

Abstract
Suicide gene therapy is an attractive strategy to selectively destroy cancer cells while minimizing unnecessary toxicity to normal cells. Since this idea was first introduced more than two decades ago, numerous studies have been conducted and significant developments have been made to further its application for mainstream cancer therapy. Major limitations of the suicide gene therapy strategy that have hindered its clinical application include inefficient directed delivery to cancer cells and the poor prodrug activation capacity of suicide enzymes. This review is focused on efforts that have been and are currently being pursued to improve the activity of individual suicide enzymes towards their respective prodrugs with particular attention to the application of nucleotide metabolizing enzymes in suicide cancer gene therapy. A number of protein engineering strategies have been employed and our discussion here will center on the use of mutagenesis approaches to create and evaluate nucleotide metabolizing enzymes with enhanced prodrug activation capacity and increased thermostability. Several of these studies have yielded clinically important enzyme variants that are relevant for cancer gene therapy applications because their utilization can serve to maximize cancer cell killing while minimizing the prodrug dose, thereby limiting undesirable side effects.
Keywords: Bystander effect, cancer, enzyme engineering, mutagenesis, prodrug, suicide gene therapy
INTRODUCTION
In the United States cancer is the second leading cause of death after heart disease. Standard cancer treatments include chemotherapy, radiation and surgery individually or in combination. While the goal of cancer treatment is to rapidly and effectively eradicate tumor cells without harming normal cells, chemotherapy and radiation lack the ability to limit toxicity solely to tumor sites. The effectiveness of these standard treatments is also frequently impeded by the emergence of drug- and radiation-resistant cancer cells that subsequently lead to treatment failure. In addition, chemotherapy and radiation have short-term negative side effects, such as hair loss, drastic weight loss, extreme fatigue and nausea. Long-term effects, such as infertility, organ damage and cataracts have also been commonly reported. Furthermore, exposure to chemotherapy and radiation treatments may induce genetic mutations that cause secondary cancers [1–5].
The discovery of safer and more potent tumor-specific treatments is a goal shared by cancer researchers worldwide. While numerous cancer therapeutic strategies are currently under rigorous investigation, most treatments either fail to reach clinical trials, yield disappointing results or reveal safety issues in clinical trials. One relatively new and promising treatment for cancer is suicide gene therapy. This approach is based on the successful delivery and expression of the suicide gene in tumor cells. The suicide gene encodes an enzyme with the unique ability to activate an otherwise impotent prodrug. Following suicide gene expression in transfected cells, an appropriate prodrug is administered and is converted to a cytotoxic compound by the actions of the suicide gene product Fig. (1). As most suicide genes encode enzymes belonging to the class of nucleotide metabolizing enzymes, the general mode of action of activated prodrugs is interference with DNA synthesis that consequently results in induction of apoptosis. The potency of these drugs is maximized in cancer cells due to their greater proliferative rate as compared to normal cells. Because of the prospect to preferentially deliver genes to tumor cells, this strategy has the potential to offer selective tumor killing while sparing normal cells, a feature that standard chemotherapeutic and radiotherapy approaches do not generally afford.
Fig. 1.
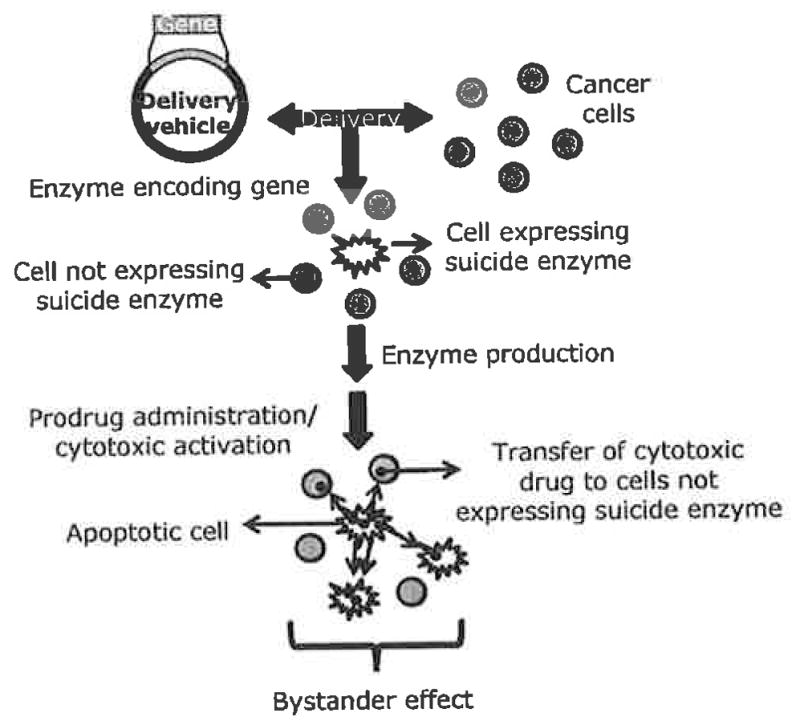
Overview of suicide gene therapy.
Although suicide gene therapy is a fairly new anti-cancer approach, the concept was originally described more than two decades ago in 1986 by Moolten [6]. He also proposed the existence of what is currently known as the bystander effect, now widely recognized as a fundamental feature of suicide gene therapy success. By definition the bystander effect is the extension of cytotoxic effects from transfected cells to non-transfected neighboring cells such that complete tumor regression is observed when only a small subpopulation of tumor cells is successfully transfected. This phenomenon is crucial to the overall effectiveness of suicide gene therapy today due to low transfection efficiencies achievable by available delivery systems. The bystander effect is thought to occur via two major mechanisms: local and immune-mediated [7]. The local mechanism involves the killing of untransfected nearby cells due to the transfer of toxic materials or suicide enzymes through gap junctions [8–10], apoptotic vesicles [11] or through diffusion of soluble toxic metabolites [12, 13]. Gap junctions are important in cell-cell interactions and are responsible for the transfer of ions, nucleotides and small molecules to adjacent cells [14]. The transfer of toxic drugs through gap junctions, however, may not be available in certain types of tumors that down regulate intracellular gap junction communication and display disorganized and non-functional gap junctions [7. 15], To address this problem, several studies have increased the expression of connexins, the building blocks of gap junctions, and demonstrated that enhanced bystander and cell killing effects were achieved [16, 17].
Accumulating evidence from in vivo experiments suggests that the immune-mediated bystander effect plays an important role in tumor regression as well. The presence of inflammatory infiltrates, chemokines, and cytokines have been found elevated in regressing tumors of immunocompetent animals receiving suicide gene therapy treatment [18–23]. These cytokines and chemokines further induce the production of immune-regulatory molecules able to stimulate a more robust anti-cancer effect and additionally, because death of transfected cells is through apoptosis, numerous inflammatory signals may be released to evoke a potent immune response.
The success of suicide gene therapy therefore appears to rely on several key factors. First, the suicide enzyme must be selectively expressed in tumor cells or at least be expressed at significantly higher levels within tumor cells as compared to normal cells, a factor mainly reliant upon the vector or delivery vehicle. Second, the suicide enzyme should have high catalytic activity towards the prodrug in order to rapidly and efficiently convert the prodrug to a cytotoxic form even when present at low concentrations. Third, the prodrug should be tolerable and non-toxic to normal cells. Fourth, the expression of the suicide enzyme itself should not result in any significant toxicity. And finally, a strong bystander effect should take place. In reality, however, such perfect suicide enzyme/prodrug combinations do not exist to date. Wild type (wt) suicide enzymes often display poor prodrug converting abilities, thus necessitating administration of high prodrug doses to achieve clinically beneficial results, potentially causing undesired negative side effects. Several approaches have been reported to overcome this limitation including the use of multiple gene copies to yield higher expression levels [24], synthesis of novel prodrugs for gene therapy [25], other chemotherapeutic drugs to modulate the nucleoside metabolizing pathway [26], multimodal therapies [27], fusion suicide enzymes [28–30] and mutant suicide genes with improved activity towards clinically relevant prodrugs [31–35]. Moreover, while Herpes Simplex Virus thymidine kinase (HSVTK) is widely used, it is advantageous to optimize current suicide genes and to search for alternative/novel suicide genes for two key reasons: 1) not all cancers are equally responsive to the same drug and 2) should treatment with one suicide gene fail, alternate suicide genes that the immune system has not been exposed to previously could be used in additional rounds of therapy to ablate tumors.
Since the first description of suicide gene therapy in 1986, hundreds of clinical trials have been conducted worldwide that explore various elements of suicide gene therapy strategies. As of June 2007, three of these trials had advanced to phase III multicenter programs [36, 37] and there are more than twenty suicide gene therapy/prodrug systems described in the literature [38]. While other literature reports the general strategy of suicide gene therapy, this review will be dedicated to describing the extensive efforts that have been pursued to optimize several suicide enzymes using protein engineering strategies Table (1).
Table 1.
Suicide Enzyme Information and Characteristics
| Enzyme | Source-Gene | Subunit mass/Active form | Natural substrate | Prodrug | Variants/Mutants | Drug inhibitory action | References |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Herpes Simplex Virus Thymidine Kinase (HSVTK) | Herpes Simplex Virus 1 (HSV-1) Thymidine Kinase (TK) | 45kDa homodimer | Thymidine | GCV, ACV | Mutant 30 | 1 | 52, 56–60 |
| Mutant 75 | 1 | 52, 56, 58 | |||||
| SR39 | 1 | 33, 58–65 | |||||
| A168H | 1 | 66 | |||||
| A167Y | 1 | 66, 69–72 | |||||
| Q125N | 1, 2 | 68, 71, 72 | |||||
|
| |||||||
| Bacterial Cytosine Deaminase (bCD) | Escherichia coli –codA | 48kDa homohexamer | Cytosine | 5-FC | D314 mutants | 1, 2, 4 | 31, 34, 44, 74, 79 |
| bCD1525 | 1, 2, 4 | 31 | |||||
|
| |||||||
| Yeast Cytosine Deaminase (yCD) | Saccharomyces cerevisiae –fcy1 | 18kDa homodimer | Cytosine | 5-FC | yCD triple | 1, 2, 4 | 32, 81 |
| D92E | 1, 2, 4 | 32 | |||||
|
| |||||||
| Drosophila melanogaster Deoxynucleoside Kinase (Dm-dNK) | Drosophilamelanogaster –dNK | 29kDa hom | All four deo | AZT, ddC | MuD | 1, 5 | 84–86 |
| CdA | B5 | 1 | 85, 87 | ||||
| F-AraA | B10 | 1, 3 | 85 | ||||
| GCV, AraG | M88R | 1 | 87–90 | ||||
| D4T | HDHD-12, HD-16 | 1, 5 | 91 | ||||
| ddT | R4.V3 | 1 | 92 | ||||
|
| |||||||
| Deoxycytidine Kinase (dCK) | Homo sapiens –dCK | 30.5kDa homodimer | Deoxycytidine | dFdC, AraC, L-dT | DMMA, DMLA | 1, 3 | 100 |
| AZT | EpTK6 | 1, 3, 5 | 101 | ||||
| dFdC, AraC | Ser-74 | 1, 3 | 102, 103 | ||||
|
| |||||||
| Purine Nucleoside Phosphorylase (PNP) | Escherichia coli -deoD | 150kDa homodimer | Purine ribonucleosides | Me(talo)-MeP-R | M64V | 1, 4 | 115, 117, 118 |
1. DNA synthesis
2. Thymidylate synthetase
3. Ribonucleotide reductase
4. RNA/protein synthesis
5. Reverse transcriptase
ENZYME ENGINEERING
Oligonucleotide-directed, random and recombination mutagenesis strategies are three cornerstones of protein engineering methodologies that have been widely used by many investigators as a means to understand and to alter enzyme function [39–42]. Oligonucleotide-directed mutagenesis can generally be divided into two methods: site-directed and random mutagenesis. Site-directed mutagenesis is usually done to introduce rationally chosen single amino acid substitutions. This technique is relatively easy to perform and commercial kits are available (e.g., Stratagene QuikChange® Mutagenesis Kit). The ability to create enzymes with significantly altered substrate preferences, however, often requires restructuring the active site to accommodate nucleoside analog structures and cannot always be predicted. Analysis of single amino acid substitutions, and/or a combination of multiple substitutions can be laborious and time consuming [43]. White some beneficial mutants may be generated using this technique [44], some of the most phenotypically desirable mutants (in this case, those with the most improvement towards their prodrugs) have been discovered by random mutagenesis and almost always contain multiple mutations [31, 33, 41]. Random mutagenesis is a powerful method used to introduce multiple random mutations within a target region or across the entire open reading frame and is used to generate large random sequence libraries containing millions of variants [39, 42,43].
Error prone PCR, one such random mutagenesis method, is a relatively straightforward technique used to introduce random mutation(s) across the entire open reading frame of the gene of interest. This approach requires no structural or mechanistic information and can be used in sequential rounds of mutagenesis to diversify amino acid substitutions. The low fidelity of Taq DNA polymerase combined with an increased concentration of MgCl2, the addition of MnCl2, and increased and/or unbalanced dNTP levels are key elements to perform error prone PCR [40, 42]. This technique can be easily done and many kits are commercially available (e.g., Stratagene’s GeneMorph and Clontech’s Diversify PCR Random Mutagenesis Kit) [40], DNA shuffling may also be used in combination with error prone PCR to provide a degree of recombination among mutations in the PCR pool. DNA shuffling is initiated by digesting the entire PCR-amplified gene fragment pool with DNase I to generate short fragments that are then subjected to primerless PCR wherein the fragments prime each other and lead to the generation of a recombined full-length gene. This approach can be repeated in several rounds and consequently results in an increased library size of mutants and an enhanced opportunity to obtain phenotypically desirable mutants [42].
A more directed approach, region-specific or random sequence mutagenesis, is used to target specific residues thought to be important in catalysis or substrate binding for mutagenesis. The first step in performing region-specific random mutagenesis is to design one or two overlapping random oligonucleotides. The random oligonucleotides are designed to introduce mutations within the target region and to exclude amino acid residues that are conserved (or immutable) in sequence alignments. Once annealed and extended with DNA polymerase, the ends are then digested with restriction enzymes to allow directional ligation into the plasmid vector containing the rest of the gene of interest in which the targeted region has been excised. Following transformation into the appropriate E. coli strain, the bacterial transformants are plated on selective media.
With both site-directed and random mutagenesis strategies, the ability to rapidly and efficiently identify functional variants is greatly facilitated by the use of genetic complementation or positive selection schemes. Especially in the case of random mutagenesis where large numbers (millions) of mutants are generated, the establishment of a positive selection system allows for the facile identification of only active mutants without the necessity to screen mutants individually, a very labor intensive task. E. coli is predominantly used because it yields high numbers of transformants, grows rapidly and numerous strains with genetic deficiencies are available. For example, a thymidine kinase deficient E. coli has been widely used to select functional plasmid-borne HSVTK variants when plated onto selective media. With the establishment of a positive selection system, the inclusion of the prodrug in the selective medium can provide the means to select for variants with activity towards the prodrug (negative selection). The prodrug concentration is set to allow for E. coli harboring the wt gene to grow and any variant with improved prodrug activation capacity, will succumb. In this fashion one can identify functional mutants by positive selection and then test those for prodrug activity by negative selection using prodrug-containing plates.
In the follow sections we discuss six distinct suicide enzymes that have been manipulated through mutagenesis to alter their respective prodrug activity for suicide gene therapy applications.
SUICIDE GENE/PRODRUG THERAPY FOR CANCER
Herpes Simplex Virus Type 1 Thymidine Kinase (HSVTK)
Herpes Simplex Virus type 1 thymidine kinase (HSVTK, EC 2.7.1.21), a homodimer with a subunit molecular mass of 45kDa Table 1 Fig. (3A), is responsible for the phosphorylation of thymidine, deoxycytidine, deoxythymidylate (dTMP) as well as various pharmaceutically important pyrimidine and guanosine analogs. Of particular note, HSVTK is responsible for the initial and rate limiting phosphorylation of the antiviral guanosine analogs acyclovir (ACV) and ganciclovir (GCV). Once monophosphorylated these analogs can be further phosphorylated by endogenous enzymes (guanylate kinase and nucleoside diphosphokinase) before being incorporated into nascent DNA to cause double strand destabilization and, subsequently, cell death Fig. (2)[45–47]. Clinically, GCV has been successfully used since the 1980s to treat cytomegalovirus (CMV) infections while ACV is widely used to treat and control alpha herpes infections such as those that cause cold sores and genital lesions. In 1986 Moolten first suggested the use of HSVTK in combination with GCV as a novel suicide gene/prodrug system for cancer treatment [6] and two recent review articles highlight advances that have been made using the HSVTK/GCV approach [48, 49]. One advantage of HSVTK is that it exhibits approximately 1000-fold higher activity towards GCV than cytosolic thymidine kinase, making it an excellent candidate for suicide gene therapy [6, 50]. ACV was initially considered a more suitable prodrug because of its low toxicity, however, the Km value of HSVTK towards ACV is extremely high (Km ~400μM) compared to that of GCV (47μM) which reduces its effective application in suicide gene therapy strategies [51]. Today GCV is the prodrug of choice for suicide gene therapy applications with HSVTK although there are still dose limitations with the use of this prodrug.
Fig. 3.

Crystal structures of suicide enzymes. Residues targeted by mutagenesis are highlighted in red and the substrate of each enzyme is highlighted in orange. (A) HSVTK (PDB: 1K12), (B) bCD (PDB: 1K70), (C) yCD (PDB: 1P60), (D) Dm-dNK (PDB: 10T3), (E) dCK (PDB: 2A30), (F) E. coli PNP (PDB: 1PK7).
Fig. 2.
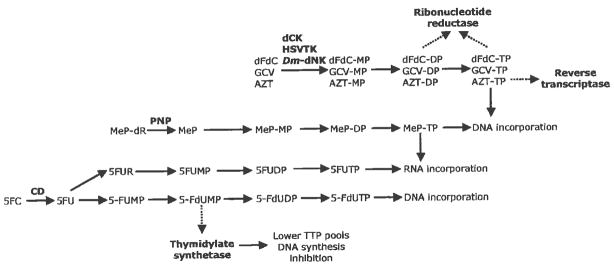
Prodrug activation pathways and mechanisms of action. A representative prodrug for each suicide enzyme and their corresponding mechanism(s) of action are denoted in this figure. CD, cytosine deaminase; HSVTK, Herpes Simplex Virus thymidine kinase; dCK, deoxycytidine kinase; PNP, E. coli purine nucleoside phosphorylase; Dm-dNK, Drosophila melanogaster deoxynucleoside kinase; 5FC, 5-fluorocytosine; MeP-dR, 9-(2-deoxy-β-D-arabinofuranosyl)-6-methylpurine; GCV, ganciclovir; dFdC, gemcitabine; AZT, azidothymidine. Dotted lines = enzyme inhibition.
Despite the more favorable kinetic parameters with GCV compared to ACV, the Km value of HSVTK for GCV is approximately 100-fold higher than its Km value for thymidine (Km=47μM versus Km=0.5μM) [51]. Because of competition for the active site, clinically relevant doses of GCV are necessarily high and result in undesirable side effects such as severe bone marrow toxicity and myelosuppression in patients [7]. As a means to overcome this, several studies were directed at creating novel HSVTK variants with increased activity towards GCV, so that lower doses of this prodrug could be used to achieve efficient tumor cell death [33, 52–54].
Alignment studies between thymidine kinases from members of the Herpesviridae family reveal six highly conserved motifs. Two motifs (Sites 3, residues 162–164 and 4, residues 170–172) were suggested to play a role in substrate binding and residues neighboring these conserved sites were targeted for random sequence mutagenesis studies [55]. From a library of over 1 million, two key HSVTK mutants containing 5 and 4 amino acid substitutions, mutant 30 (L159I/I160L/F161A/A168Y/L169F) and mutant 75 (I160L/F161L/A168V/L169M), respectively, were isolated and characterized. Initial results using baby hamster kidney (BHK) tk− cells stably transfected with mutants 30 or 75 revealed a 4.5- and 43-fold decrease in IC50 for GCV, respectively, when compared to wt HSVTK [52]. In a separate study, rat C6 glioma cells stably transfected with mutant 30 showed a significant decrease in IC50 value of 0.025μM for GCV when compared to the IC50 value exhibited by wt enzyme (30μM for GCV) whereas mutant 75 was unable to significantly improve the sensitivity of cells to GCV, relative to wt HSV-transfected cells [56]. Kinetic parameters for mutants 30 and 75 suggest two distinct mechanisms for the observed difference in cell line specific prodrug-mediated cell killing. When thymidine is used as a substrate, mutant 30 displays a 3,000-fold reduction in overall enzyme catalytic efficiency compared to wt enzyme. In contrast, mutant 75 exhibits a Km for thymidine similar to that of wt HSVTK and a 5-fold lower Km for GCV. While mutant 75 demonstrates a more robust GCV-mediated cell killing in BHK tk− transfected cells, this did not translate to cells containing a functional endogenous thymidine kinase. Because both endogenous thymidine and administered GCV compete for the enzyme active site, it is important to consider the ratio of specificity constants for both substrates. Taking this into consideration, mutant 75 displays a 14-fold higher value than wt HSVTK whereas mutant 30 displays a 67-fold improvement in relative catalytic activity towards GCV. Therefore, mutant 30 was the focus of further characterization. From in vivo studies a 10-fold decrease in GCV concentration was shown to be sufficient to achieve tumor growth inhibition in nude mice bearing mutant 30 transfected tumor rat C6 cells compared to those expressing wt HSVTK [56]. Further in vivo bystander experiments with C6 tumors containing an 80:20 ratio of vector transfected to wt HSVTK. or mutant 30 expressing cells reiterated the superior antitumor activity of mutant 30. In this study mutant 30 expressing tumors demonstrated a significant impairment in tumor growth in mice treated with GCV (5mg/kg) relative to the saline control while wt type HSVTK expressing tumors displayed no bystander activity at the GCV concentration used [56], Other studies using mutant 30 in vitro demonstrated high sensitivity in many different tumor cell lines towards GCV with the brain cancer cell line, SKMG-4 being the most sensitive [57].
While mutant 30 demonstrated significant improvement in the ability to sensitize transfected cells to GCV at low concentrations, additional improvements in the kinetic parameters toward the prodrugs was suggested as a means to provide more clinically relevant reductions in GCV doses. Towards that end, a restricted or semi-random library was generated using select codons at 5 residues within the putative active site. From that mutagenesis and subsequent in vitro studies, rat C6 glioma cells stably transfected with mutant SR39 (L159I/I160F/F161L/A168F/L169M) exhibited the greatest GCV sensitivity among seven identified active HSVTK variants. The observed low IC50 value of 0.017μM with SR39 corresponds to a 294-fold reduction in prodrug dose relative to wt HSVTK for GCV. In vivo xenograft tumor studies demonstrated that SR39 was effective at inhibiting tumor growth, requiring a 10-fold lower GCV concentration (0.5mg/kg) needed to see growth inhibition of tumors expressing wt HSVTK (5mg/kg) [33]. This low GCV dose (0.5mg/kg) is a significant decrease from reported doses used in other in vivo mouse experiments (up to 300mg/kg/day) [7]. Mutant SR39 has also shown to be an effective suicide gene for metastatic prostate cancer where it demonstrated increased sensitivity towards GCV as compared to wt HSVTK or mutants 30 and 75 [58]. In a xenograft tumor model using immunodeficient (SCID) mice injected with metastatic prostate CL1 cells expressing the suicide enzymes and treated with 20mg/kg GCV, SR39 showed a 63% reduction in tumor growth in comparison with tumors expressing HSVTK or mutants 30 or 75 [58]. Mutant SR39 as well as mutant 30 have also been shown to increase tumor cell killing of various cell lines following treatment with GCV as indicated by lower IC50 values when compared to wt HSVTK. Wiewrodt et al. (2003) reported a reduction in IC50 values displayed by mutant 30 and SR39 for GCV ranging from 1.1- to 5.9-fold lower than wt HSVTK in several cell lines including rat glioma (C6), human mesothelioma (I-45, REN, LRK, H513, H2052), human NSCLC (A549), cervical carcinoma (C33A) and mouse fibroblast tumors (EJ62), that also correlated with xenograft tumor studies in SCID mice and in vivo bystander experiments using pretransduced tumors containing a 95:5 ratio of untransduced cells to cells infected with wt HSVTK, mutant 30 or mutant SR39 expressing adenoviral vectors [59]. Ardiani et al. (2009) compared the bystander activity of mutant 30 and mutant SR39 in vitro and demonstrated that both mutants displayed significant and similar bystander activity at a GCV concentration where co-cultures containing vector transfected and wt HSVTK expressing rat C6 cells remained unresponsive. Further in vivo bystander experiments demonstrated that wt HSVTK and mutant 30 were unable to exhibit sufficient bystander activity while mutant SR39 induced a strong bystander effect at the low GCV (1mg/kg) dose used [60]. In addition to its application for suicide gene therapy strategies, mutant SR39 was previously reported to have improved sensitivity for non-invasive imaging in positron emission tomography (PET) [61–65].
Balzarini et al. (2006) employed site-directed mutagenesis to target residues in the active site as a means to understand the influence of particular side chains on substrate interactions and catalysis and to alter substrate preference. One such mutant, A168H abolished thymidine kinase activity while preserving GCV activity approximately 4-fold higher than that of wt HSVTK and preliminary reports indicated that A168H transfected OST TK− cells were more sensitive to several prodrugs compared to parental cells although it was not noted to what degree [66]. An extension of those studies employed a splice corrected and codon-optimized version of A168H, TK.007, that demonstrated cell growth inhibition of several cancer cell lines in the presence of GCV with IC50 values ranging from 25 to 100-fold lower than for the splice corrected HSVTK [67]. Significant in vivo tumor growth inhibition with TK.007 transduced cells was also observed using a human glioblastoma xenograft mouse model following GCV administration of 10mg/kg/day for five days. In bystander experiments using 50mg/kg daily GCV doses, mixtures of 10% or 50% TK.007 transduced cell tumors exhibited tumor growth inhibition compared to controls [67].
Another candidate for site-directed mutagenesis was the active site residue Q125 due to its ability to form hydrogen bonds with substrates. To test the role played by this residue in HSVTK activity, Hinds et al. (2000) created three Q125 mutants (Q125E, Q125N and Q125D) [68]. Among these three mutants, HSVTK variant Q125N retained both thymidine and GCV activity although much reduced from wt HSVTK with a 10-fold reduction in kcat/Km for thymidine and a 4-fold reduction in the kcat/Km for GCV [68]. In other studies, Q125N showed GCV phosphorylation comparable to wt HSVTK in mouse embryonic fibroblast NIH3T3 cells and human colorectal HCT116 cells. Furthermore, Q125N resulted in the selective abolishment of HSVTK thymidylate kinase activity toward thymidine-MP and bromovinylde-oxyuridine-monophosphate (BVDU-MP). Computational modeling of the HSVTK active site led Balzarini et al. (2002) to create the site-specific mutant A167Y. This HSVTK variant is reported to display a complete loss in pyrimidine nucleoside kinase activity but maintains purine nucleoside kinase activity towards analogs like GCV [66, 69, 70]. Since intracellular accumulation of BVDU-MP results in the inhibition of thymidylate synthetase, the enzyme responsible for converting dUMP to dTMP, a new approach was proposed to take advantage of two mechanisms of drug action by using HSVTK-Q125N and HSVTK-A167Y with prodrugs BVDU and GCV [71, 72].
While many studies have been devoted to HSVTK, only a few have identified variants with significantly altered substrate preferences and, of those, only a small subset has been evaluated in vitro and in vivo. Of those, HSVTK SR39 has garnered the most attention as a therapeutically important contributor in reducing GCV concentrations necessary for tumor growth inhibition and as an important PET imaging agent.
Escherichia coli Cytosine Deaminase
Cytosine deaminase (CD; EC 3.5.4.1) is an enzyme in the pyrimidine salvage pathway that catalyzes the deamination of cytosine to form uracil and ammonia [73]. CD from E. coli (bCD) is a hexamer of 48kDA subunits with a catalytic metal iron Table 1, Fig. (3B). This enzyme is absent in mammals and uniquely present in fungi and bacteria. It is used in suicide gene therapy because of its ability to deaminate the anti-fungal drug, 5-fluorocytosine (5FC), to 5-fluorouracil (5FU), a potent anti-neoplastic drug [74]. 5FC was originally synthesized in 1957 as a potential antitumor agent but was later found to be inactive against tumors [75]. It was not until 1993 when the bCD gene (codA) became available, that the use of bCD in combination with 5FC became a promising gene therapy approach for cancer [76]. 5FU, the active anti-metabolite of 5FC, has been widely used in first line chemotherapeutic regimens to treat colorectal, breast and prostate cancers. In cells, 5FU is converted into several active metabolites: 5-fluoro-2′-deoxyuridine 5′-monophosphate (5FdUMP), 5-fluorodeoxy uridine-triphosphate (5FdUTP) and 5-fluorouridine-triphosphate (5FUTP), all of which cause cell apoptosis via different mechanisms Fig. (2) [77]. The major mechanism of action is inhibition of thymidylate synthetase (TS) by 5FdUMP, which results in disruption of dTTP synthesis and ultimately, DNA synthesis and repair [75]. 5FdUTP can be misincorporated during DNA synthesis and repaired by the base excision repair enzyme uracil-N-glycosylase (UNG). However, repair is generally futile and results in further false-nucleotide incorporations that cause DNA strand breaks and cell death [77]. Finally, 5FUTP can also be mis incorporated into RNA synthesis and interrupt normal RNA processing and function [77].
One of the advantages of utilizing the CD/5FC strategy in suicide gene therapy compared to HSVTK/GCV is that the bystander effect mediated by this system functions independent of gap junctions [7]. This is because 5FU is a small, uncharged molecule and therefore able to diffuse across cellular membranes. An additional advantage of the CD/5FC system, when compared to direct 5FU chemotherapy treatment, that can be clinically beneficial in patients is the ability of 5FC to cross the blood-brain barrier, a unique property that 5FU lacks [75]. Furthermore, 5FU has radiosensitizing properties, that serves to augment drug potency when combined with radiotherapy [78]. Despite these advantages, the presence of bacteria in the intestinal flora can be problematic as they can deaminate 5FC that can lead to normal cell killing and unwanted side effects, such as severe bone marrow depression and gastrointestinal complications [75]. Additionally, the poor activity of bCD towards 5FC (a 36-fold lower preference for 5FC compared to cytosine) serves to exacerbate this problem since high doses of 5FC are needed to achieve significant tumor killing.
Several mutagenesis studies aimed to obtain bCD variants with improved activity towards 5FC and to identify what residues may be responsible for this relative specificity have been done in an attempt to resolve these problems [31, 44, 74]. Mahan et al. (2004) performed a site-directed mutagenesis study targeting residues 310–320 since this region has been shown to undergo a conformational change upon substrate binding and contains several amino acids in direct contact with the substrate. In that study, residues 310–320 were individually replaced by alanine (alanine scanning mutagenesis), and analyzed for enhanced 5FC activity relative to wt bCD in E. coli in a negative selection assay. One bCD mutant, D314A, demonstrated a significant decrease in cytosine activity (17-fold) as well as a slight increase in activity toward 5FC (2-fold) [44], In a separate study, error prone PCR was used to introduce mutations randomly throughout the entire bCD open reading frame [74], The mutant that conferred the highest degree of sensitivity to 5FC in the negative selection system contained two amino acid substitutions, Q102R and D314G, the latter of which was identified as being solely responsible for the enhanced sensitivity. To further investigate the role of D314 in substrate selectivity, several other substitutions were made at this position. All mutants from this later study, except mutant D314S, were inactive towards cytosine as assessed by genetic complementation [74]. Kinetic studies indicated that bCD mutants D314G and D314S displayed a 2.5- and 4.2-fold shift in substrate preference towards 5FC, respectively [44]. However in vitro analysis using rat C6 glioma cells revealed that all three bCD mutants (D314G, D314A, and D314S) provided modest benefit with only a 2- to 4-fold decrease in IC50 for 5FC as compared to wt bCD transfectants [31]. Since mutant D314A appeared to exhibit the highest shift in substrate preference towards 5FC, Buchsbaum et al. (2007) constructed adenoviral vectors encoding mutant D314A (AdbCD-D314A) and used the construct to treat several human glioma cell lines in combination with 5FC and radiation treatment. Results from in vitro studies indicated a more potent tumor cytotoxicity effect in all three tested human glioma cell lines (D54MG, U87MG, and U251MG) in comparison with the use of wt bCD (AdbCD-wt). In all cell lines, AdbCD-D314A demonstrated a reduction in IC50 values for 5FC of approximately 7- to 33-fold lower than that of AdbCD-wt [34]. Significant tumor growth inhibition was seen in mice receiving adenoviral vectors expressing D314A, 500mg/kg/d 5FC, and irradiation with 2Gy when compared to mice receiving adenoviral vectors expressing wt enzyme and the same amount of 5FC and radiation dose. When the radiation dose was increased to 5Gy in the mice receiving D314A and 500mg/kg/d 5FC, 50% tumor reduction was observed. The same AdbCD-D314A vectors were also used to treat pancreatic cancer cells and similar results with mutant D314A displaying increased tumor killing in vitro and in vivo were seen [79]. Deng et al. (2011) showed similar improvements using human colon cancer xenograft models after lentiviral delivery of bCD-D314A and 5FC treatments [80].
More recently, using structural information as well as previous mutagenesis results. Fuchita et al. (2009) targeted two regions lining the active site of bCD (residues 149–159 and 310–320) for random mutagenesis in an effort to identify mutants with the capacity to confer enhanced sensitivity to 5FC in vitro and in vivo [31]. From this double site-targeted library containing over 1.35 million transformants, three bCD variants were identified that conferred sensitivity towards 5FC in E. coli and were further analyzed biochemically and in three different cancer cell lines. Kinetic and in vitro studies revealed that all mutants showed improvement in 5FC activity. One particular mutant (bCD1525) containing three amino acid substitutions (V152A/F316C/D317G) displayed the greatest sensitivity with an approximately 19-fold shift in substrate preference toward 5FC. Results from prodrug sensitivity assays demonstrate that bCDl525 confers increased sensitivity to 5FC in all cell lines examined, albeit to varying degrees in the different cell lines. In rat C6 glioma transfectants, a modest reduction in IC50 of approximately 2.7-fold was observed compared to wt bCD-transfected cells. The IC50 value of mutant bCD1525-transfected human prostate DU145 cells was 5-fold lower than wt bCD-transfectants. bCD1525 displayed the greatest reduction in IC50 value in human colorectal HCT116 transfectants at 17-fold lower values than wt bCD transfected cells. Furthermore, bCD1525 displayed a robust bystander effect in every cell line examined [31].
Fuchita et al. (2009) reported that significant tumor growth inhibition was observed in a xenograft tumor model using nude mice harboring bCD 1525-transfected tumors compared to mice harboring wt bCD-transfected tumors when 5FC was administered. Throughout the course of 5FC administration, wt bCD tumors increased in volume by 30.8-fold whereas the bCD1525 tumors increased by only 5.7-fold. In an attempt to understand the structural changes incurred by the three amino acid substitutions found in bCD1525, X-ray crystallography experiments were carried out. Interestingly, the D317G mutation found in bCD1525 allows D314 to swing away from the fluorinated substrate and appears to recapitulate the cavity observed in the D314A structure [31]. Thus the improved kinetic parameters towards 5FC are likely a reflection of the structural changes observed with substitutions of smaller residues at positions D314 or D317 to accommodate 5FC. Taken together, the kinetic, in vitro, in vivo and structural studies indicate that bCD1525 is the most suitable choice of bCD gene for gene therapy applications.
Saccharomyces cerevisiae Cytosine Deaminase
Saccharomyces cerevisiae or yeast cytosine deaminase (yCD, EC 3.5.4.1) is a homodimer of 17.5kDa subunits Table 1, Fig. (3C) and has been shown to be more active towards 5FC than bCD (22-fold lower Km) with a slightly better catalytic efficiency (kcat/Km) toward 5FC relative to its natural substrate cytosine. Yet, yCD is thermolabile at physiological temperatures with a half-life of around 4 hrs, a characteristic that may preclude its full therapeutic potential [81]. To extend the half-life and enhance the stability of yCD, Korkegian et al. (2005) took a computational approach to thermostabilizing yCD by utilizing an energy function that evaluates the fitness of a particular sequence for a given fold and a search algorithm in the program Rosetta Design. A series of point mutations were predicted to thermostabilize yCD and initial experimental studies identified two interacting mutations, A23Land I140L to be key to its thermostabilization. The mutant with these two substitutions was designated yCDdouble while an additional distant mutation, VI081, was added onto yCDdouble to create the mutant yCDtriple. Both of these mutants displayed increased apparent melting temperatures (Tm). longer half-life activity and similar catalytic efficiency relative to wt yCD [81]. Of the two mutants, yCDtriple displayed the greatest improvement in thermostability with a 10°C increase in Tm and a 30-fold increase in half-life (117 hr) at 50°C relative to wt yCD. The thermostabilization of yCDdouble had a lesser effect with a 6°C and 5-fold (21 hr) increase in Tm and half-life, respectively. Structural studies of yCDdouble and yCDtriple found that the mutated residues packed more tightly in the enzyme’s hydrophobic core and created stabilizing interactions, such as hydrophobic packing amongst neighboring residues, to allow for increased yCD thermostability [81].
In an attempt to improve the catalytic efficiency of yCD towards 5FC, Stolworthy et al. (2008) created a large library of yCD variants by regio-specific random mutagenesis and the identification of functional variants by genetic complementation in E. coli. Three single site mutants (D92E, M93L and I98L) were found to confer the highest degree of 5FC sensitivity in negative complementation assays. Of these three, D92E conferred the greatest reduction in IC50 (~30%) for 5FC in rat C6 glioma cells. These mutants were then overlaid onto the thermostabilized mutant yCDtriple and tested in vitro using C6 cells to investigate the combined effects of these mutations in terms of their ability to confer enhanced 5FC sensitivity. Surprisingly, none of the overlaid mutants enhanced 5FC-mediated cytotoxicity but rather negated the effects of yCDtriple. The thermostabilized mutant yCDtrjple alone displayed the greatest enhancement in sensitivity, with an approximate 50% reduction in IC50 value relative to wt yCD, indicating that the primary limiting behavior of yCD may be its thermostability. A combination of enzyme kinetics, thermostability studies and structural information revealed that the D92E mutation failed to significantly influence the kinetic parameters of yCD but rather increased its thermostability by forming a stabilizing interaction at the dimer interface of the enzyme. This interaction potentially reduced the flexibility of the protein backbone and may be the reason why the overlaid mutant yCDtriple/D92E failed to synergistically amplify 5FC sensitivity in vitro. Both yCDtriple and D92E were later tested in vivo using a xenograft tumor model and mice bearing yCDtriple-expressing tumors treated with 500mg/kg/day 5FC displayed the greatest reduction in tumor growth [32]. These results indicate that yCDtriple is the most efficient yCD variant tested to date.
Drosophila melanogaster Deoxyribonucleoside Kinase (Dm-dNK)
Drosophila melanogaster deoxyribonucleoside kinase (Dm-dNK; EC 2.7.1.145) is a 29kDa homodimeric, multisubstrate kinase Table 1, Fig. (3D) able to phosphorylate all four natural deoxyribonucleosides required for DNA synthesis. In addition to its broad substrate specificity, Dm-dNK. exhibits higher catalytic rates toward these natural deoxynucleosides and several nucleoside analogs as compared to mammalian deoxynucleoside kinases [82]. These distinctive characteristics make Dm-dNK a unique candidate for suicide gene therapy applications. Zheng et al. (2000) reported that the TK deficient osteosarcoma 143B and MIA PaCa-2 adenocarcinoma human cell lines transduced with a Dm-dNK expressing retroviral vector demonstrated up to 400- to 6,400-fold increases in sensitivity, respectively, towards a number of pyrimidine and purine nucleoside analogs compared to untransduced cells. This enhanced sensitivity was more prominent when pyrimidine analogs were used as Dm-dNK naturally prefers pyrimidine substrates [83]. Bystander effect studies using the combination of Dm-dNK and the pyrimidine analog 2′-bromovinyl-2′-deoxyuridine (BVDU) revealed that the presence of 10% Dm-dNK expressing cells could cause a significant bystander effect (>50% cell killing) in mixed cultures of 143B osteosarcoma cells. However, this combination was unable to induce a bystander effect in MIA PaCa-2 cells, a cell line with low connexin expression levels and therefore presumably unable to efficiently transfer activated BVDU via gap junctions [3,4].
As a means to manipulate the substrate specificity of Dm-dNK, and further enhance cancer cell prodrug sensitivity, multiple enzyme engineering strategies have been implemented to create Dm-dNK mutants with improved kinetic properties for nucleoside analog activation. Knecht et al. (2000) created a series of first generation mutants via high frequency random mutagenesis and thymidine kinase genetic complementation in E. coli Of the thirteen reported mutants, the double mutant MuD (N45D/N64D) was chosen for further molecular characterization due to its ability to lower lethal dose (LD100) values in the tdk deficient E. coli KY895 strain for all four nucleoside analogs tested. Kinetic analyses predicted a 324-fold and 28-fold increase in specificity towards the prodrugs azidothymidine (AZT) and dideoxycytidine (ddC), respectively. This increased specificity was due to a decreased activity towards natural substrates concomitant with decreased feedback regulation levels by dTTP [84].
In an attempt to further improve kinetic properties for nucleoside analog activation Knecht et al. (2007) performed DNA shuffling using MuD and other first generation mutant genes. Based on kinetic analysis results, two mutants from this library, B5 (V84A/N210D/L239P) and B10 (N45D/N64D/N210D/L239P), were transduced into multiple human cancer cell lines and evaluated for optimal suicide gene/prodrug combinations. Human 143B osteosarcoma cells transduced with mutant B5 were 9-fold more sensitive to the cytotoxic effects of cladribine (CdA) relative to wt Dm-dNK transduced cells. A 470-fold difference in sensitivity was seen when IC50 values were compared between mutant B5 transduced cells and the parental cell line. Human U87MG glioblastoma cells transduced with mutant B10 were ~1000-fold more sensitive to fludarabine (F-AraA) relative to wt Dm-dNK transduced cells. The success of these mutants in combination with their respective prodrugs can be attributed to their decreased ability to phosphorylate natural pyrimidine substrates, which wt Dm-dNK naturally prefers, while maintaining similar kcat/Km ratios for the tested purine analogs [85].
Structure-function analysis of the Dm-dNK MuD variant was performed to elucidate the specific function of the two mutated residues, N45D and N64D. Results suggest the N64D mutation is responsible for enzymatic changes as the introduction of a charged aspartate residue at this location destabilizes the LID region, a flexible loop structure important in ATP binding and catalysis. This destabilization is thought to provide space for more bulky 3′-substituents, like those found in AZT, while negatively influencing feedback inhibitor interactions, such as those with dTTP [86]. The V84A mutation found in mutant B5 defines its altered substrate specificity as this amino acid exchange mimics the alanine found at the same position in human deoxycytidine kinase, and may be responsible for the increased preference towards deoxycytidine and its analogs [87]. The role of the additional substitutions found in mutant B5 is unclear although it has been suggested that these mutations may impart a general effect on Dm-dNK sensitivity to nucleoside analogs [85].
A separate study was performed by Knecht et al. (2002) to modify Dm-dNK to display a preference for purine substrates. A comparison between the three-dimensional structures of Dm-dNK, human deoxyguanosine kinase, and HSVTK was used to identify three amino acid residues (V84, M88, A110) lining the substrate binding pocket of Dm-dNK as candidates for involvement in purine specificity. Mutations at these residues converted Dm-dNK from a mainly pyrimidine specific to purine specific kinase with expanded activity towards multiple purine analogs [87]. Solaroli et al. (2007) further characterized these engineered enzymes in terms of their ability to sensitize 143B osteosarcoma cells to various purine analogs, including GCV and nelarabine (AraG). In these studies, cells transduced with the most promising mutant, M88R, acquired more than 10-fold and ~100-fold lower IC50 levels for GCV and AraG, respectively, compared to wt Dm-dNK transduced cells. The increased sensitivity to these purine analogs has been attributed to the arginine substitution of methionine at position 88 as this reflects the same position in human deoxyguanosine kinase that has been shown to be responsible for purine deoxynucleoside selectivity [87]. Although Dm-dNK the 20 amino acid C-terminal truncated form of has been reported to increase the catalytic rate of Dm-dNK, the truncated form of mutant M88R did not increase cancer cell sensitivity to either GCV or AraG [88, 89]
Gerth et al. (2007) reported a recombination mutagenesis study aimed at creating humanized enzymes with increased activity towards nucleoside analogs. Hybrid enzymes of Dm-dNK and human thymidine kinase 2 (hTK2) were produced via the non-homologous recombination techniques ITCHY and SCRATCHY and then screened for thymidine kinase activity using genetic complementation. Two of the most active chimeras, HDHD-12 and HD-16, were tested for activity to multiple nucleoside analogs. Of greatest interest was their novel activity towards the anti-HIV prodrug 2′,3′-didehydro-3′-deoxythymidine (d4T), an activity that neither wt Dm-dNK nor hTK2 possess, and was also two orders of magnitude greater than all reported natural or engineered deoxynucleoside kinases [90]. The reason for this novel activity was unclear from available structural information.
Liu et al. (2009) designed and validated a novel FACS-based approach to screening for functional Dm-dNK mutants with altered substrate specificity to the prodrug dideoxythymidine (ddT). Combined with conventional random mutagenesis and DNA shuffling techniques, this approach identified a Dm-dNK variant, R4.V3 (T85M/E172V/Y179F/H193Y), with a 20-fold increased substrate preference for ddT over thymidine. Both active site mutations El72V and Y179F were determined to discriminate against natural substrates by both removing favorable hydrogen-bonding interactions in the Dm-dNK binding pocket and optimizing the pocket for the hydrophobic 2′,3′-dideoxyribose moiety of ddT. While the H193Y mutation was found neutral, the T85M mutation was argued to provide favorable binding interactions for the fluorescent analog of ddT selected for during screening by enlarging the Dm-dNK substrate binding pocket. Reversion of this mutation allowed for a 10-fold improvement in catalytic performance for ddT [91]. A more in depth coverage of the multiple strategies used and recent mutagenesis efforts performed by the Lutz group is the focus of a recent review [92].
While the unique properties of the majority of the aforementioned Dm-dNK mutants have shown promise in vitro, these mutants have yet to be further characterized in an in vivo or clinical setting. For example, although advancements have been made to improve Dm-dNK activity towards GCV, the HSVTK variant SR39 remains the suicide gene of choice with GCV. Nonetheless, the future use of Dm-dNK variants for suicide gene therapy applications may augment the cytotoxic potential of current strategies by providing a range of new suicide gene/prodrug combinations.
Human Deoxycytidine Kinase (dCK)
Human deoxycytidine kinase (dCK; EC 2.7.1.74) is a 30.5kDa homodimeric enzyme Table 1, Fig. (3E) in the salvage pathway of deoxyribonucleosides and is responsible for activating all natural deoxyribonucleosides, excluding thymidine, as precursors for DNA synthesis. Due to its broad substrate specificity, dCK is able to activate multiple nucleoside analogs effective against different types of cancer. However, wt dCK is intrinsically a relatively poor catalyst with low turnover rates and prodrug activation is dependent on its expression levels [93]. Indeed, nucleoside analogs that are efficient substrates of dCK, such as cytarabine (AraC), fludarabine (F-AraA), cladribine (CdA), and gemcitabine (dFdC) Fig. (2), are effective anti-leukemic agents as lymphoblasts have been shown to have high dCK expression levels whereas cancer cells lacking dCK activity are resistant to these same analogs. To overcome this resistance, and restore drug sensitivity, Stegmann et al. (1995) transfected an AraC resistant rat leukemic cell line with a vector encoding the wt dCK gene. IC50 levels for the resistant cell line were >10mM, while dCK transfected cells fully restored drug sensitivity to levels comparable to the parental cell line (IC50 ~0.85μM) [94]. In other studies, dCK has been used as a candidate suicide enzyme against different cancer cell types including, but not limited to, 9L (glioma), Panel (pancreatic carcinoma), MCF-7 (breast carcinoma) and HT-29 (colon carcinoma) cells [95–97]. Similar to the bystander effect with HSVTK and GCV, gap junctions and direct cell-to-cell contact are key factors for the bystander effect of nucleoside analogs activated by dCK [96, 98].
As a means to improve the catalytic efficiency of dCK, and in turn further improve drug sensitivity and selectivity, Sabini et al. (2007) exploited structural information of dCK to mutate three active site residues to those found in Dm-dNK. The designed A100V/R104M/D133A triple mutant displayed a 50-fold increase in efficiency toward its natural substrate deoxycytidine and a 4-fold increase in efficiency toward the prodrug gemcitabine [93]. Harza et al. (2009) sought to better understand the differences in substrate specificity and enzymatic activity between dCK and Dm-dNK, or more specifically why the Dm-dNK enzyme is more efficient than dCK and displays thymidine kinase activity. These studies further emphasized the importance of dCK residues R104 and D133 in substrate specificity and revealed that site-directed mutations at these residues can not only render dCK more active but also endow this enzyme with novel thymidine kinase activity. On a D133A background, residue R104 was mutated to a hydrophobic residue, glutamine, or lysine as these residues were predicted to accommodate the C5-methyl group present in the thymine base. Of the double mutants (DM) tested for thymidine kinase activity, both DMMA (R104M/D133A) and DMLA (R104L/D133A) were more efficient in converting natural deoxynucleoside substrates, including thymidine, along with a number of tested nucleoside analogs. In terms of relevance to suicide gene therapy applications is the novel catalytic activity of these mutants toward the L-form of thymidine (L-dT) with DMMA phosphorylating the L-form twice as fast as the D-form. Structural analysis of DMMA in complex with L-dT revealed that the R104M mutation facilitated penetration of L-dT deeper into the nucleoside binding site by making space for the C5 methyl group of the thymine base. Furthermore, the concomitant mutation of D133A was found necessary to compensate for the removal of the charged R104 residue [99].
Separate random and site-saturation mutagenesis studies done by Iyidogan et al. (2008) reiterated the key importance of dCK residues R104 and D133 in substrate specificity. Published prior to reports describing the DMMA and DMLA mutants, these studies used error prone PCR random mutagenesis and genetic complementation in E. coli or site-saturation mutagenesis using degenerate primers targeting codons for residues R104 and D133 to enhance the catalytic activity and tailor the substrate specificity of dCK. The created mutants, all carrying R104/D133 mutations, showed significant thymidine kinase and nucleoside analog activity and the six mutants tested for nucleoside analog activity exhibited up to a 100-fold increase in AZT turnover and up to a 30-fold increase in ddT activity. Of the mutants tested for nucleoside analog activity, mutant epTK6 (D47E/R104Q/D133G/N163I/F242L) displayed a broader specificity and elevated turnover for all natural and nucleoside analog substrates. This activity was later linked to the two active site residues R104 and D133 as reversion of the remaining distant mutations to wt dCK showed little impact on catalytic activity [100].
Taking a different approach to increase dCK catalytic activity, McSorley et al. (2008) targeted amino acid residue S74 based on a previous study by Smal et al. (2006) that demonstrated the importance of S74 phosphorylation in the control of dCK activity [101]. To mimic S74 phosphorylation, S74D and S74E mutants were generated. Kinetic analyses of mutant S74E showed the most marked changes in kcat values with a 11-fold and 3-fold increase for the cytidine analogs gemcitabine and AraC, respectively. Specific enzymatic activity in HEK 293T cell lysates was interpreted to be 10-fold higher in cells expressing the S74E mutant compared to wt dCK expressing cells, suggesting that phosphorylation may be a mechanism for modifying and enhancing the catalytic activity of the dCK. enzyme [102].
Although the previously described dCK mutants displayed an overall increased catalytic activity toward their tested nucleoside analogs, many displayed an unfavorable relative specificity compared to deoxycytidine suggesting that competition for the active site may limit their efficacy in cells. Indeed several attempts have been made to generate novel dCK variants with improved gemcitabine activity for gene therapy applications, yet it appears that wt dCK. is sufficiently active towards clinically relevant prodrugs such that making any kinetic improvement may not translate to reduced IC50 values.
Escherichia coli Purine Nucleoside Phosphorylase
Escherichia coli purine nucleoside phosphorylase (EC 2.4.2.1; E. coli PNP) is a homohexameric enzyme with a molecular weight of about 150kDa Table 1, Fig. (3F) that catalyzes the reversible phosphorolysis of 6-amino and 6-oxopurine (2′-deoxy) ribonucleosides to (2′-deoxy)ribose-l-phosphate and purine base [103], Additionally, E. coli PNP can cleave nontoxic adenosine analogs such as 9-(2-deoxy-β-D-ribofuranosyl)-6-methylpurine (MeP-dR), 2-fluoro-2′-deoxyadenosine (F-dAdo) and 9-(β-D-arabinofuranosyl)-2-fluoroadenine (fludarabine or F-araA) to the adenine analogs 6-methylpurine (MeP) or 2-fluoroadenine (F-Ade). The distinctive nucleoside analog activity of E. coli PNP makes it a unique suicide enzyme candidate and has therefore been developed as a suicide gene therapy strategy for the treatment of solid tumors [103–105]. The adenine analogs, MeP and F-Ade, formed by E. coli PNP are further activated to cytotoxic ATP analogs in the cytosol by adenine phosphoribosyltransferase, are then incorporated into RNA, and in turn inhibit both RNA and protein synthesis, killing both proliferating and non-proliferating tumor cells [106]. As tumors grow at variable rates, the cytotoxic activity of these analogs against non-proliferating cells facilitates complete tumor ablation. Additionally, MeP and F-Ade are 10- and 1000-fold more potent against human leukemic CEM cells than 5FU, respectively [107]. Moreover, MeP and F-Ade are membrane permeable and can thus readily diffuse to adjacent tumor cells, resulting in robust bystander killing effects [108]. Low level expression of E. coli PNP, even in less than 1% of transfected tumor cells, was shown to lead to complete bystander killing of non-transfected tumor cells following MeP-dR dosing [109].
The E. coli PNP system has demonstrated exceptional in vivo anticancer activities in a variety of tumor cells, such as gliomas, ovarian, pancreatic, prostate and bladder cancers [103, 104, 108–112]. However, this approach is limited because the prodrug can be cleaved by endogenous PNP present in the intestinal flora, leading to systemic toxicity [111, 113, 114]. One strategy to reduce this toxicity is to create an anti-cancer prodrug that would be selectively cleaved by an engineered PNP but not by either E. coli PNP present in the intestinal flora or by any human enzyme. Secrist et al. (1999) found analogs with 5′-modifications in the ribose ring, such as 9-(6-deoxy-α-L-talofuranosyl)-6-methylpurine (Me(talo)-MeP-R), 9-(6-deoxy-β-D-allofuranosyl)-6-methylpurine [Me(allo)-MeP-R], and 9-α-L-lyxofuranosyladenine (lyxo-Ado) are poorly cleaved by E. coli PNP compared to MeP-dR [115]. Computer modeling studies of the E. coli PNP/Me (talo)-MeP-R complex suggested that poor cleavage results from a steric clash between the C6′ methyl group of Me (talo)-MeP-R and the side chain of methionine 64 (M64), an important residue involved in ribose binding. To relieve this clash, smaller hydrophobic residues were introduced to replace M64 and several mutants (M64A, M64V, M64I, M64Q) appeared to create the required space for the C6′ methyl group of Me(talo)-MeP-R [116].
Bennett et al. (2003) further analyzed the PNP M64 mutants with four 5′-modified prodrugs including Me(talo)-MeP-R, Me(allo)-MeP-R, lyxo-Ado and 9-(5′-5′-di-C-methyl-β-D-ribofuranosyl)-6-methylpurine (5′,5′-dimethyl- MeP-R) in search of the most effective enzyme/prodrug combination [114]. Results suggested that mutant M64V cleaved the modified nucleoside analogs more efficiently than wt E. coli PNP. Of greatest interest was the superior activity of mutant M64V with analog Me(talo)-MeP-R, where mutant M64V displayed a kcat/Km value approximately 140-fold greater than that of the wt enzyme. In vitro studies using human astrocytoma D54 cells transfected with PNP M64V showed that these cells were killed at a low concentration of Me(talo)-MeP-R (20μM). Further in vivo evaluation resulted in tumor regression for atleast 60 days when mice were treated with l00mg/kg/day of Me(talo)-MeP-R given on three consecutive days. The maximum tolerated dose (MTD) of Me(talo)-MeP-R was only 4-fold greater than that of MeP-dR (from 100 mg/kg/day to 400 mg/kg/day given on 3 consecutive days), a value less than the difference in catalytic rates between Me(talo)-MeP-R and MeP-dR with wt E. coli PNP (about 1000-fold). This suggests a mechanism of toxicity not associated with E. coli PNP as being a limiting factor for Me(talo)-MeP-R potency. The three best substrates of M64V, Me(talo)-MeP-R, 9-[6-deoxy-α-L-talofuranosyl]-2-F-adenine (Me(talo)-F-Ado) and 9-α-L-lyxofuranosyl-2-F-adenine (lyxo-F-Ade), were further evaluated in vivo. Results from those studies indicated that Me(talo)-MeP-R toxicity was due to its cleavage to MeP by a bacterial enzyme not likely to be E. coli PNP, and that the toxicity of the two purine analogs was due to their cleavage to F-Ade by mammalian methylthioadenosine phosphorylase [114]. Silamkoti et al. (2005) also evaluated the activity of M64V towards a novel series of MeP nucleoside derivatives with substitutions at 5′-position in the ribose ring. Results showed the mutant to cleave three compounds with similar structure, including 9-H-purine-6-amine-9-(2-deoxy-α-L-lyxofuranosyl)-2-fluoro, 9-H-purine-6-amine-2-fluoro-9-α-L-lyxofuranosyl and 9-H-Purine-9-α-L-lyxofuranosyl-6-methyl with 12-, 30- and 50-fold greater activity, respectively, compared to E. coli PNP [117].
While steps have been made to enhance the efficacy and lower the toxicity of the E. coli PNP based suicide gene therapy strategy, its success appears to be limited by side toxicity caused by enzymes found in the natural flora of the gut or endogenous enzymes that are capable of cleaving the tested adenine analogs. Therefore, the future success of this strategy may rely on the design of adenine analogs that are poorly cleaved by wt E. coli PNP or other endogenous enzymes but are efficiently activated by designed E. coli PNP variants. Nonetheless, the aforementioned studies describe an alternative strategy to create a suicide enzyme/prodrug system via the synthesis of prodrug analogs that are poorly activated by wt suicide enzyme and the design of variants with activity catered to the synthesized analog.
CONCLUSION
Tremendous hurdles in the feasibility and therapeutic response of gene therapy have gradually been overcome in the past two decades. However, despite the vast preclinical information available that demonstrates the efficacy of suicide gene therapy to treat cancer, successes observed in preclinical studies have yet to be fully attained in clinical settings. Many limiting factors are likely responsible for this discrepancy, including insufficient gene transfer, rapid clearance of delivery vectors by the hosts’ immune system, and poor activity of wt suicide enzymes towards their respective prodrugs. Nevertheless, suicide gene therapy is maintaining momentum because it offers much safer and less toxic treatments than chemotherapy. However, it is apparent that further advances to improve suicide gene therapy must be addressed before it becomes a mainstream therapeutic approach for treating cancer. As focused on this review, one approach to address this problem is to improve the suicide enzyme itself. When engineered suicide enzymes with improved activity towards their prodrugs are used to replace wt enzymes, lower doses of prodrugs could be administered to obtain the same or even better tumor cytotoxicity and, subsequently minimize unwanted side effects in patients. Similarly, utilizing a fusion gene has also proven to be a more effective means to kill tumor cells in vitro and in vivo [28, 29, 118], Additionally, with the generation of higher levels of active antimetabolites, greater and broader bystander effects should take place, ultimately leading to an enhanced reduction in tumor cell burden. Towards that end, the application of optimized suicide enzymes will likely improve the clinical outcome of suicide gene therapy in cancer patients and compensate, in part, for inefficiencies of current delivery methods.
Although suicide gene therapy may be used as a single treatment for cancer therapy, it is reasonable to state that the field has been gradually moving towards combining suicide gene therapy with standard radiation, chemotherapy and/or surgery approaches. By combining suicide gene therapy with these treatments, reduced doses of chemotherapeutic/radiation-based therapies can be administered to achieve complete tumor ablation while minimizing toxicity to normal cells. Advances in the gene delivery field have allowed complicated and refined techniques to be performed to enhance the transduction efficiency, safety, and tumor-specific cytotoxicity of current delivery vectors. Sophisticated delivery vehicles will continue to be generated as a means to overcome the barriers of adequate gene delivery for transduction efficiency and safety. With the future use of such vectors and/or carrier cells to deliver optimized enzyme/prodrug combinations to achieve superior prodrug-mediated tumor cytotoxicity and bystander effect, it is anticipated that significant and reproducible clinical success using suicide gene therapy strategies will be realized sooner rather than later. These approaches will also serve to extend the applicability of improved suicide genes beyond the treatment of solid tumors to the elimination of graft versus host disease following allogeneic hematopoietic stem cell transplantation, as safety genes and in non-invasive imaging technologies such as positron emission tomography (PET).
Acknowledgments
Declared none.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Relling MV, Rubnitz JE, Rivera GK, et al. High incidence of secondary brain tumours after radiotherapy and antimetabolites. The Lancet. 1999;354(9172):34–9. doi: 10.1016/S0140-6736(98)11079-6. [DOI] [PubMed] [Google Scholar]
- 2.Kaldor J, Day N, Pettersson F, et al. Leukemia following chemotherapy for ovarian cancer. N Engl J Med. 1990;322(1):1–6. doi: 10.1056/NEJM199001043220101. [DOI] [PubMed] [Google Scholar]
- 3.Curtis R, JB, Stovall M, et al. Risk of leukemia after chemotherapy and radiation treatment for breast cancer. N Engl J Med. 1992;326(26):1745–51. doi: 10.1056/NEJM199206253262605. [DOI] [PubMed] [Google Scholar]
- 4.Travis LB, Curtis RE, Glimelius B, et al. Bladder and kidney cancer following cyclophosphamide therapy for non-Hodgkin’s lymphoma. J Natl Cancer Inst. 1995;87(7):524–31. doi: 10.1093/jnci/87.7.524. [DOI] [PubMed] [Google Scholar]
- 5.Travis LB, Holowaty EJ, Bergfeldt K, et al. Risk of leukemia after platinum-based chemotherapy for ovarian cancer. N Engl J Med. 1999;340(5):351–7. doi: 10.1056/NEJM199902043400504. [DOI] [PubMed] [Google Scholar]
- 6.Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res. 1986;46(10):5276–81. [PubMed] [Google Scholar]
- 7.Greco O, Dachs GU. Gene directed enzyme/prodrug therapy of cancer: historical appraisal and future prospectives. J Cell Physiol. 2001;187(1):22–36. doi: 10.1002/1097-4652(2001)9999:9999<::AID-JCP1060>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 8.Elshami A, Saavedra A, Zhang H, et al. Gap junctions play a role in the ‘bystander effect’ of the herpes simplex virus thymidine kinase/ganciclovir system in vitro. Gene Ther. 1996;3(1):85–92. [PubMed] [Google Scholar]
- 9.Mesnil M, Yamasaki H. Bystander effect in herpes simplex virus thymidine kinase/ganciclovir cancer gene therapy: role of gapjunctional intercellular communication. Cancer Res. 2000;60(15):3989–99. [PubMed] [Google Scholar]
- 10.Mesnil M, Piccoli C, Tiraby G, Willecke K, Yamasaki H. Bystander killing of cancer cells by herpes simplex virus thymidine kinase gene is mediated by connexins. Proc Natl Acad Sci U S A. 1996;93(5):1831–6. doi: 10.1073/pnas.93.5.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freeman SM, Abboud CN, Whartenby KA, et al. The “bystander effect”: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993;53(21):5274–83. [PubMed] [Google Scholar]
- 12.Huber BE, Austin EA, Richards CA, Davis ST, Good SS. Metabolism of 5-fluorocytosine to 5-fluorouracil in human colorectal tumor cells transduced with the cytosine deaminase gene: significant antitumor effects when only a small percentage of tumor cells express cytosine deaminase. Proc Natl Acad Sci U S A. 1994;91(17):8302–6. doi: 10.1073/pnas.91.17.8302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lawrence TS, Rehemtulla A, Ng EY, Wilson M, Trosko JE, Stetson PL. Preferential cytotoxicity of cells transduced with cytosine deaminase compared to bystander cells after treatment with 5-flucytosine. Cancer Res. 1998;58(12):2588–93. [PubMed] [Google Scholar]
- 14.Prise KM, O’Sullivan JM. Radiation-induced bystander signalling in cancer therapy. Nat Rev Cancer. 2009;9(5):351–60. doi: 10.1038/nrc2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carystinos GD, Katabi MM, Laird DW, et al. Cyclic-AMP induction of gap junctional intercellular communication increases bystander effect in suicide gene therapy. Clin Cancer Res. 1999;5(1):61–8. [PubMed] [Google Scholar]
- 16.Tanaka T, Yamasaki H, Mesnil M. Induction of a bystander effect in HeLa cells by using a bigenic vector carrying viral thymidine kinase and connexin32 genes. Mol Carcinog. 2001;30(3):176–80. doi: 10.1002/mc.1026. [DOI] [PubMed] [Google Scholar]
- 17.Estin D, Li M, Spray D, Wu JK. Connexins are expressed in primary brain tumors and enhance the bystander effect in gene therapy. Neurosurgery. 1999;44(2):361–8. doi: 10.1097/00006123-199902000-00068. [DOI] [PubMed] [Google Scholar]
- 18.Kuriyama S, Kikukawa M, Masui K, et al. Cytosine deaminase/5-fluorocytosine gene therapy can induce efficient antitumor effects and protective immunity in immunocompetent mice but not in athymic nude mice. Int J Cancer. 1999;81(4):592–7. doi: 10.1002/(sici)1097-0215(19990517)81:4<592::aid-ijc15>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 19.Kuriyama S, Kikukawa M, Masui K, et al. Cancer gene therapy with HSV-tk/GCV system depends on T-cell-mediated immune responses and causes apoptotic death of tumor cells in vivo. Int J Cancer. 1999;83(3):374–80. doi: 10.1002/(sici)1097-0215(19991029)83:3<374::aid-ijc13>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 20.Nishiyama T, Kawamura Y, Kawamoto K, et al. Antineoplastic effects in rats of 5-fluorocytosine in combination with cytosine deaminase capsules. Cancer Res. 1985;45(4):1753–61. [PubMed] [Google Scholar]
- 21.Consalvo M, Mullen CA, Modesti A, et al. 5-Fluorocytosine-induced eradication of murine adenocarcinomas engineered to express the cytosine deaminase suicide gene requires host immune competence and leaves an efficient memory. J Immunol. 1995;154(10):5302–12. [PubMed] [Google Scholar]
- 22.Vile RG, Castleden S, Marshall J, Camplejohn R, Upton C, Chong H. Generation of an anti-tumour immune response in a non-immunogenic tumour: HSVtk killing in vivo stimulates a mononuclear cell infiltrate and a Th 1-like profile of intratumoural cytokine expression. Int J Cancer. 1997;71(2):267–74. doi: 10.1002/(sici)1097-0215(19970410)71:2<267::aid-ijc23>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 23.Caruso M, Panis Y, Gagandeep S, Houssin D, Salzmann JL, Klatzmann D. Regression of established macroscopic liver metastases after in situ transduction of a suicide gene. Proc Natl Acad Sci USA. 1993;90(15):7024–8. doi: 10.1073/pnas.90.15.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim YG, Bi W, Feliciano ES, Drake RR, Stambrook PJ. Ganciclovir-mediated cell killing and bystander effect is enhanced in cells with two copies of the herpes simplex virus thymidine kinase gene. Cancer Gene Ther. 2000;7(2):240–6. doi: 10.1038/sj.cgt.7700113. [DOI] [PubMed] [Google Scholar]
- 25.Davies LC, Friedlos F, Hedley D, et al. Novel fluorinated prodrugs for activation by carboxypeptidase G2 showing good in vivo antitumor activity in gene-directed enzyme prodrug therapy. J Med Chem. 2005;48(16):5321–8. doi: 10.1021/jm0502182. [DOI] [PubMed] [Google Scholar]
- 26.Boucher PD, Ostruszka LJ, Shewach DS. Synergistic enhancement of herpes simplex virus thymidine kinase/ganciclovir-mediated cytotoxicity by hydroxyurea. Cancer Res. 2000;60(6):1631–6. [PubMed] [Google Scholar]
- 27.Boucher PD, Im MM, Freytag SO, Shewach DS. A novel mechanism of synergistic cytotoxicity with 5-fluorocytosine and ganciclovir in double suicide gene therapy. Cancer Res. 2006;66(6):3230–7. doi: 10.1158/0008-5472.CAN-05-3033. [DOI] [PubMed] [Google Scholar]
- 28.Willmon CL, Krabbenhoft E, Black ME. A guanylate kinase/HSV-1 thymidine kinase fusion protein enhances prodrug-mediated cell killing. Gene Ther. 2006;13(17):1309–12. doi: 10.1038/sj.gt.3302794. [DOI] [PubMed] [Google Scholar]
- 29.Erbs P, Regulier E, Kintz J, et al. In vivo cancer gene therapy by adenovirus-mediated transfer of a bifunctional yeast cytosine deaminase/uracil phosphoribosyltransferase fusion gene. Cancer Res. 2000;60(14):3813–22. [PubMed] [Google Scholar]
- 30.Tiraby M, Cazaux C, Baron M, Drocourt D, Reynes J, Tiraby G. Concomitant expression of E. coli cytosine deaminase and uracil phosphoribosyltransferase improves the cytotoxicity of 5-fluorocytosine. FEMS Microbiol Lett. 1998;167(1):41–9. doi: 10.1111/j.1574-6968.1998.tb13205.x. [DOI] [PubMed] [Google Scholar]
- 31.Fuchita M, Ardiani A, Zhao L, Serve K, Stoddard BL, Black ME. Bacterial cytosine deaminase mutants created by molecular engineering show improved 5-fluorocytosine-mediated cell killing in vitro and in vivo. Cancer Res. 2009;69(11):4791–9. doi: 10.1158/0008-5472.CAN-09-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stolworthy TS, Korkegian AM, Willmon CL, et al. Yeast cytosine deaminase mutants with increased thermostability impart sensitivity to 5-fluorocytosine. J Mol Biol. 2008;377(3):854–69. doi: 10.1016/j.jmb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Black ME, Kokoris MS, Sabo P. Herpes simplex virus-1 thymidine kinase mutants created by semi-random sequence mutagenesis improve prodrug-mediated tumor cell killing. Cancer Res. 2001;61(7):3022–6. [PubMed] [Google Scholar]
- 34.Kaliberov SA, Market JM, Gillespie GY, et al. Mutation of Escherichia coli cytosine deaminase significantly enhances molecular chemotherapy of human glioma. Gene Ther. 2007;14(14):1111–9. doi: 10.1038/sj.gt.3302965. [DOI] [PubMed] [Google Scholar]
- 35.Kokoris MS, Black ME. Characterization of herpes simplex virus type 1 thymidine kinase mutants engineered for improved ganciclovir or acyclovir activity. Protein Sci. 2002;11(9):2267–72. doi: 10.1110/ps.2460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dachs GU, Tupper J, Tozer GM. From bench to bedside for gene-directed enzyme prodrug therapy of cancer. Anticancer Drugs. 2005;16(4):349–59. doi: 10.1097/00001813-200504000-00001. [DOI] [PubMed] [Google Scholar]
- 37.Norris JS, Norris KL, Holman DH, El-Zawahry A, Keane TE, Dong J-y, et al. The present and future for gene and viral therapy of directly accessible prostate and squamous cell cancers of the head and neck. Future Oncol. 2005;1(1):115–23. doi: 10.1517/14796694.1.1.115. [DOI] [PubMed] [Google Scholar]
- 38.Niculescu-Duvaz I, Springer C. Introduction to the background, principles, and state of the art in suicide gene therapy. Mol Bio-technol. 2005;30(1):71–88. doi: 10.1385/MB:30:1:071. [DOI] [PubMed] [Google Scholar]
- 39.Lutz S, Patrick WM. Novel methods for directed evolution of enzymes: quality, not quantity. Curr Opin Biotechnol. 2004;15(4):291–7. doi: 10.1016/j.copbio.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 40.Neylon C. Chemical and biochemical strategies for the randomization of protein encoding DNA sequences: library construction methods for directed evolution. Nucl Acids Res. 2004;32(4):1448–59. doi: 10.1093/nar/gkh315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bloom JD, Meyer MM, Meinhold P, Otey CR, MacMillan D, Arnold FH. Evolving strategies for enzyme engineering. Curr Opin Struct Biol. 2005;15(4):447–52. doi: 10.1016/j.sbi.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 42.Devanathan S, Willmon CL, Mahan SD, Black ME. Engineering enzymes for improved cancer gene therapy. Res Adv in Cancer. 2002;2:315–26. [Google Scholar]
- 43.Encell LP, Landis DM, Loeb LA. Improving enzymes for cancer gene therapy. Nat Biotechnol. 1999;17(2):143–7. doi: 10.1038/6142. [DOI] [PubMed] [Google Scholar]
- 44.Mahan SD, Ireton GC, Stoddard BL, Black ME. Alanine-scanning mutagenesis reveals a cytosine deaminase mutant with altered substrate preference. Biochemistry. 2004;43(28):8957–64. doi: 10.1021/bi049720z. [DOI] [PubMed] [Google Scholar]
- 45.Field AK, Davies ME, DeWitt C, et al. 9-([2-hydroxy-1-(hydroxymethyl)ethoxy]methyl)guanine: a selective inhibitor of herpes group virus replication. Proc Natl Acad Sci U S A. 1983;80(13):4139–43. doi: 10.1073/pnas.80.13.4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng YC, Grill SP, Dutschman GE, Nakayama K, Bastow KF. Metabolism of 9-(1,3-dihydroxy-2-propoxymethyl)guanine, a new anti- herpes virus compound, in herpes simplex virus-infected cells. J Biol Chem. 1983;258(20):12460–4. [PubMed] [Google Scholar]
- 47.Ashton W, Karkas J, Field A, Tolman R. Activation by thymidine kinase and potent antiherpetic activity of 2′-nor-2′-deoxyguanosine (2′NDG) Biochem Biophys Res Commun. 1982;108(4):1716–21. doi: 10.1016/s0006-291x(82)80109-5. [DOI] [PubMed] [Google Scholar]
- 48.Candolfi M, Kroeger K, Muhammad A, et al. Gene therapy for brain cancer: combination therapies provide enhanced efficacy and safety. Curr Gene Ther. 2009;9(5):409–21. doi: 10.2174/156652309789753301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Määttä A, Samaranayake H, Pikkarainen J, Wirth T, Ylä-Herttuala S. Adenovirus mediated herpes simplex virus-thymidine kinase/ganciclovir gene therapy for resectable malignant glioma. Curr Gene Ther. 2009;9(5):356–67. doi: 10.2174/156652309789753365. [DOI] [PubMed] [Google Scholar]
- 50.Singhal S, Kaiser L. Cancer chemotherapy using suicide genes. Surg Oncol Clin N Am. 1998;7(3):505–36. [PubMed] [Google Scholar]
- 51.Black M. Enzyme and pathway engineering for suicide gene therapy. Genet Eng (N Y) 2001;23:113–27. doi: 10.1007/0-306-47572-3_7. [DOI] [PubMed] [Google Scholar]
- 52.Black M, Newcomb T, Wilson H, Loeb L. Creation of drug-specific herpes simplex virus type 1 thymidine kinase mutants for gene therapy. Proc Natl Acad Sci U S A. 1996;93(8):3525–9. doi: 10.1073/pnas.93.8.3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mercer KE, Ahn CE, Coke A, Compadre CM, Drake RR. Mutation of herpesvirus thymidine kinase to generate ganciclovir-specific kinases for use in cancer gene therapies. Protein Eng. 2002;15(11):903–11. doi: 10.1093/protein/15.11.903. [DOI] [PubMed] [Google Scholar]
- 54.Likar Y, Dobrenkov K, Olszewska M, et al. A new acycloguanosine-specific supermutant of herpes simplex virus type 1 thymidine kinase suitable for PET imaging and suicide gene therapy for potential use in patients treated with pyrimidine-based cytotoxic drugs. J Nucl Med. 2008;49(5):713–20. doi: 10.2967/jnumed.107.046425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Black M, Loeb L. Identification of important residues within the putative nucleoside binding site of HSV-1 thymidine kinase by random sequence selection: analysis of selected mutants in vitro. Biochemistry. 1993;32:11618–26. doi: 10.1021/bi00094a019. [DOI] [PubMed] [Google Scholar]
- 56.Kokoris M, Sabo P, Adman E, Black ME. Enhancement of tumor ablation by a selected HSV-1 thymidine kinase mutant. Gene Ther. 1999;6(8):1415–26. doi: 10.1038/sj.gt.3300966. [DOI] [PubMed] [Google Scholar]
- 57.Qiao J, Black ME, Caruso M. Enhanced ganciclovir killing and bystander effect of human tumor cells transduced with a retroviral vector carrying a herpes simplex virus thymidine kinase gene mutant. Hum Gene Ther. 2000;11(11):1569–76. doi: 10.1089/10430340050083298. [DOI] [PubMed] [Google Scholar]
- 58.Pantuck AJ, Matherly J, Zisman A, et al. Optimizing prostate cancer suicide gene therapy using herpes simplex virus thymidine kinase active site variants. Hum Gene Ther. 2002;13(7):777–89. doi: 10.1089/10430340252898966. [DOI] [PubMed] [Google Scholar]
- 59.Wiewrodt R, Amin K, Kiefer M, et al. Adenovirus-mediated gene transfer of enhanced herpes simplex virus thymidine kinase mutants improves prodrug-mediated tumor cell killing. Cancer Gene Ther. 2003;10(5):353–64. doi: 10.1038/sj.cgt.7700589. [DOI] [PubMed] [Google Scholar]
- 60.Ardiani A, Sanchez-Bonilla M, Black M. Fusion enzymes containing HSV-1 thymidine kinase mutants and guanylate kinase enhance prodrug sensitivity in vitro and in vivo. Cancer Gene Ther. 2009;17(2):86–96. doi: 10.1038/cgt.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gambhir SS, Bauer E, Black ME, et al. A mutant herpes simplex virus type 1 thymidine kinase reporter gene shows improved sensitivity for imaging reporter gene expression with positron emission tomography. Proc Natl Acad Sci U S A. 2000;97(6):2785–90. doi: 10.1073/pnas.97.6.2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pantuck AJ, Berger F, Zisman A, et al. CL1-SR39: A noninvasive molecular imaging model of prostate cancer suicide gene therapy using positron emission tomography. J Urol. 2002;168(3):1193–8. doi: 10.1016/S0022-5347(05)64624-1. [DOI] [PubMed] [Google Scholar]
- 63.Yang H, Berger F, Tran C, Gambhir SS, Sawyers CL. MicroPET imaging of prostate cancer in LNCAP-SR39TK-GFP mouse xenografts. Prostate. 2003;55(1):39–47. doi: 10.1002/pros.10208. [DOI] [PubMed] [Google Scholar]
- 64.Yaghoubi SS, Barrio JR, Namavari M, Satyamurthy N, Phelps ME, Herschman HR, et al. Imaging progress of herpes simplex virus type 1 thymidine kinase suicide gene therapy in living subjects with positron emission tomography. Cancer Gene Ther. 2005;12(3):329–39. doi: 10.1038/sj.cgt.7700795. [DOI] [PubMed] [Google Scholar]
- 65.Jacobs AH, Rueger MA, Winkeler A, Li H, Vollmar S, Waerzeggers Y, et al. Imaging-guided gene therapy of experimental gliomas. Cancer Res. 2007;67(4):1706–15. doi: 10.1158/0008-5472.CAN-06-2418. [DOI] [PubMed] [Google Scholar]
- 66.Balzarini J, Liekens S, Solaroli N, El Omari K, Stammers DK, Karlsson A. Engineering of a single conserved amino acid residue of herpes simplex virus type 1 thymidine kinase allows a predominant shift from pyrimidine to purine nucleoside phosphorylation. J Biol Chem. 2006;281(28):19273–9. doi: 10.1074/jbc.M600414200. [DOI] [PubMed] [Google Scholar]
- 67.Preuß E, Muik A, Weber K, Otte J, von Laer D, Fehse B. Cancer suicide gene therapy with TK.007: superior killing efficiency and bystander effect. J Mol Med. 2011;89(11):1113–24. doi: 10.1007/s00109-011-0777-8. [DOI] [PubMed] [Google Scholar]
- 68.Hinds TA, Compadre C, Hurlburt BK, Drake RK. Conservative mutations of glutamine-125 in herpes simplex virus type 1 thymidine kinase result in a ganciclovir kinase with minimal deoxypyrimidinc kinase activities. Biochemistry. 2000;39(14):4105–11. doi: 10.1021/bi992453q. [DOI] [PubMed] [Google Scholar]
- 69.Degreve B, Esnouf R, De Clercq E, Balzarini J. Selective abolishment of pyrimidine nucleoside kinase activity of herpes simplex virus type 1 thymidine kinase by mutation of alanine-167 to tyrosine. Mol Pharmacol. 2000;58(6):1326–32. doi: 10.1124/mol.58.6.1326. [DOI] [PubMed] [Google Scholar]
- 70.Balzarini J, Liekens S, Esnouf R, De Clercq E. The A167Y mutation converts the herpes simplex virus type 1 thymidine kinase into a guanosine analogue kinase. Biochemistry. 2002;41(20):6517–24. doi: 10.1021/bi0255930. [DOI] [PubMed] [Google Scholar]
- 71.Drake RR, Wilbert TN, Hinds TA, Gilbert KM. Differential ganciclovir-mediated cell killing by glutamine 125 mutants of herpes simplex virus type 1 thymidine kinase. J Biol Chem. 1999;274(52):37186–92. doi: 10.1074/jbc.274.52.37186. [DOI] [PubMed] [Google Scholar]
- 72.Degreve B, Esnouf R, De Clercq E, Balzarini J. Mutation of Glnl25 to Asn selectively abolishes the thymidylate kinase activity of herpes simplex virus type 1 thymidine kinase. Mol Pharmacol. 2001;59(2):285–93. doi: 10.1124/mol.59.2.285. [DOI] [PubMed] [Google Scholar]
- 73.Ireton GC, McDermott G, Black ME, Stoddard BL. The structure of Escherichia coli cytosine deaminase. J Mol Biol. 2002;315(4):687–97. doi: 10.1006/jmbi.2001.5277. [DOI] [PubMed] [Google Scholar]
- 74.Mahan SD, Ireton GC, Knoeber C, Stoddard BL, Black ME. Random mutagenesis and selection of Escherichia coli cytosine deaminase for cancer gene therapy. Protein Eng Des Sel. 2004;17(8):625–33. doi: 10.1093/protein/gzh074. [DOI] [PubMed] [Google Scholar]
- 75.Vermes A, Guchelaar HJ, Dankert J. Flucytosine: a review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J Antimicrob Chemother. 2000;46(2):171–9. doi: 10.1093/jac/46.2.171. [DOI] [PubMed] [Google Scholar]
- 76.Austin EA, Huber BE. A first step in the development of gene therapy for colorectal carcinoma: cloning, sequencing, and expression of Escherichia coli cytosine deaminase. Mol Pharmacol. 1993;43(3):380–7. [PubMed] [Google Scholar]
- 77.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330–8. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 78.Kievit E, Nyati MK, Ng E, et al. Yeast cytosine deaminase improves radiosensitization and bystander effect by 5-fluorocytosine of human colorectal cancer xenografts. Cancer Res. 2000;60(23):6649–55. [PubMed] [Google Scholar]
- 79.Kaliberova L, Manna D, Krendelchtchikova V, Black M, Buchsbaum D, Kaliberov S. Molecular chemotherapy of pancreatric cancer using novel mutant bacterial cytosine deaminase gene. Mol Cancer Ther. 2008;7(9):2845–54. doi: 10.1158/1535-7163.MCT-08-0347. [DOI] [PubMed] [Google Scholar]
- 80.Deng L-Y, Wang J-P, Gui Z-F, Shen L-Z. Antitumor activity of mutant bacterial cytosine deaminase gene for colon cancer. World J Gastoenterol. 2011;17(24):2958–64. doi: 10.3748/wjg.v17.i24.2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Korkegian A, Black ME, Baker D, Stoddard BL. Computational thermostabilization of an enzyme. Science. 2005;308(5723):857–60. doi: 10.1126/science.1107387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Munch-Petersen B, Piskur J, Sondergaard L. Four deoxynucleoside kinase activities from Drosophila melanogaster are contained within a single monomeric enzyme, a new multifunctional deoxynucleoside kinase. J Biol Chem. 1998;273(7):3926–31. doi: 10.1074/jbc.273.7.3926. [DOI] [PubMed] [Google Scholar]
- 83.Zheng X, Johansson M, Karlsson A. Retroviral transduction of cancer cell lines with the gene encoding Drosophila melanogaster multisubstrate deoxyribonucleoside kinase. J Biol Chem. 2000;275(50):39125–9. doi: 10.1074/jbc.M006212200. [DOI] [PubMed] [Google Scholar]
- 84.Knecht W, Munch-Petersen B, Piskur J. Identification of residues involved in the specificity and regulation of the highly efficient multisubstrate deoxyribonucleoside kinase from Drosophila melanogaster. J Mol Biol. 2000;301(4):827–37. doi: 10.1006/jmbi.2000.3990. [DOI] [PubMed] [Google Scholar]
- 85.Knecht W, Rozpedowska E, Le Breton C, Willer M, Gojkovic Z, Sandrini MPB, et al. Drosophila deoxyribonucleoside kinase mutants with enhanced ability to phosphorylate purine analogs. Gene Ther. 2007;14(17):1278–86. doi: 10.1038/sj.gt.3302982. [DOI] [PubMed] [Google Scholar]
- 86.Welin M, Skovgaard T, Knecht W, Zhu C, Berenstein D, Munch-Petersen B, et al. Structural basis for the changed substrate specificity of Drosophila melanogaster deoxyribonucleoside kinase mutant N64D. FEBS J. 2005;272(14):3733–42. doi: 10.1111/j.1742-4658.2005.04803.x. [DOI] [PubMed] [Google Scholar]
- 87.Knecht W, Sandrini M, Johansson K, Eklund H, Munch-Petersen B, Piskur J. A few amino acid substitutions can convert deoxyribonucleoside kinase specificity from pyrimidines to purines. EMBO J. 2002;21(7):1873–80. doi: 10.1093/emboj/21.7.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Munch-Petersen B, Knecht W, Lenz C, Sondergaard L, Piskur J. Functional expression of a multisubstrate deoxyribonucleoside kinase from Drosophila melanogaster and its C-terminal deletion mutants. J Biol Chem. 2000;275(9):6673–9. doi: 10.1074/jbc.275.9.6673. [DOI] [PubMed] [Google Scholar]
- 89.Solaroli N, Johansson M, Balzarini J, Karlsson A. Enhanced toxicity of purine nucleoside analogs in cells expressing Drosophila melanogaster nucleoside kinase mutants. Gene Ther. 2007;14(1):89–62. doi: 10.1038/sj.gt.3302835. [DOI] [PubMed] [Google Scholar]
- 90.Gerth ML, Lutz S. Non-homologous recombination of deoxyribonucleoside kinases from human and Drosophila melanogaster yields human-like enzymes with novel activities. J Mol Biol. 2007;370(4):742–51. doi: 10.1016/j.jmb.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu L, Li Y, Liotta D, Lutz S. Directed evolution of an orthogonal nucleoside analog kinase via fluorescence-activated cell sorting. Nucl Acids Res. 2009;37(13):4472–81. doi: 10.1093/nar/gkp400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lutz S, Liu L, Liu Y. Engineering kinases to phosphorylate nucleoside analogs for antiviral and cancer therapy. Chimia (Aarau) 2009;63(11):737–44. doi: 10.2533/chimia.2009.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sabini E, Ort S, Monnerjahn C, Konrad M, Lavie A. Structure of human dCK suggests strategies to improve anticancer and antiviral therapy. Nat Struct Biol. 2003;10(7):513–9. doi: 10.1038/nsb942. [DOI] [PubMed] [Google Scholar]
- 94.Stegmann AP, Honders WH, Willemze R, Ruiz van Haperen VW, Landegem JE. Transfection of wild-type deoxycytidine kinase (dck) cDNA into an AraC- and DAC-resistanl rat leukemic cell line of clonal origin fully restores drug sensitivity. Blood. 1995;85(5):1188–94. [PubMed] [Google Scholar]
- 95.Hapke DM, Stegmann AP, Mitchell BS. Retroviral transfer of deoxycytidine kinase into tumor cell lines enhances nucleoside toxicity. Cancer Res. 1996;56(10):2343–7. [PubMed] [Google Scholar]
- 96.Manome Y, Wen P, Dong Y, Tanaka T, Mitchell BS, Kufe D, et al. Viral vector transduction of the human deoxycytidine kinase cDNA sensitizes glioma cells to the cytotoxic effects of cytosine arabinoside in vitro and in vivo. Nat Med. 1996;2(5):567–73. doi: 10.1038/nm0596-567. [DOI] [PubMed] [Google Scholar]
- 97.Rejiba S, Bigand C, Parmentier C, Hajri A. Gemcitabine-based chemogene therapy for pancreatic cancer using Ad-dCK::UMK GDEPT and TS/RR siRNA strategies. Neoplasia. 2009;11(7):637–50. doi: 10.1593/neo.81686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cottin S, Ghani K, de Campos-Lima P, Caruso M. Gemcitabine intercellular diffusion mediated by gap junctions: new implications for cancer therapy. Mol Cancer. 2010;9(1):141. doi: 10.1186/1476-4598-9-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hazra S, Sabini E, Ort S, Konrad M, Lavie A. Extending thymidine kinase activity to the catalytic repertoire of human deoxycytidine kinase. Biochemistry. 2009;48(6):1256–63. doi: 10.1021/bi802062w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Iyidogan P, Lutz S. Systematic exploration of active site mutations on human deoxycytidine kinase substrate specificity. Biochemistry. 2008;47(16):471–20. doi: 10.1021/bi800157e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Smal C, Vertommeu D, Bertrand L, et al. Identification of in vivo phosphorylation sites on human deoxycytidine kinase: role of Ser-74 in the control of enzyme activity. J Biol Chem. 2006;281(8):4887–93. doi: 10.1074/jbc.M512129200. [DOI] [PubMed] [Google Scholar]
- 102.McSorley T, Ort S, Hazra S, Lavie A, Konrad M. Mimicking phosphorylation of Ser-74 on human deoxycytidine kinase selectively increases catalytic activity for dC and dC analogues. FEBS Lett. 2008;582(5):720–4. doi: 10.1016/j.febslet.2008.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kikuchi E, Menendez S, Ozu C, et al. Delivery of replication-competent retrovirus expressing Escherichia coli purine nucleoside phosphorylase increases the metabolism of the prodrug, fludarabine phosphate and suppresses the growth of bladder tumor xenografts. Cancer Gene Ther. 2007;14(3):279–86. doi: 10.1038/sj.cgt.7701013. [DOI] [PubMed] [Google Scholar]
- 104.Sophie D, Séverine W, Marc A, Amor H. Transcriptional tumor-selective promoter targeting of E. coli purine nucleoside phosphorylase for pancreatic cancer suicide gene therapy. J Gene Med. 2005;7(5):672–80. doi: 10.1002/jgm.701. [DOI] [PubMed] [Google Scholar]
- 105.Sorscher E, Peng S, Bebok Z, Allan P, Bennett L, Parker W. Tumor cell bystander killing in colonic carcinoma utilizing the Escherichia coli DeoD gene to generate toxic purines. Gene Ther. 1994;1(4):233–8. [PubMed] [Google Scholar]
- 106.Tai C-K, Wang W, Lai Y-H, et al. Enhanced efficiency of prodrug activation therapy by tumor-selectively replicating retrovirus vectors armed with the Escherichia coli purine nucleoside phosphorylase gene. Cancer Gene Ther. 2010;17(9):614–23. doi: 10.1038/cgt.2010.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Parker WB, Allan PW, Shaddix SC, et al. Metabolism and metabolic actions of 6-methylpurine and 2-fluoroadenine in human cells. Biochem Pharmacol. 1998;55(10):1673–81. doi: 10.1016/s0006-2952(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 108.Hughes BW, Wells AH, Bebok Z, et al. Bystander killing of melanoma cells using the human tyrosinase promoter to express the Escherichia coli purine nucleoside phosphorylase gene. Cancer Res. 1995;55(15):3339–45. [PubMed] [Google Scholar]
- 109.Gadi V, Alexander S, Kudlow J, Allan P, Parker W, Sorscher E. In vivo sensitization of ovarian tumors to chemotherapy by expression of E. coli purine nucleoside phosphorylase in a small fraction of cells. Gene Ther. 2000;7(20):1738–43. doi: 10.1038/sj.gt.3301286. [DOI] [PubMed] [Google Scholar]
- 110.Deharvengt S, Wack S, Uhring M, Aprahamian M, Hajri A. Suicide gene/prodrug therapy for pancreatic adenocarcinoma by E.coli purine nucleoside phosphorylase and 6-methylpurine 2′-deoxyriboside. Pancreas. 2004;28(2):e54–64. doi: 10.1097/00006676-200403000-00020. [DOI] [PubMed] [Google Scholar]
- 111.Bharara S, Sorscher EJ, Gillespie GY, et al. Antibiotic-mediated chemoprotection enhances adaptation of E.coli PNP for herpes simplex virus-based glioma therapy. Hum Gene Ther. 2005;16(3):339–47. doi: 10.1089/hum.2005.16.339. [DOI] [PubMed] [Google Scholar]
- 112.Rosetta MW, Allison D, Dale JV, et al. Gene-directed enzyme prodrug therapy for prostate cancer in a mouse model that imitates the development of human disease. J Gene Med. 2004;6(1):43–54. doi: 10.1002/jgm.474. [DOI] [PubMed] [Google Scholar]
- 113.Lubin M, Lubin A. Selective killing of tumors deficient in methylthioadenosine phosphorylase: a novel strategy. PLoS One. 2009;4(5):e5735. doi: 10.1371/journal.pone.0005735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bennett EM, Anand R, Allan PW, et al. Designer gene therapy using an Escherichia coli purine nucleoside phosphorylase/prodrug system. Chem Biol. 2003;10(12):1173–81. doi: 10.1016/j.chembiol.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 115.Secrist J, Parker W, Allan P, et al. Gene therapy of cancer: activation of nucleoside prodrugs with E.coli purine nucleoside phosphorylase. Nucleosides Nucleotides. 1999;18(4–5):745–57. doi: 10.1080/15257779908041562. [DOI] [PubMed] [Google Scholar]
- 116.Parker W, Allan P, Ealick S, et al. Design and evaluation of 5′-modified nucleoside analogs as prodrugs for an E.coli purine nucleoside phosphorylase mutant. Nucleosides Nucleotides Nucleic Acids. 2005;24(5–7):387–92. doi: 10.1081/ncn-200059807. [DOI] [PubMed] [Google Scholar]
- 117.Silamkoti A, Allan P, Hassan A, et al. Synthesis and biological activity of 2-fluoro adenine and 6-methyl purine nucleoside analogs as prodrugs for suicide gene therapy of cancer. Nucleosides Nucleotides Nucleic Acids. 2005;24(5–7):881–5. doi: 10.1081/ncn-200059237. [DOI] [PubMed] [Google Scholar]
- 118.Gopinath P, Ghosh S. Apoptotic induction with bifunctional E. coli suicide gene therapy is synergized by curcumin treatment in vitro. Mol Biotechnol. 2008;39:39–48. doi: 10.1007/s12033-007-9026-3. [DOI] [PubMed] [Google Scholar]