126, a specific MAPK kinase (MEK)1/2 inhibitor that prevents p42/44 MAPK phosphorylation (P-p42/44). PRB and its S294 phosphorylated moiety were examined to determine P-p42/44-dependent early and late events that might affect PR isoform down-regulating mechanisms. As expected, U0126 inhibited P-p42/44 to similar extent in both PRB and PRA cell lines (Fig. 3A). After 1 h, the agonist-induced as well as antagonist-induced pS294-PRB remained unchanged in the presence of U0126 (Fig. 3A, upper panels), indicating that an unknown kinase distinct from p42/44 targets S294, as previously suggested for agonist ligand (28, 29). Similar results were obtained for PRA and its pS130 species (Fig. 3, lower panels). Surprisingly, after 6 h, inhibition of P-p42/44 specifically triggered degradation of RU486-bound PRB with a parallel decrease in pS294-PRB without altering basal or agonist-bound PRB levels (Fig. 3A, middle panels). In sharp contrast, U0126 did not affect RU486-bound PRA level. At longer time period (24 h), although P-p42/44 inhibition further enhanced RU486-bound PRB degradation, small decrease in ligand-free as well as R5020-bound PRB was also observed (Fig. 3A, right panels), indicating that P-p42/44 also enhances the agonist-bound PRB stability but to a lesser extent as compared with RU486. Quantitative RT-PCR analysis showed that PRB mRNA levels were unchanged after U0126 treatment (Supplemental Fig. 4), indicating that p42/44 control PRB stability at posttranslational levels. Similar P-p42/44-dependent stabilization of RU486-bound PRB was observed when Ishikawa PRB cells were cultured in serum-free medium (Supplemental Fig. 5). Surprisingly, U0126 treatment did not affect basal or ligand-bound PRA levels even after 24 h, indicating that PRB but not PRA turnover is negatively controlled by P-p42/44 in a ligand-sensitive manner (Fig. 3A, right panels). To substantiate P-p42/44 as well as ligand specificity for PRB stabilization at shorter time points (6 h), Ishikawa PRB cells were exposed to increasing concentrations of U0126 under constant amounts of R5020 or RU486 (Fig. 3B). In contrast to vehicle or R5020 treatment, degradation of RU486-bound PRB (and pS294-PRB species) occurred as a function of P-p42/44 inhibition with a decrease in overall PRB upshift. This strongly indicates that RU486-induced PRB stabilization is controlled by p42/44 activity in a dose-dependent manner. These results demonstrate that RU486, when compared with R5020 or vehicle, strongly facilitates P-p42/44-dependent phosphorylation of PRB on a residue other than S294 resulting in slower PRB degradation.
Fig. 3.

Phosphorylated p42/44 MAPK stabilize PRB but not PRA in a ligand-dependent manner. A, Ishikawa PRB or PRA cells were pretreated with dimethylsulfoxide (DMSO) or U0126 (10 μm) during 30 min and then incubated without or with R5020 (10−8 m) or RU486 (10−8 m) during 1, 6, or 24 h. Whole cell extracts were immunoblotted using either phospho-specific (pS294-PRB, pS130-PRA) or anti-PR antibody (PRB, PRA). From the same immunoblot, either total p42/p44 or their phosphorylated species (P-p42/44) were analyzed using the corresponding antibodies. B, Ishikawa PRB cells were pretreated without or with U0126 (5, 10, or 20 μm) during 30 min and then treated without or with R5020 (10−8 m) or RU486 (10−8 m) during 6 h. Whole cell extracts were immunoblotted as in A.
P42/44 MAPK control proteasome-dependent turnover of ligand-bound PRB in endometrial and mammary cancer cells
To further analyze the p42/44-dependent mechanism of PRB stabilization, we asked whether this mechanism could be also functional in breast cancer cells. For this, MDA-MB-231 PRB cells were treated or not by RU486 and U0126 for 24 h. In contrast to Ishikawa cells, basal PRB level increased after inhibition of p42/44 activity (Fig. 4A). However, RU486-bound PRB was degraded after U0126 treatment as in Ishikawa cells, indicating that ligand-specific p42/44-dependent mechanism controlling PRB stability is conserved in both cell types.
Fig. 4.

P42/44 MAPK control proteasome-dependent turnover of ligand-bound PRB. A, MDA-MB-231 cells (MDA) stably expressing PRB were treated with vehicle or RU486 (10−8 m) during 24 h, and whole cell extracts were immunoblotted as in Fig. 3. B, MDA-MB-231-PRB or Ishikawa-PRB cells were pretreated with dimethylsulfoxide (DMSO) or U0126 (10 μm) during 30 min and then incubated without or with progesterone (P4) (10−8 m) during 24 h. Whole cell extracts were immunoblotted as in A. C, MDA-MB-231 PRB cells were pretreated with DMSO or U0126 (10 μm) during 30 min and then incubated with vehicle or R5020 (10−8 m) or RU486 (10−8 m) during 24 h. Immunofluorescence analysis was performed as described in Materials and Methods using anti-PR antibody, and images were obtained for an identical time exposure. D, Ishikawa PRB cells were pretreated without or with MG132 (5 μm) and/or U0126 (10 μm) during 30 min and then treated without or with R5020 (10−8 m) or RU486 (10−8 m) during 6 h. Immunoblot analysis was performed as in A. Cell lysates from the same RU486-treated cells were also immunoblotted for either GR detection using anti-GR antibody or tubulin as loading control (inset).
Our previous results in Fig. 3A showed that at delayed time points, P-p42/44 inhibition also accelerated R5020-bound PRB degradation. Therefore, we asked whether ligand-specific p42/44 control of PRB stability is relevant for the natural ligand progesterone known to induce slower PRB turnover than synthetic progestin R5020. As shown in Fig. 4B, progesterone-bound PRB degradation was enhanced by U0126 in both MDA and Ishikawa cells, thus indicating that p42/44 activity also slows down progesterone-induced PRB turnover.
To verify whether association of RU486 and U0126 had provoked any change in subcellular localization of PRB that might intervene in PRB stabilization, immunofluorescence studies in MDA-MB-231 PRB cells demonstrated that PRB remained mainly localized in the nuclei in all conditions (Fig. 4C). As expected, the agonist stimulated PRB degradation, whereas RU486 provoked a strong PRB nuclear retention. Specific inhibition of p42/44 resulted in RU486-bound PRB degradation consistent with Western blot analyses (Fig. 4A), thus strengthening the important role of P-p42/44 signaling cascade in ligand-bound PRB stabilization.
Given that PR is degraded via proteasomes, we wondered whether inhibition of RU486-bound PRB ubiquitination could be reversed by ubiquitin overexpression. Ishikawa PRB cells were transiently transfected with control or HA-ubiquitin encoding vector during 24 h and treated with vehicle or R5020 or RU486 during 6 h. As expected, ubiquitin overexpression decreased basal PRB levels. However, RU486-bound PRB (and pS294-PRB species) underwent much slower degradation as compared with R5020-induced PRB turnover (Supplemental Fig. 6). We next examined the contribution of P-p42/44 in the control of such processes by using proteasome inhibitors. We found that MG132, as well as lactacystin (data not shown), strongly enhanced P-p42/44 in Ishikawa cells without affecting total p42/44 levels (Fig. 4D), as was already reported for other cell lines (30). As expected, MG132 exposure resulted in PRB accumulation in vehicle as well as in hormonal conditions. Interestingly, however, P-p42/44 inhibition partially impaired PRB accumulation under MG132 (Fig. 4D, lanes 3 vs. 4, 7 vs. 8, and 11 vs. 12) and lactacystin exposure (data not shown), indicating that proteasome inhibitors stabilize PRB by activating p42/44 in addition to the blockade of proteolytic functions of proteasome. To rule out the possibility that U0126 might interfere with proteasome activity, we examined the expression of glucocorticoid receptor (GR), another nuclear receptor belonging to the same nuclear receptor subfamily as PR and also degraded by proteasomes. In the presence of RU486, also a powerful antagonist of GR, U0126 treatment did not induce degradation of RU486-bound GR, nor inhibited GR accumulation by MG132 (Fig. 4D, inset), demonstrating that P-p42/44 selectively controls PRB stability without affecting general proteasome activity.
Collectively, these results demonstrate that P-p42/44-dependent mechanism slows down the proteasome-dependent turnover rate of ligand-bound PRB in mammary as well as in endometrial cells. This stabilizing mechanism is potentiated by RU486 as compared with progestins and is also functional with the natural ligand progesterone. Therefore, p42/44 MAPK act as brakes for proteasome-dependent turnover of PRB in a ligand-sensitive manner.
Phosphorylated p42/p44 inhibit function of a down-regulating protein partner
To analyze the impact of P-p42/44-dependent phosphorylation on PRB turnover independently of transcriptional and translational events, we preincubated Ishikawa-PRB cells with cycloheximide alone or in combination with U0126 and then treated with vehicle or R5020 or RU486 (Fig. 5). Surprisingly, we found that presynthesized PRB was highly stabilized after a 24-h treatment by cycloheximide, to a level similar for each ligand condition (lanes 3, 7, and 11). Of note, the strong impact of progestin on PRB degradation as well as RU486-bound PRB degradation in the presence of U0126 was fully abolished when neosynthesis was turned off. This strongly suggested that both agonist-induced as well as antagonist-induced PRB down-regulation requires de novo synthesis of down-regulating protein partner(s). Intermediary patterns were also analyzed at shorter time points (data not shown), mainly showing that agonist-induced PRB down-regulation was inhibited as early as 6 h after cycloheximide treatment. Cycloheximide abrogated the degradation of agonist-bound pS294-PRB, which is known to be directed to the proteasome pathway (lane 7 vs. 5). This indicates that the putative down-regulating factor might preferentially target the pS294-PRB species. We noted that cycloheximide decreased the level of RU486-induced pS294-PRB (lane 11 vs. 9). By blocking neosynthesis of the ligand-specific kinase targeting S294, cycloheximide might interrupt the delayed pS294 processes (6–24 h) induced by RU486 without affecting early processes (1–2 h) initiated by agonist as shown in Fig. 2C. We may thus hypothesize that agonist ligand induces interaction of pS294-PRB with down-regulating factor(s) and that RU486 might specifically inhibit this step. Furthermore, we observed that cycloheximide led to increased P-p42/44 levels (but not total p42/44) that might contribute toward PRB protein stabilization (lanes 3, 7, and 11) consistent with our previous findings showing that P-p42/44 stabilizes PRB. Cotreatment of cells with U0126 partially restored degradation of presynthesized PRB (lanes 3 vs. 4, 7 vs. 8, and 11 vs. 12), supporting that P-p42/44 might inhibit interaction with a protein partner required for PRB turnover. Differential effects of ligands on PRB stability might result from their respective ability to control kinetics of at least two phosphorylation events having opposite effects on PR stability, one targeting S294 of PRB independently of MAPK (accelerating turnover) and the other involving a p42/44-dependent kinase activity targeting phosphorylation site other than S294 that inhibits pS294-PRB degradation.
Fig. 5.

Ligand-induced PRB degradation requires protein neosynthesis. Ishikawa PRB cells were pretreated without or with 100 μg/ml cycloheximide (CHX) and/or U0126 (10 μm) during 30 min and then treated without or with R5020 (10−8 m) or RU486 (10−8 m) during 24 h. Immunoblot analyses were performed as in Fig. 3A.
p42/44 MAPK differentially impact PRB transcriptional activity
Because proteasome-dependent turnover of PRB has been shown to be coupled to its transcriptional activity, we asked whether p42/44-dependent stabilization of ligand-bound PRB could impact transcription of progesterone-responsive genes. In both MDA-MB-231 PRB cells and Ishikawa PRB cells, inhibition of p42/44 activity dramatically decreased PRB-mediated reporter gene transcription in response to progesterone and R5020 (Fig. 6A). The partial agonistic effect of RU486 was similarly diminished after U0126 treatment. This shows that p42/44 facilitates PRB transcriptional activity from synthetic promoters.
Fig. 6.
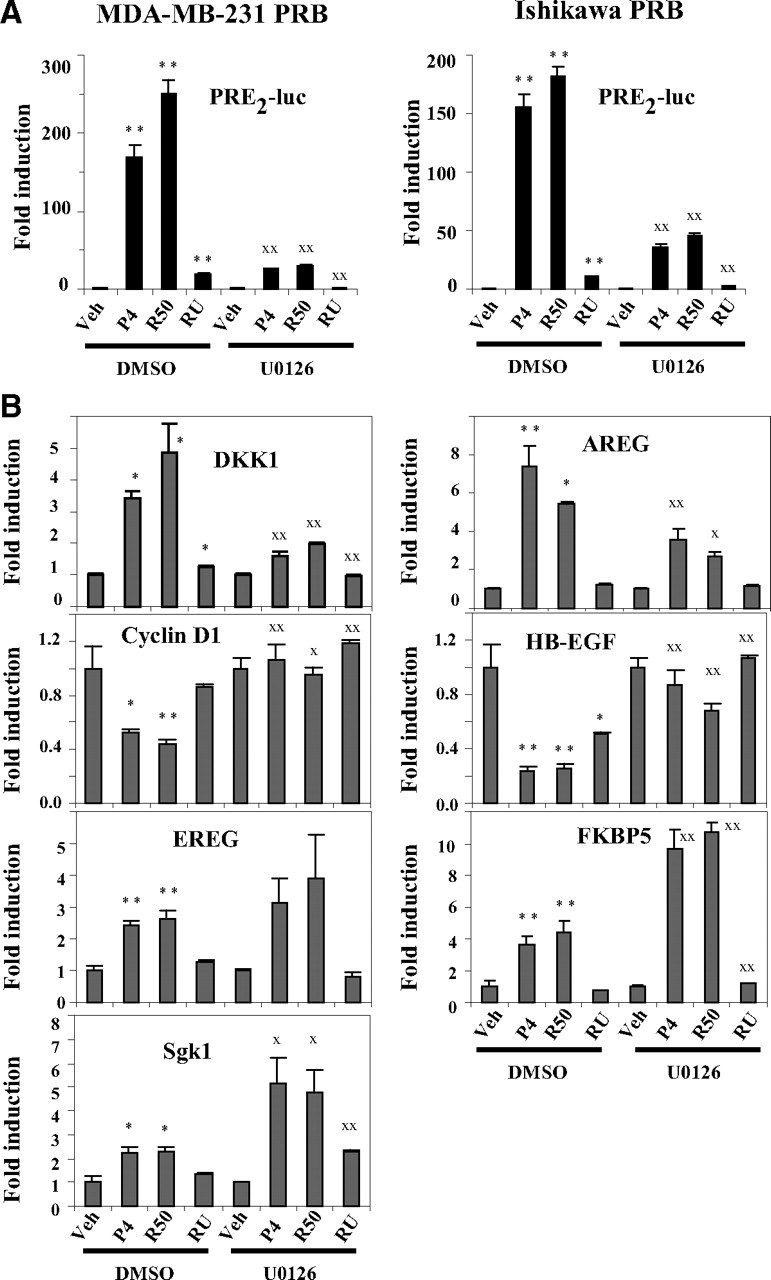
Phosphorylated p42/p44 differentially influence PRB transcriptional activity. A, MDA-MB-231-PRB or Ishikawa PRB cells were transiently transfected with PRE2-luciferase vector during 24 h, pretreated with dimethylsulfoxide (DMSO) or U0126 (10 μm) during 30 min, and then incubated with vehicle (Veh) or progesterone (P4) (10−8 m) or R5020 (R50) (10−8 m) or RU486 (RU) (10−8 m) during 24 h. Luciferase activity was determined and normalized to total protein concentration. The data (mean ± sem) from six independent cell cultures are set to 1 for ligand-free condition from DMSO or U0126-treated cells, and fold induction by hormone is presented. Statistical significance is shown by asterisks for ligand-induced transactivation as compared with vehicle or by XX when similar ligand condition is compared between DMSO or U0126-pretreated cells. B, MDA-MB-231 PRB cells were incubated with U0126 and hormones as above during 6 h, and quantitative real time PCR analysis was performed for indicated gene transcripts. The data (mean ± sem) from three independent cell cultures measured in duplicate are set to 1 for ligand-free condition from DMSO or U0126-treated cells, and fold induction by hormone is presented. Statistical significance is shown as in A. DKK1, Dickkopf homolog 1; AREG, amphiregulin; EREG, epiregulin; Sgk1, serum and glucocorticoid-regulated kinase 1. *, P ≤ 0.05; X, P ≤ 0.05; **, P ≤ 0.01; XX, P ≤ 0.01.
Given that PRB-mediated transcription of endogenous genes involves promoter-dependent recruitment of coregulators, we examined the impact of MAPK signaling on ligand-dependent transcription of various PRB target genes. MDA-MB-231 cells were preincubated with U0126 and treated with vehicle, progesterone, R5020, or RU486 during 6 h. As shown in Fig. 6B, P-p42/44 inhibition differentially influenced ligand-regulated transcription of PRB target genes. Similar to inhibitory effect of U0126 on reporter gene transcription, agonist-induced Dickkopf homolog 1 and amphiregulin gene transcription was dramatically reduced after U0126 treatment. Likewise, U0126 reversed the agonist ligand-dependent transcriptional repression of cyclin D1 and heparin-binding (HB) epidermal growth factor (EGF)-like growth factor (HB-EGF) genes. Moreover, RU486 also decreased HB-EGF gene transcription that was inhibited by U0126. However, U0126 did not alter PRB-mediated epiregulin gene transcription. Interestingly, P-p42/44 inhibition strongly enhanced ligand-induced transcription of FKBP5 and serum and glucocorticoid-regulated kinase 1 genes. These results show that p42/44 MAPK fine tune PRB mediated transcription depending on target gene promoter context and influence transcription of both up-regulated as well as down-regulated PRB target genes. Thus, p42/44 not only stabilize ligand-bound PRB but also play a major role in modulating as well as selecting PRB-mediated transcriptional response to ligands.
MEK kinase (MEKK)1 stabilizes PRA through phosphorylated-p38 MAPK
Our findings that P-p42/44 stabilizes PRB but not PRA suggest that distinct MAPK cascades could selectively control PR isoforms stabilities. To test this hypothesis, we transiently transfected PRB or PRA expressing Ishikawa cells by a vector encoding constitutively active MEKK1 (cMEKK1) and treated with agonist or antagonist ligands for 6 or 24 h. cMEKK1 primarily phosphorylates p38 and c-Jun-N-terminal kinase (JNK)/stress-activated protein kinase and to a lesser extent p42/44 MAPK (Fig. 7A, inset). cMEKK1 increased basal PRB levels and concomitantly S294 phosphorylated species after 6 h (Supplemental Fig. 7) as well as 24 h (Fig. 7A). However, a much more pronounced increase in total PRA and pS130-PRA levels was observed under both vehicle as well as ligand conditions (Fig. 7A and Supplemental Fig. 7), suggesting that high MEKK1 activity preferentially stabilized PRA in Ishikawa cells. Such stabilizing effect was observed at lower extent by decreasing cMEKK1 amount, showing dose dependency of the mechanism (Supplemental Fig. 8). We then aimed at identifying the specific MEKK1-downstream MAPK, possibly involved in the regulation of PRA stability. Ishikawa PRA cells were pretreated with specific inhibitors of P-p42/p44 (U0126), phosphorylated p38 MAPK (P-p38) (PD169316), or phosphorylated JNK MAPK (P-JNK) (SP600125) and transfected with cMEKK1 expression vector. After 24 h, the phosphorylation status of MAPK was examined (Fig. 7B, inset). Although U0126 and SP600125 inhibited P-p42/44 and P-JNK, respectively, it is not surprising that PD169316 did not inhibit MEKK1-induced p38 phosphorylation. Indeed, in contrast to U0126 or SP600125 that selectively inhibit the phosphorylation of p-42/44 or JNK, respectively, PD169316 is known to selectively inhibit the kinase activity of the phosphorylated p38 without hindering upstream kinases to phosphorylate p38 (31, 32). Increased phospho p-38 levels in the presence of PD169316 (Fig. 7B, inset) are most likely due to blockade of negative feedback loop of dephosphorylation of p38 MAPK by MAPK phosphatases (33, 34). As shown in Fig. 7B, MEKK1-dependent increase in PRA stability was clearly impaired in cells treated with PD169316 but not by U0126 or SP600125, suggesting that P-p38 pathway is implicated in the regulation of PRA stability. To strengthen our argument for p38-dependent stabilization of PRA, Ishikawa PRA cells were cotransfected with control or cMEKK1 vector along with specific small interfering RNA (siRNA) against both p42 and p44 or p38 MAPK. Results presented in Fig. 7C demonstrate that p38 but not p42/44 siRNA clearly inhibited increase in PRA stability by cMEKK. These observations along with our previous findings provided first evidence that distinct MAPK differentially regulate PR isoforms stability.
Fig. 7.

MEKK1-induced PRA stabilization is impaired by p38 inhibition. A, Ishikawa PRB or PRA cells were transiently transfected with either empty control (Ctl) vector or cMEKK1 expression vector during 24 h and then treated without or with R5020 (10−8 m) or RU486 (10−8 m) during 24 h. Whole cell extracts were immunoblotted, and P-p42/44, P-p38, and P-JNK MAPK levels were detected by specific antibodies (inset). B, Ishikawa PRA cells were pretreated with vehicle (V) or U0126 (U0) (10 μm) or PD169316 (PD) (10 μm) or SP600125 (SP) (10 μm) during 30 min and then transfected with empty or MEKK1 expression vector during 24 h. Immunoblot analysis was performed as above, and normalized PRA band intensities are presented as fold increase as compared with PRA levels in control cells (inset). Statistical significance is represented by asterisks when comparison is done between control or cMEKK1 condition and by XX when selective MAPK inhibition is compared with nontreated MEKK1-transfected cells. C, Ishikawa PRA cells were cotransfected with control or cMEKK1 vector along with either control or specific siRNA against both p42 and p44 or p38 MAPK during 24 h as described in Materials and Methods. Cells were then incubated in 5% fetal calf serum containing medium for another 48 h before performing immunoblot analysis.
PRA/PRB expression ratio is controlled by distinct MAPK
The selective MAPK control of PR isoforms stabilities prompted us to examine the impact of MAPK on PRA/PRB expression ratio when both isoforms are coexpressed in the same cells, i.e. in conditions where ligand-bound PRA and PRB can interact as heterodimers and can compete for their proteasome-mediated turnover. In Ishikawa PRAB cells coexpressing both PR isoforms, MEKK1 stabilized basal PRA at much higher level than PRB (Fig. 8A), indicating that basal PRA turnover is selectively and highly sensitive to p38 MAPK activities even in the presence of PRB. Such effect led to a strong increase of ligand-free PRA/PRB ratio from 0.3 to 1. As expected (22, 29), basal as well as MEKK1-induced pS294-PRB levels were higher as compared with pS130-PRA levels in cells coexpressing both PR isoforms. This cell-based model enabled us to investigate the relative contribution of P-p42/44 and P-p38 MAPK in regulating PRB or PRA stabilities under MEKK1 stimulation and thus in controlling PRA/PRB expression ratio at posttranslational level. P-p42/44 inhibition using U0126 (Fig. 8B) or p42/44 knockdown by specific siRNA (Supplemental Fig. 9) selectively but not exclusively decreased PRB stability. Such preferential decrease in PRB levels after p42/44 inhibition resulted in increased PRA/PRB ratio under vehicle and R5020 exposure but not in RU486-treated cells (Fig. 8B). In contrast, PRA/PRB ratio drastically decreased after PD169316 treatment, irrespective of ligand conditions, consistent with impaired PRA stabilization upon P-p38 inhibition. Moreover, inhibition of P-JNK by SP600125 enhanced PRB stability, thus decreasing PRA/PRB ratio. However, PRA expression was also slightly decreased by U0126, particularly in vehicle and R5020-treated cells, suggesting that p42/44 specificity of PRB might be conferred to the PRA:PRB heterodimer. Furthermore, variation of pS294-PRB and pS130-PRA levels was correlated with ligand-induced changes in total PRB and total PRA levels under selective inhibition of MAPK. These results indicate that S294-PRB and S130-PRA are targeted by a kinase distinct from p42/44, p38, or JNK MAPK. Of interest, the differential impact of distinct MAPK pathways on PR isoforms stability, i.e. P-p42/44 for PRB and P-p38 for PRA, also varies to different extent depending on the nature of PR ligand (agonist or antagonist). For a given status of MAPK activities, ligand treatment led to higher PRA stability as compared with PRB, resulting in increased PRA/PRB ratio. In contrast, for a given ligand condition, p38 or p42/44 MAPK selectively controlled PRA or PRB stabilities, resulting in overall upshift or downshift in PRA/PRB ratio. Such mechanisms controlling PRA/PRB expression ratio might play a crucial role in hormonal responsiveness in progesterone target tissues.
Fig. 8.
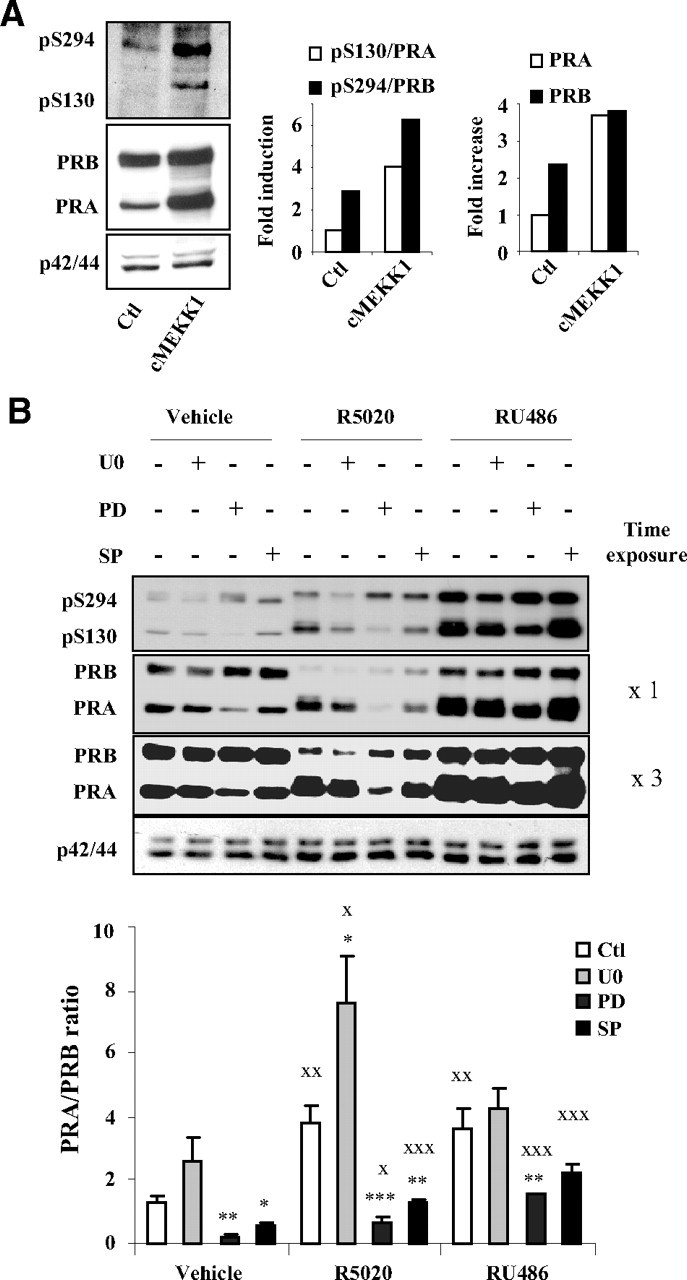
Distinct MAPK control PRA/PRB expression ratio. A, Ishikawa cells stably coexpressing PRA and PRB (Ishikawa PRAB) were transiently transfected with empty control (Ctl) vector or cMEKK1 expression vector during 24 h, and pS294-PRB or pS130-PRA levels or total PRB or total PRA levels were detected by immunoblotting whole cell extracts using phospho-specific or total PR or p42/44 antibodies. Band intensities were quantified, and pS130/PRA or pS294/PRB (middle panel) as well as total PRA and PRB levels (right panel) under basal and cMEKK1 conditions are presented. B, Ishikawa PRAB cells were pretreated with vehicle control (Ctl) or selective inhibitors of p42/44 (U0126) (UO) or p38 (PD169316) (PD) or JNK (SP600125) (SP) activity. Cells were then transfected with MEKK1 expression vector in the presence of vehicle or R5020 (10−8 m) or RU486 (10−8 m) during 24 h. Whole cell extracts were immunoblotted as in Fig. 7B. Band densities corresponding to PRA and PRB were quantified from at least two nonsaturating exposures of the same immunoblot (two film exposures are shown). PRA/PRB expression ratio was calculated for each ligand and inhibitor condition from three independent cell cultures and presented as mean ± sem. Under a given ligand condition, the effect of selective MAPK inhibition as compared with nontreated cells is shown by asterisks. Statistical significance is shown by XX when the effect of ligand is compared with vehicle under a given MAPK inhibition. *, P ≤ 0.05; X, P ≤ 0.05; **, P ≤ 0.01; XX, P ≤ 0.01; ***, P ≤ 0.001; XXX, P ≤ 0.001.
Discussion
The putative functional link between agonist-induced PRB phosphorylation and down-regulation has been extensively analyzed by other laboratories (11). Agonist ligands induce PRB phosphorylation at multiple sites in the N-terminal region, notably at serine residues 102, 294, and 345 (8), whereas other residues are phosphorylated in the ligand-free PRB (35). Although RU486 induces phosphorylation of identical sites as compared with agonist (8), it was shown that RU486 has either no effect on PRB down-regulation (36) or induces PR down-regulation through much slower kinetics than agonist (21). We have recently reported that steroid receptor coactivator 1 was degraded by the proteasome in a PRB-dependent manner that was also inhibited by RU486 (37). To explore the role of PR phosphorylation on its degradation, mutagenesis experiments revealed that substitution of S294 by an alanine (S294A) led to PRB stabilization, suggesting that PRB down-regulation is mainly addressed by the Ser294 site (11). However, in stably transfected T47D cells, PRB-S294A mutant underwent ligand-induced turnover, although to a lesser extent as compared with wild-type PRB (38). We have thus considered that PR stability/turnover might also be governed by pS294-independent mechanisms. Here, we demonstrate that RU486 promotes PRB or PRA protein stabilization despite inducing pS294 or pS130 (equivalent serine residue on PRA), respectively, indicating that RU486 interferes in downstream events of pS294- or pS130-signaled PR isoforms down-regulation. Our data do not correlate with previous reports using T47D-YB cells (stably expressing PRB) or in HeLa cells transiently transfected with PRB expression vector showing that P-p42/44 MAPK accelerate PRB degradation (11, 28). Furthermore, it was reported that EGF- but not progestin-induced pS294 requires p42/44 MAPK activity (14). However, in the same study, it was shown that EGF, despite inducing pS294, increased PRB stability in T47D-YB cells, consistent with our observations. Increased pS294-PRB levels observed after EGF in this previous report might in part be due to PRB accumulation by p42/44 activation. Similarly, enhancing p42/44 activity by MEKK1 was reported to induce pS294 and accelerated PRB turnover in transiently transfected HeLa cells (14). Our results in cells stably expressing PRB show that p42/44 MAPK increase PRB stability that might in part account for increased pS294-PRB species. In support of our results, it has been recently described that degradation of androgen receptor (AR) is enhanced after p42/p44 inhibition by U0126 in prostate cancer LNCaP cells (39). It thus seems very likely that such p42/44 MAPK-dependent-stabilizing effect might be conserved for this nuclear receptor subfamily.
The N-terminal BUS domain of PRB, containing several PEST-like sequences (enriched in proline, glutamate, serine, and threonine) that might initiate turnover process as degron signals (40), accounts for increased turnover rate of PRB than PRA. BUS domain can also confer ligand-dependent down-regulating properties to other nuclear receptors, such as estrogen receptor and AR (41). This property corresponds to the N-end rule for protein degradation as defined by Varshavsky (42). Furthermore, the BUS domain is involved in N-C-terminal intramolecular interactions via two LXXLL motifs similar to nuclear receptor boxes present in coactivator sequences that interact with nuclear receptors (43), accounting for native PRB conformation that is distinct from PRA. Mutations of these sequences abolish the agonist-induced PRB turnover (41) and decrease the reporter gene transcriptional activity similar to that exhibited by PRA. It has been shown that PR-interacting proteins having associated ubiquitin E3-ligase activity, such as breast cancer 1 (23) and E6-associated protein (44), selectively control PRA or PRB turnover, indicating that differential regulatory proteins are involved in PR isoform down-regulation. Involvement of such molecular partners is very likely, because we found that agonist-induced PRB down-regulation was completely abrogated by blocking protein neosynthesis. Recently, it was demonstrated that RU486-bound PRB conformation, in conjunction with PR coregulatory protein Jun dimerization protein-2, exposes protein interaction surfaces that are distinct from those presented by agonist ligand (45). In agreement with these studies, our results indicate that unique conformation of RU486-bound PRB might strongly facilitate stabilizing effects of p42/44-dependent phosphorylation (on a residue other than S294), which impedes interaction with coregulatory proteins implicated in PRB turnover. This p42/44-dependent phosphorylation also occurs upon agonist binding but with a more discrete stabilizing effect as compared with RU486. Such differences might be due to distinct conformations induced by ligands in PRB N-terminal domain. Although agonist ligand might strongly favor interaction of pS294-PRB with putative ubiquitin-ligase(s), RU486-bound PRB might be refractory to such interactions by favoring the stabilizing effect of the p42/44-dependent phosphorylation. As shown by the surprising effect of U0126 in restoring fast RU486-PRB turnover, this interaction is directly inhibited by p42/44-dependent phosphorylation(s). Whether p42/p44 target a PRB-interacting down-regulatory protein is less likely given the ligand sensitivity of the mechanism and the lack of PRB electrophoretic upshift under P-p42/44 inhibition. Nevertheless, we could not rule out the possibility that MAPK-dependent phosphorylation(s) of PR molecular partner(s) may also play a role in determining PR stability.
We have studied the impact of p42/44 on PRB stabilization and its transcriptional activity. Although p42/44 MAPK inhibition dramatically reduced transcriptional activity from exogenous promoter, differential effects were observed on endogenous gene transcription. Inhibition of P-p42/44 reversed the ligand-induced transcriptional activation (Dickkopf homolog 1 and amphiregulin genes) or repression (cyclin D1 and HB-EGF genes). Certain genes might be insensitive to MAPK inhibition (epiregulin), whereas transcription of a gene subset (FKBP5 and serum and glucocorticoid-regulated kinase 1) was highly potentiated by inhibition of p42/44 activity. This shows that p42/44 MAPK fine tune PRB-mediated transcription depending on target gene promoter context and influence transcription of both up-regulated as well as down-regulated PRB target genes. It was shown previously that HB-EGF and cyclin D1 expression increased after progestin treatment in T47D cells, (38, 46). However, in MDA-MB-231 cells, we found that both agonist (progesterone or R5020) and antagonist (RU486) ligands decreased cyclin D1 and HB-EGF expression similar to antiproliferative effects of these ligands (data not shown). It has been reported that progesterone decreases HB-EGF transcription in epithelial cells, whereas in stromal cells, HB-EGF transcription is increased by progesterone (47). Consistent with our results, it was shown that progestin inhibits proliferation of MDA-MB-231 cells stably expressing both PR isoforms (48) and that progesterone decreased cyclin D1 expression as early as 4 h after hormonal treatment (49). The differences in T47D (luminal) and MDA-MB-231 (basal epithelial) cells for these PR target genes regulations might result from differential PR signaling and/or differential expression of coregulatory proteins. Diverse transcriptional effects after p42/44 inhibition does not support that U0126 could artifactually shut down PRB activity through nonspecific effects. MAPK-dependent extracellular signaling might thus selectively influence PRB-mediated transcription depending on various parameters linked to both target gene promoter context and dynamics of proteasome-dependent PRB turnover. MAPK inhibitors have been recently shown to promote the interaction of corepressor silencing mediator of retinoid and thyroid receptors (SMRT) with antagonist-bound AR (50). Moreover, combined treatment of LNCaP prostate cancer cells by p42/p44 inhibitor and AR antagonist cyproterone acetate inhibits AR-mediated transcription as well as agonist-induced cell proliferation. These features are similar to what we obtained with RU486 and U0126 in MDA-MB231 cells. Because SMRT corepressor also mediates RU486-bound PRB transcriptional repression (51), enhancement of SMRT interaction for PRB after p42/44 inhibition remains to be proven.
Although p42/p44 stabilizes PRB, p38 MAPK selectively enhances PRA stabilization, irrespective of ligand, through an unidentified site other than S130. PR upshift after ligand exposure is mainly attributed to PR phosphorylation at S345, a MAPK consensus residue (52). Given that PRB degradation was enhanced after U0126 treatment, a role of S345 in such stabilizing mechanism seems likely. Lack of BUS domain in PRA structure might allow p38-dependent phosphorylation that might be inaccessible in PRB due to conformational differences in PR isoforms. In Ishikawa cells coexpressing PRA and PRB, MEKK1 stimulation increased basal PRA/PRB expression ratio that was further enhanced by agonist as well as antagonist ligands. Although PRB was able to confer p42/p44 sensitivity to PRA:PRB heterodimer, PRB remained refractory to p38-dependent PRA-stabilizing effect. These observations highly support that distinct MAPK-mediated extracellular signaling can highly influence PRA/PRB expression ratio. PRA and PRB regulate common as well as distinct target gene subsets (4, 41), and disruption of relative PR isoforms expression is reported in both breast and endometrial cancers (2, 13). Variations in PRA/PRB expression ratio leading to a change in PR isoforms homo- and heterodimers balance might thus be a critical determinant of PR target gene selection and/or disordered transcriptional regulation resulting in altered cellular response to hormonal stimuli that might contribute toward pathogenesis. Our results highlight that imbalance in PRA/PRB ratio frequently associated with carcinogenesis might be a direct consequence of disorders in MAPK signaling. Using p42/44 selective inhibitors in mammary oncotherapy, because was previously proposed to decrease PRB transcriptional activity (24), might indirectly favor PRA stability/signaling to the detriment of PRB. In contrast, we propose that p38 inhibitors might help to rescue normal PRA/PRB balance in cancer cells overexpressing PRA.
In sum, our results, summarized in Fig. 9, reveal that p38 and p42/44 MAPK selectively control PRA and PRB stabilities. We propose that the BUS domain encompasses a down-regulation tag conferring to PRB a fast agonist-inducible turnover that is negatively controlled by p42/44 MAPK targeting PRB on a residue distinct from S294. PRB stabilization by RU486 might be due to enhancement of this p42/p44 control resulting in downstream inhibition of interaction with (or function of) mandatory down-regulating partner(s). Given the conformational differences between PRA and PRB, p38 MAPK selectively targets PRA, leading to its stabilization. Extracellular stimuli, such as EGF or proinflammatory cytokines, which preferentially activate p42/44 or p38 MAPK, respectively, may lead to opposite variations in PRA/PRB expression ratio at posttranslational level. Changes in extracellular signaling in these cells might strongly influence PRA/PRB ratio and lead to dramatic shift in selection of PR target gene subsets, thus switching cellular responses to hormonal/growth factor stimuli. This might be of broad concern for designing pharmacological intervention in breast cancers regarding combination of selective MAPK inhibitors along with antiprogestins.
Fig. 9.

MAPK-dependent control of PRA/PRB expression ratio. Control of PRA and PRB stabilities and PRA/PRB ratio by p38 and p42/44 MAPK activities is schematically summarized. Progesterone (Prog) and specific MAPK inhibitors are indicated (U0, U0126; PD, PD169316).
Materials and Methods
Cell culture and reagents
Human endometrial cancer cell lines Ishikawa PRA, Ishikawa PRB, Ishikawa PRAB engineered to stably express either, or both PR isoforms (PRA, PRB, PRA, and PRB) were kindly provided by L. J. Blok (Erasmus University, Rotterdam, The Netherlands) (25). Human breast cancer cells MDA-MB-231 stably expressing PRB were kindly provided by A. Gompel (Université Paris Descartes, Paris, France) (26). All cell lines were routinely cultured in DMEM with glutamine, enriched with 10% fetal calf serum (FCS) (BioWest, Denver, CO), and supplemented with antibiotics (penicillin 100 UI/ml, streptomycine 100 μg/ml) (PAA Laboratories GmbH, Cölbe, Germany). For each experiment, cells were preincubated in steroid-free medium containing 5% dextran-coated charcoal-treated serum without antibiotics for at least 24 h before hormonal treatment. Progesterone, R5020, RU486 and inhibitors for MEK1/2 (U0126), phospho-p38 (PD169316) and phospho-JNK (SP600125) MAPK, proteasome (MG132), and protein neosynthesis (cycloheximide) were purchased from Sigma (St. Louis, MO).
Immunoblotting
For whole cell protein extraction, cells were rinsed twice with PBS and lysed by scrapping in extraction buffer [0.1% (vol/vol) Triton X-100, 50 mm Tris-HCl (pH 7.5), 5 mm EDTA, 150 mm NaCl, 0.2% (wt/vol) NaF, and 1.3% (wt/vol) sodium pyrophosphate] containing phosphatases and proteases inhibitors mixture (Sigma). Lysates were clarified by centrifugation at 16,000 × g for 15 min in a refrigerated microfuge. Soluble proteins were quantified using the bicinchoninic acid assay kit (Interchim, Montluçon, France), and equal amounts of protein were mixed with 1/3 volume of 3× Laemmli sample buffer [187.5 mm Tris-HCl (pH 6.8), 15% (vol/vol) β mercapto-ethanol, 30% (vol/vol) glycerol, 6% (vol/vol) sodium dodecyl sulfate, and 0.03% (wt/vol) bromophenol blue] and heated at 95 C for 5 min for denaturation. Equal amounts of protein were resolved by SDS-PAGE (7.5 or 10% acrylamide) and transferred on polyvinylidene fluoride membrane. Primary antibody solutions were prepared in Tris-buffered saline with Tween 20 containing 5% fat skimmed dry milk at the final dilution of 1:3000 for PRA and PRB phospho S294-specific antibody (Affinity BioReagent, Golden, CO), 1:500 for anti-PRB-specific mouse monoclonal antibody Let 126 (53), 1:10,000 for mouse monoclonal anti-PRA and anti-PRB antibody (NCL-L-PGR-312/2; Novocastra Laboratories, Newcastle upon Tyne, UK), 1:3000 for phospho-specific or total p38, p42/p44, or JNK MAPK antibodies (Cell Signaling Technology, Beverly, MA), 1:250 for anti-GR antibody (AbC10-G015; AbCys S.A., Paris, France), or 1:10,000 for anti-α-tubulin antibody (Sigma). The membranes were immersed in primary antibody solution on a rotator either at 4 C overnight or at room temperature during 1 h. Incubation with horseradish peroxidase-conjugated goat antimouse or antirabbit secondary antibody solution (Vector Laboratories, Burlingame, CA) was prepared in Tris-buffered saline with Tween 20 5% skimmed dry milk at 1:15,000 dilutions. Membranes were then incubated for 1 h at room temperature. Target proteins were detected using ECL Plus reagent (GE Healthcare, Princeton, NJ) and visualized by chemiluminescence. Bands corresponding to target proteins were quantified by scanning films obtained for several nonsaturating time exposures, using MacBiophotonics ImageJ 1.43s software and were normalized to either tubulin or total p42/p44 loading control.
Immunoprecipitation assays
Parental Ishikawa cells were transfected in a 100-mm plate with HA-ubiquitin and PRB expression vectors (54) during 48 h in steroid-free medium. Cells were treated with MG132 (5 μm) during 30 min before treatment with vehicle or R5020 (10−8 m) or RU486 (10−8 m) during 4 h in 5% steroid-free FCS-containing medium. Cells were lysed at 4 C in 500 μl lysis buffer, cell debris was pelleted by centrifugation (14,000 × g, 15 min, 4 C), and the supernatant was obtained. One milligram of total protein was immunoprecipitated using anti-PR antibody (C-19; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and Protein G Magnetic Beads (Millipore, Bedford, MA) according to the manufacturer's instructions. Bound immunocomplexes were boiled in Laemmli buffer and resolved on 7.5% acrylamide gel as described above. Anti-HA (12CA5; Roche Diagnostics, Indianapolis, IN) or anti-PR antibody (NCL-L-PGR-312/2; Novocastra Laboratories) was used for the detection of ubiquitinated or total PRB, respectively.
Real-time quantitative RT-PCR
Hormone-treated cells were rinsed twice with PBS, and total RNA was extracted using TRIZOL reagent (Invitrogen, Carlsbad, CA) as described previously (54). One microgram of total RNA was treated with DNase I Amplification Grade (Invitrogen) and reverse transcribed using cDNA RT kit from Applied Biosystems (Foster City, CA). cDNA thus obtained was diluted 10-fold, and 1/20th fraction of the cDNA preparation was amplified by PCR using the Power SYBR Green PCR Master Mix (Applied Biosystems). Primers (300 nm) sequences are presented in Supplemental Table 1. Reaction parameters were set to 95 C for 10 min, 40 cycles at 95 C for 15 sec, and 60 C for 1 min on ABI 7300 Sequence Detector (Applied Biosystems). A dissociation curve was also obtained to verify primer pair specificity. For standards preparation, amplicons were purified after agarose gel electrophoresis, subloned in pGEMT-easy (Promega, Madison, WI), and then sequenced for verification of the amplification product. These plasmid-amplicons were linearized and used for standardization of real-time quantitative PCR. All samples were analyzed in duplicate from at least three independent cell cultures. The relative expression level of each gene transcript was normalized with 18S RNA level of the corresponding sample.
Transient transfection
cMEKK1 expression vector (55) was kindly provided by M. H. Cobb (University of Texas, Southwestern Medical Center, Dallas, TX). Transfections were performed using LipofectAMINE 2000 according to the manufacturer's recommendations (Invitrogen). The cells were plated at 1.2 × 106 per well in six-well plates and then transiently transfected with control or HA-ubiquitin or MEKK1 expression vector during indicated time periods in phenol red-free medium containing 2.5% steroid-depleted FCS. The cells were then treated with ethanol (vehicle) or R5020 (10−8 m) or RU486 (10−8 m) for indicated durations in steroid-free medium. For siRNA transfection experiments, cells were cotransfected with control or cMEKK1 expression vector (1 μg) along with either of the following siRNA (SignalSilence; Cell Signaling Technology): control (no. 6568), p42 and p44 (no. 6560), or p38 (nos. 6564 or 6243) MAPK (100 nm) using Lipofectamine 2000.
Luciferase reporter gene assays
MDA-MB-231 PRB or Ishikawa PRB cells were cultured in steroid-free medium and transfected with PRE2-TATA-luciferase reporter gene (100 ng) and β-galactosidase (10 ng) plasmids in 96-well plates. After 24 h of transfection, cells were incubated with vehicle or progesterone (10−8 m) or R5020 (10−8 m) or RU486 (10−8 m) for 24 h. Cells were collected with the Passive Lysis Buffer (Promega). Luciferase activity was measured with a luminometer (Victor; PerkinElmer, Waltham, MA) and normalized with either β-galactosidase activity or total protein concentration. The data are presented as means ± se of six independent cell cultures (n = 6).
Immunocytochemical assays
Cells were seeded in 24-well plates, fixed with 4% paraformaldehyde, and permeabilized 30 min with PBS containing 0.5% Triton X-100. Cells were then incubated with primary anti-PR antibody (Novocastra Laboratories) overnight at 4 C and for 30 min with an Alexa Fluor 488-coupled antimouse IgG secondary antibody. Fluorescent cells were analyzed with an Olympus Provis AX70 microscope (Olympus, Tokyo, Japan). Pictures acquisition was performed at ×20 magnitude for 160 msec with imaging Qcapture Pro version 5.1 (QImaging, Inc., Surrey, Canada).
Statistical analysis
Data are expressed as mean ± sem. Nonparametric Mann-Whitney test for transactivation studies or unpaired t test for quantitative analysis of Western blotting images was used to determine significant differences between groups using the computer software Prism 4 (GraphPad Software, San Diego, CA). Statistical significance is indicated at P ≤ 0.05, 0.01, and 0.001.
Acknowledgments
We thank Dr. M. H. Cobb (University of Texas, Southwestern Medical Center, Dallas, TX) for providing MEKK1 expression vector, Dr. L. J. Blok (Erasmus University, Rotterdam, The Netherlands) for Ishikawa cells lines, Dr. A Gompel (Université Paris Descartes, Paris, France) for MDA-MB-231 cell lines, Dr. Say Viengchareun for useful discussions and technical assistance, Meriem Messina for plasmid preparations, and Luc Outin for image quantifications.
This work was supported by grants from Institut National de la Santé et de la Recherche Médicale, the Université Paris-Sud 11, and Association pour la Recherche sur le Cancer. J.A.K. is on study leave from the Department of Physiology and Pharmacology, University of Agriculture, Faisalabad, Pakistan, and is a recipient of doctoral scholarship from Higher Education Commission, Islamabad, Pakistan, and a fellowship from La Ligue Contre le Cancer, France. C.B. is recipient of a fellowship from the Conseil Régional de la Martinique.
Disclosure Summary: The authors have nothing to disclose.
NURSA Molecule Pages†:
Nuclear Receptors: PR;
Ligands: Progesterone | R5020 | RU486.
Annotations provided by Nuclear Receptor Signaling Atlas (NURSA) Bioinformatics Resource. Molecule Pages can be accessed on the NURSA website at www.nursa.org.
- AR
- Androgen receptor
- BUS
- B-upstream segment
- cMEKK1
- constitutively active MEKK1
- EGF
- epidermal growth factor
- FCS
- fetal calf serum
- FKBP5
- FK506 binding protein 5
- GR
- glucocorticoid receptor
- HB
- heparin binding
- HB-EGF
- HB EGF-like growth factor
- JNK
- c-Jun-N-terminal kinase
- MEK
- MAPK kinase
- MEKK
- MEK kinase
- P-JNK
- phosphorylated JNK MAPK
- P-p38
- phosphorylated p38 MAPK
- P-p42/44
- p42/44 MAPK phosphorylation
- PR
- progesterone receptor
- pS294
- serine-294 phosphorylation
- siRNA
- small interfering RNA
- SMRT
- silencing mediator of retinoid and thyroid receptor.
References
- 1. Kastner P , Krust A , Turcotte B , Stropp U , Tora L , Gronemeyer H , Chambon P. 1990. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J 9:1603–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mote PA , Bartow S , Tran N , Clarke CL. 2002. Loss of co-ordinate expression of progesterone receptors A and B is an early event in breast carcinogenesis. Breast Cancer Res Treat 72:163–172 [DOI] [PubMed] [Google Scholar]
- 3. Sartorius CA , Melville MY , Hovland AR , Tung L , Takimoto GS , Horwitz KB. 1994. A third transactivation function (AF3) of human progesterone receptors located in the unique N-terminal segment of the B-isoform. Mol Endocrinol 8:1347–1360 [DOI] [PubMed] [Google Scholar]
- 4. Richer JK , Jacobsen BM , Manning NG , Abel MG , Wolf DM , Horwitz KB. 2002. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem 277:5209–5218 [DOI] [PubMed] [Google Scholar]
- 5. Conneely OM , Mulac-Jericevic B , Lydon JP , De Mayo FJ. 2001. Reproductive functions of the progesterone receptor isoforms: lessons from knock-out mice. Mol Cell Endocrinol 179:97–103 [DOI] [PubMed] [Google Scholar]
- 6. Pratt WB , Toft DO. 1997. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev 18:306–360 [DOI] [PubMed] [Google Scholar]
- 7. Beck CA , Zhang Y , Altmann M , Weigel NL , Edwards DP. 1996. Stoichiometry and site-specific phosphorylation of human progesterone receptor in native target cells and in the baculovirus expression system. J Biol Chem 271:19546–19555 [DOI] [PubMed] [Google Scholar]
- 8. Beck CA , Zhang Y , Weigel NL , Edwards DP. 1996. Two types of anti-progestins have distinct effects on site-specific phosphorylation of human progesterone receptor. J Biol Chem 271:1209–1217 [DOI] [PubMed] [Google Scholar]
- 9. Dennis AP , Lonard DM , Nawaz Z , O'Malley BW. 2005. Inhibition of the 26S proteasome blocks progesterone receptor-dependent transcription through failed recruitment of RNA polymerase II. J Steroid Biochem Mol Biol 94:337–346 [DOI] [PubMed] [Google Scholar]
- 10. Daniel AR , Faivre EJ , Lange CA. 2007. Phosphorylation-dependent antagonism of sumoylation derepresses progesterone receptor action in breast cancer cells. Mol Endocrinol 21:2890–2906 [DOI] [PubMed] [Google Scholar]
- 11. Lange CA , Shen T , Horwitz KB. 2000. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci USA 97:1032–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Graham JD , Roman SD , McGowan E , Sutherland RL , Clarke CL. 1995. Preferential stimulation of human progesterone receptor B expression by estrogen in T-47D human breast cancer cells. J Biol Chem 270:30693–30700 [DOI] [PubMed] [Google Scholar]
- 13. Arnett-Mansfield RL , deFazio A , Wain GV , Jaworski RC , Byth K , Mote PA , Clarke CL. 2001. Relative expression of progesterone receptors A and B in endometrioid cancers of the endometrium. Cancer Res 61:4576–4582 [PubMed] [Google Scholar]
- 14. Shen T , Horwitz KB , Lange CA. 2001. Transcriptional hyperactivity of human progesterone receptors is coupled to their ligand-dependent down-regulation by mitogen-activated protein kinase-dependent phosphorylation of serine 294. Mol Cell Biol 21:6122–6131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pierson-Mullany LK , Lange CA. 2004. Phosphorylation of progesterone receptor serine 400 mediates ligand-independent transcriptional activity in response to activation of cyclin-dependent protein kinase 2. Mol Cell Biol 24:10542–10557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Weigel NL , Moore NL. 2007. Kinases and protein phosphorylation as regulators of steroid hormone action. Nucl Recept Signal 5:e005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dressing GE , Hagan CR , Knutson TP , Daniel AR , Lange CA. 2009. Progesterone receptors act as sensors for mitogenic protein kinases in breast cancer models. Endocr Relat Cancer 16:351–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lange CA. 2008. Challenges to defining a role for progesterone in breast cancer. Steroids 73:914–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reid G , Hübner MR , Métivier R , Brand H , Denger S , Manu D , Beaudouin J , Ellenberg J , Gannon F. 2003. Cyclic, proteasome-mediated turnover of unliganded and liganded ERα on responsive promoters is an integral feature of estrogen signaling. Mol Cell 11:695–707 [DOI] [PubMed] [Google Scholar]
- 20. Klijn JG , Setyono-Han B , Foekens JA. 2000. Progesterone antagonists and progesterone receptor modulators in the treatment of breast cancer. Steroids 65:825–830 [DOI] [PubMed] [Google Scholar]
- 21. el-Ashry D , Oñate SA , Nordeen SK , Edwards DP. 1989. Human progesterone receptor complexed with the antagonist RU 486 binds to hormone response elements in a structurally altered form. Mol Endocrinol 3:1545–1558 [DOI] [PubMed] [Google Scholar]
- 22. Clemm DL , Sherman L , Boonyaratanakornkit V , Schrader WT , Weigel NL , Edwards DP. 2000. Differential hormone-dependent phosphorylation of progesterone receptor A and B forms revealed by a phosphoserine site-specific monoclonal antibody. Mol Endocrinol 14:52–65 [DOI] [PubMed] [Google Scholar]
- 23. Poole AJ , Li Y , Kim Y , Lin SC , Lee WH , Lee EY. 2006. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science 314:1467–1470 [DOI] [PubMed] [Google Scholar]
- 24. Faivre EJ , Lange CA. 2007. Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol Cell Biol 27:466–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smid-Koopman E , Blok LJ , Kühne LC , Burger CW , Helmerhorst TJ , Brinkmann AO , Huikeshoven FJ. 2003. Distinct functional differences of human progesterone receptors A and B on gene expression and growth regulation in two endometrial carcinoma cell lines. J Soc Gynecol Investig 10:49–57 [PubMed] [Google Scholar]
- 26. Petit E , Courtin A , Kloosterboer HJ , Rostène W , Forgez P , Gompel A. 2009. Progestins induce catalase activities in breast cancer cells through PRB isoform: correlation with cell growth inhibition. J Steroid Biochem Mol Biol 115:153–160 [DOI] [PubMed] [Google Scholar]
- 27. Qiu M , Lange CA. 2003. MAP kinases couple multiple functions of human progesterone receptors: degradation, transcriptional synergy, and nuclear association. J Steroid Biochem Mol Biol 85:147–157 [DOI] [PubMed] [Google Scholar]
- 28. Qiu M , Olsen A , Faivre E , Horwitz KB , Lange CA. 2003. Mitogen-activated protein kinase regulates nuclear association of human progesterone receptors. Mol Endocrinol 17:628–642 [DOI] [PubMed] [Google Scholar]
- 29. Narayanan R , Edwards DP , Weigel NL. 2005. Human progesterone receptor displays cell cycle-dependent changes in transcriptional activity. Mol Cell Biol 25:2885–2898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen JJ , Huang WC , Chen CC. 2005. Transcriptional regulation of cyclooxygenase-2 in response to proteasome inhibitors involves reactive oxygen species-mediated signaling pathway and recruitment of CCAAT/enhancer-binding protein δ and CREB-binding protein. Mol Biol Cell 16:5579–5591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mathay C , Giltaire S , Minner F , Bera E , Hérin M , Poumay Y. 2008. Heparin-binding EGF-like growth factor is induced by disruption of lipid rafts and oxidative stress in keratinocytes and participates in the epidermal response to cutaneous wounds. J Invest Dermatol 128:717–727 [DOI] [PubMed] [Google Scholar]
- 32. Samuvel DJ , Jayanthi LD , Bhat NR , Ramamoorthy S. 2005. A role for p38 mitogen-activated protein kinase in the regulation of the serotonin transporter: evidence for distinct cellular mechanisms involved in transporter surface expression. J Neurosci 25:29–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Patterson KI , Brummer T , O'Brien PM , Daly RJ. 2009. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J 418:475–489 [DOI] [PubMed] [Google Scholar]
- 34. Farooq A , Zhou MM. 2004. Structure and regulation of MAPK phosphatases. Cell Signal 16:769–779 [DOI] [PubMed] [Google Scholar]
- 35. Ward RD , Weigel NL. 2009. Steroid receptor phosphorylation: assigning function to site-specific phosphorylation. Biofactors 35:528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kahmann S , Vassen L , Klein-Hitpass L. 1998. Synergistic enhancement of PRB-mediated RU486 and R5020 agonist activities through cyclic adenosine 3′,5′-monophosphate represents a delayed primary response. Mol Endocrinol 12:278–289 [DOI] [PubMed] [Google Scholar]
- 37. Amazit L , Roseau A , Khan JA , Chauchereau A , Tyagi RK , Loosfelt H , Leclerc P , Lombès M , Guiochon-Mantel A. 2011. Ligand-dependent degradation of SRC-1 is pivotal for progesterone receptor transcriptional activity. Mol Endocrinol 25:394–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Skildum A , Faivre E , Lange CA. 2005. Progesterone receptors induce cell cycle progression via activation of mitogen-activated protein kinases. Mol Endocrinol 19:327–339 [DOI] [PubMed] [Google Scholar]
- 39. Agoulnik IU , Bingman WE , Nakka M , Li W , Wang Q , Liu XS , Brown M , Weigel NL. 2008. Target gene-specific regulation of androgen receptor activity by p42/p44 mitogen-activated protein kinase. Mol Endocrinol 22:2420–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Varshavsky A. 1997. The ubiquitin system. Trends Biochem Sci 22:383–387 [DOI] [PubMed] [Google Scholar]
- 41. Tung L , Abdel-Hafiz H , Shen T , Harvell DM , Nitao LK , Richer JK , Sartorius CA , Takimoto GS , Horwitz KB. 2006. Progesterone receptors (PR)-B and -A regulate transcription by different mechanisms: AF-3 exerts regulatory control over coactivator binding to PR-B. Mol Endocrinol 20:2656–2670 [DOI] [PubMed] [Google Scholar]
- 42. Varshavsky A. 1997. The N-end rule pathway of protein degradation. Genes Cells 2:13–28 [DOI] [PubMed] [Google Scholar]
- 43. Dong X , Challis JR , Lye SJ. 2004. Intramolecular interactions between the AF3 domain and the C-terminus of the human progesterone receptor are mediated through two LXXLL motifs. J Mol Endocrinol 32:843–857 [DOI] [PubMed] [Google Scholar]
- 44. Ramamoorthy S , Dhananjayan SC , Demayo FJ , Nawaz Z. 2010. Isoform-specific degradation of PR-B by E6-AP is critical for normal mammary gland development. Mol Endocrinol 24:2099–2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wardell SE , Narayanan R , Weigel NL , Edwards DP. 2010. Partial agonist activity of the progesterone receptor antagonist RU486 mediated by an amino-terminal domain coactivator and phosphorylation of serine400. Mol Endocrinol 24:335–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Daniel AR , Qiu M , Faivre EJ , Ostrander JH , Skildum A , Lange CA. 2007. Linkage of progestin and epidermal growth factor signaling: phosphorylation of progesterone receptors mediates transcriptional hypersensitivity and increased ligand-independent breast cancer cell growth. Steroids 72:188–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Z , Funk C , Roy D , Glasser S , Mulholland J. 1994. Heparin-binding epidermal growth factor-like growth factor is differentially regulated by progesterone and estradiol in rat uterine epithelial and stromal cells. Endocrinology 134:1089–1094 [DOI] [PubMed] [Google Scholar]
- 48. Lin VC , Ng EH , Aw SE , Tan MG , Ng EH , Chan VS , Ho GH. 1999. Progestins inhibit the growth of MDA-MB-231 cells transfected with progesterone receptor complementary DNA. Clin Cancer Res 5:395–403 [PubMed] [Google Scholar]
- 49. Lin VC , Woon CT , Aw SE , Guo C. 2003. Distinct molecular pathways mediate progesterone-induced growth inhibition and focal adhesion. Endocrinology 144:5650–5657 [DOI] [PubMed] [Google Scholar]
- 50. Eisold M , Asim M , Eskelinen H , Linke T , Baniahmad A. 2009. Inhibition of MAPK-signaling pathway promotes the interaction of the corepressor SMRT with the human androgen receptor and mediates repression of prostate cancer cell growth in the presence of antiandrogens. J Mol Endocrinol 42:429–435 [DOI] [PubMed] [Google Scholar]
- 51. Liu Z , Auboeuf D , Wong J , Chen JD , Tsai SY , Tsai MJ , O'Malley BW. 2002. Coactivator/corepressor ratios modulate PR-mediated transcription by the selective receptor modulator RU486. Proc Natl Acad Sci USA 99:7940–7944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang Y , Beck CA , Poletti A , Edwards DP , Weigel NL. 1995. Identification of a group of Ser-Pro motif hormone-inducible phosphorylation sites in the human progesterone receptor. Mol Endocrinol 9:1029–1040 [DOI] [PubMed] [Google Scholar]
- 53. Lorenzo F , Jolivet A , Loosfelt H , Thu vu Hai M , Brailly S , Perrot-Applanat M , Milgrom E. 1988. A rapid method of epitope mapping. Application to the study of immunogenic domains and to the characterization of various forms of rabbit progesterone receptor. Eur J Biochem 176:53–60 [DOI] [PubMed] [Google Scholar]
- 54. Georgiakaki M , Chabbert-Buffet N , Dasen B , Meduri G , Wenk S , Rajhi L , Amazit L , Chauchereau A , Burger CW , Blok LJ , Milgrom E , Lombès M , Guiochon-Mantel A , Loosfelt H. 2006. Ligand-controlled interaction of HBO1 with the N-terminal transactivating domain of progesterone receptor induces SRC-1-dependent co-activation of transcription. Mol Endocrinol 20:2122–2140 [DOI] [PubMed] [Google Scholar]
- 55. Xu S , Robbins DJ , Christerson LB , English JM , Vanderbilt CA , Cobb MH. 1996. Cloning of rat MEK kinase 1 cDNA reveals an endogenous membrane-associated 195-kDa protein with a large regulatory domain. Proc Natl Acad Sci USA 93:5291–5295 [DOI] [PMC free article] [PubMed] [Google Scholar]