

Abstract
Human disorders of hereditary and nonhereditary heterotopic ossification are conditions in which osteogenesis occurs outside of the skeleton, within soft tissues of the body. The resulting extraskeletal bone is normal. The aberration lies within the mechanisms that regulate cell-fate determination, directing the inappropriate formation of cartilage or bone, or both, in tissues such as skeletal muscle and adipose tissue. Specific gene mutations have been identified in two rare inherited disorders that are clinically characterized by extensive and progressive extraskeletal bone formation—fibrodysplasia ossificans progressiva and progressive osseous heteroplasia. In fibrodysplasia ossificans progressiva, activating mutations in activin receptor type-1, a bone morphogenetic protein type I receptor, induce heterotopic endochondral ossification, which results in the development of a functional bone organ system that includes skeletal-like bone and bone marrow. In progressive osseous heteroplasia, the heterotopic ossification leads to the formation of mainly intramembranous bone tissue in response to inactivating mutations in the GNAS gene. Patients with these diseases variably show malformation of normal skeletal elements, identifying the causative genes and their associated signaling pathways as key mediators of skeletal development in addition to regulating cell-fate decisions by adult stem cells.
Introduction
The formation and maintenance of tissues and organ systems depend on the coordination of cellular events and mechanisms. In human genetic diseases, the normal functions of cells are perturbed by alterations in aspects of the expression, duration of signaling, structure or interaction (or combinations thereof) of cellular proteins during embryonic development or later in life (or both). Throughout life, bone formation is normally limited to the skeletal system. During embryogenesis, most skeletal elements, such as the long bones, form through endochondral ossification, in which cartilaginous skeletal anlagen are replaced by bone, while other elements, such as the skull, ossify directly through a process described as intramembranous ossification.1
Heterotopic ossification is a pathological condition in which bone forms in nonskeletal tissues.2,3 Formation of this ectopic bone in soft tissues requires precursor cells that have the potential to differentiate into bone or cartilage (or both), a conducive tissue microenvironment, and an inducing event to initiate the cellular and molecular events that lead to bone formation. The heterotopic bone that forms is qualitatively normal endochondral or intramembranous bone, and develops through processes that parallel the events that occur during normal embryonic bone and skeletal formation, as well as those occurring in bone regeneration during fracture healing. The pathophysiology of heterotopic ossification is caused by dysregulation of cell-fate determination and in appropriate induction of the bone formation program.
Nonhereditary forms of heterotopic ossification are frequent complications of a number of common conditions, although the environmental or genetic factors that predispose some individuals to heterotopic ossification remain uncertain. Induction of nonhereditary hetero topic ossification is associated with certain types of severe tissue trauma, including injury to the spinal cord and brain, hip replacement surgery, severe burns, and high-energy war wounds; nonhereditary heterotopic ossification is also a complication of age-associated conditions such as atherosclerosis and pressure ulcers.2–7 Nonhereditary forms of heterotopic ossification have been reviewed elsewhere2,3 and will not therefore be discussed further in this article.
Two human genetic diseases of progressive and extensive heterotopic ossification are known: fibrodysplasia ossificans progressiva (FOP) and progressive osseous hetero plasia (POH).8,9 Each of these conditions is caused by mutations in a single (different) gene, which indicates that these disease-causing genes are critical components of the regulatory mechanisms that direct cell-fate decisions and bone tissue and/or organ formation. In this review, we compare and contrast the bone formation process that occurs in patients with FOP and POH, and discuss the relevance of the mutated pathways that underlie these conditions to normal bone and skeletal development.
Fibrodysplasia ossificans progressiva
FOP is a severely disabling genetic disease in which bone forms at extraskeletal sites within connective tissues such as skeletal muscle, tendon, ligament, fascia and aponeuroses.8–13 Specific skeletal malformations also occur in patients with this disease. Classic clinical features of FOP include progressive heterotopic ossification and big toe malformation (Figure 1). FOP is a rare condition, occurring at a population frequency of approximately one per 2 million individuals. Although most cases occur in individuals with no prior family history of FOP, autosomal dominant inheritance has been observed in a small number of families.14
Figure 1.
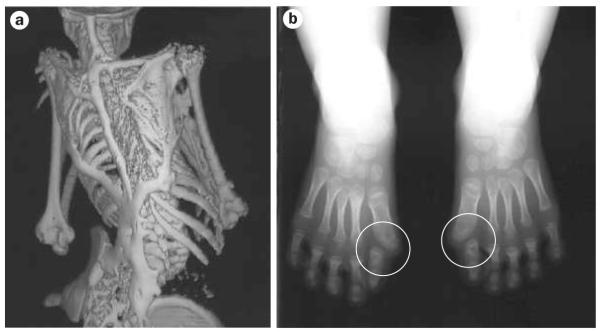
Characteristic clinical features of FOP. a | Extensive heterotopic bone formation typical of FOP seen in a three-dimensional reconstructed CT scan of the back of a 12-year-old child. Flare-ups of FOP arise and progress in a well-defined spatial pattern, resulting in ribbons, sheets and plates of bone that fuse the joints of the axial and appendicular skeletons. b | Anteroposterior radiograph of the feet of a 3-year-old child, showing symmetrical big toe malformations of metatarsals and proximal phalanges together with microdactyly, fused interphalangeal joints and hallux valgus deviations at the metatarsophalangeal joints (circled). Abbreviation: FOP, fibrodysplasia ossificans progressiva. Permission obtained for Figure 1a,b from Nature Publishing Group © Shore, E. M. et al. Nat. Genet. 38, 525-527 (2006).
FOP and mutation of ACVR1
Genetic linkage analysis and positional cloning were used to identify ACVR1 (located on chromosome 2q23–24) as the mutated gene responsible for FOP.15 Activin receptor type-1 (ACVR1; also known as activin-like kinase 2 [ALK-2]) is a bone morphogenetic protein (BMP) type I receptor. All individuals with classic features of FOP harbor the identical heterozygous single nucleotide substitution (a guanine to adenine change at position 617) that changes amino acid 206 from arginine to histidine. Codon 206 is highly conserved and occurs within the glycine–serine region of the cytoplasmic domain of ACVR1.
BMP signaling
BMPs are members of the transforming growth factor β (TGF-β) family of extracellular signaling proteins, which regulate a diverse range of cellular activities including differentiation, proliferation, apoptosis, migration, mediating positional information and stem-cell renewal.16–22 A unique function of many members of the BMP subfamily is the induction of the complete pathway of endochondral bone formation.23 BMP signaling is important in embryonic development and skeletal formation, although BMPs and their receptors are also expressed in many adult tissues, including skeletal muscles and chondrocytes.
Signal transduction through the BMP pathway is mediated through heterotetrameric receptor complexes comprising two type I and two type II serine/threonine kinase receptors (Figure 2). In addition to ACVR1, other type I receptors include BMPR-1A (also known as ALK-3) and BMPR-1B (also known as ALK-6). In response to ligand binding, type II receptors phosphorylate the cytoplasmic glycine–serine domain of type I receptors, resulting in their subsequent activation.24–29 Activated type I receptors mediate downstream signaling through BMP-pathway-specific SMAD proteins and through mitogen-activated protein kinase pathways, both of which can directly or indirectly regulate the transcription of target genes in the nucleus.24,30–32 The signaling specificity attained by type I BMP receptors is probably established through their highly regulated temporal and spatial expression, and through other mechanisms such as receptor interactions, including the existence of different combinations of receptor heterodimers within a receptor complex.24,27,30,31,33–35 In addition, BMPs act as morphogens, inducing receptor activation in a dose-dependent manner. This ability of BMPs to function as morphogens is established, in part, by the presence of BMP antagonists, which prevent BMP from binding to its receptors.36 BMP signaling is a highly regulated process that is dependent on negative and positive regulatory feedback.27
Figure 2.

Generalized schematic representation of the BMP signaling pathway. Type I and type II BMP receptors span the cell membrane and bind extracellular BMP ligand. Ligand binding to BMP heterotetrameric receptor complexes activates signaling through type II-receptor-mediated phosphorylation of the type I receptor on the GS domain. Type I receptor phosphorylation is accompanied by decreased binding to the GS domain of proteins that prevent receptor signaling in the absence of ligand binding. Activated type I receptors phosphorylate cytoplasmic signal transduction proteins such as R-SMADs and MAPKs (including JNK, ERK and p38), which, in turn, directly or indirectly regulate the transcription of target genes in the nucleus. Abbreviations: BMP, bone morphogenetic protein; BMPR, bone morphogenetic protein receptor; Co-SMAD, common-mediator SMAD; ERK, extracellular signal-regulated kinase; GS domain, glycine–serine domain; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; R-SMAD, receptor-regulated SMAD.
ACVR1 and the skeleton
ACVR1 is expressed in chondrocytes and osteoblasts, and overexpression of the constitutively active form of ACVR1—caALK2—enhances chondrogenesis, expands cartilage elements, stimulates joint fusions, and induces heterotopic ossification in animal models.37,38 FOP is associated with similar clinical findings and with dysregulation of the BMP signaling pathway.39–42 In vitro and in vivo assays support the concept that the Arg206His ACVR1 mutation in FOP is an activating mutation that induces BMP signaling in BMP-independent and BMP-responsive manners to activate downstream signaling.43,44 However, in contrast to caALK2, which causes embryonic lethality in mouse models,45,46 the FOP Arg206His ACVR1 mutation is more mildly activating, providing an explanation for its compatibility with life.44
Heterotopic ossification in FOP
The most clinically relevant feature of FOP is the episodic formation of extraskeletal bone.8,9,11–13,47 Postnatal heterotopic ossification in FOP usually begins before the age of 5 years, and proceeds in predictable temporal and spatial patterns.48,49 In the absence of trauma, which alters the natural progression of the disease, episodes of heterotopic bone formation occur in a pattern that parallels the sequence of formation of skeletal elements during embryonic development. Typically, the upper back and neck are the first parts of the skeleton to be affected; the physiological basis of this progression pattern has not been identified. Over time, ectopic bone formation in FOP is progressive, cumulative and extensive, bridging the joints of the axial and appendicular skeletons and causing near-complete immobilization of the body (Figure 1a).
Stages of FOP heterotopic ossification
Although many episodes of FOP lesion formation (also known as ‘flare-ups’; FOP flare-ups are irreversible once bone formation occurs) seem to initiate spontaneously, exacerbation of the disease is frequently stimulated by soft tissue injury, such as surgery, muscle fatigue, intramuscular injections, or preschool immunizations, or by viral illnesses.11,48–51 Whether or not an episode is associated with overt injury, the immune system seems to have an important role in triggering FOP flare-ups. Histological evaluation of the stages of FOP lesion formation has shown that a phase of tissue destruction precedes a phase of proliferation and tissue formation (Figure 3).52,53 Inflammatory cells of lymphocytic, macrophage, and mast cell origin are present in the perivascular space of early FOP lesions, deep within skeletal muscle and connective tissue.52,54,55 The response of patients with FOP is similar to, but greater in magnitude than, a normal tissue response to injury. The presence of inflammatory cells is associated with damage to skeletal muscle cells and a hypoxic microenvironment, both of which are hypothesized to trigger the fibroproliferative response in early FOP lesions.47,53,56,57
Figure 3.
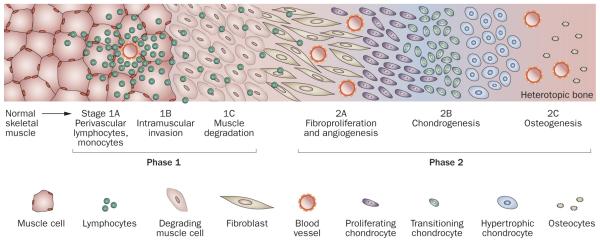
Schematic histologic representation of the stages of endochondral heterotopic ossification in FOP. Lesion formation in FOP involves inflammation and the destruction of connective tissues (phase 1) followed by a replacement phase of new tissue development (phase 2). The initial histologic evidence of lesion induction is the presence of abundant perivascular lymphocytes (stage 1A) in connective tissue such as skeletal muscle. Lymphocytes expand into the tissue (stage 1B) and loss of the connective tissue structure follows (stage 1C). As the tissue is degraded, it is rapidly replaced by fibroproliferative cells (stage 2A). Angiogenesis and vascularization occur (stage 2B), followed by chondrogenesis and osteogenesis (stage 2C) and the formation of heterotopic bone. Abbreviation: FOP, fibrodysplasia ossificans progressiva.
Early FOP lesions are highly angiogenic56 and investiga tions of FOP patients and in vivo mouse models of FOP-like heterotopic ossification have demonstrated that connective tissue progenitor cells of vascular origin contribute to multiple stages in the development of the heterotopic anlagen.57,58 An angiogenic fibroproliferative stage of FOP lesion formation is followed by the production of connective tissue with cartilage and bone through a normal sequence of endochondral ossification stages (Figure 3).47,52,53,56 In essence, one tissue is replaced by another.
The stages and events of FOP endochondral heterotopic ossification reflect the events that occur in embryonic skeletal development and in bone repair during fracture healing. Mature heterotopic bone in FOP can form bone marrow cavities and produce apparently normal bone marrow.59 The ectopic bone that forms in FOP is normal skeletal bone as defined by most criteria, including histology, biochemistry, metabolism, radiology and biomechanics.59–61 The consequence is a skeletal-like bone organ system that develops in FOP patients in response to the mutated BMP receptor.
A model for the effect of ACVR1 mutation
A working model proposes that the moderate constitutive activation of BMP signaling that results from mutation of ACVR1 in FOP alters the BMP signaling ‘set point’, thereby increasing the basal level of BMP pathway acti vity to prime cells in connective tissues to be more responsive to interactions with inducers and modulators of cell differentiation in the tissue microenvironment (Figure 4). Stimuli such as injury then trigger or mediate (or both) active episodes of bone formation in FOP patients. This model is consistent with the quiescent periods that are observed in FOP patients between active episodes of heterotopic bone formation.
Figure 4.
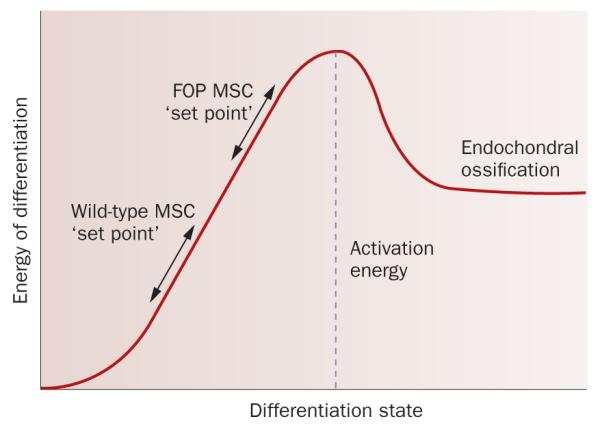
A working model for altered BMP signaling in FOP. The ACVR1 mutation that is present in the vast majority of patients with FOP is an activating mutation that can stimulate signaling, at least in part in the absence of BMP, but this activity might only be moderately ‘on’ under basal in vivo conditions, effectively raising the BMP pathway activation set-point and reducing the amount of additional activation that is needed to stimulate endochondral ossification. Triggering events, such as tissue injury and associated changes in the tissue microenvironment, enhance signaling and overcome the reduced amount of ‘activation energy’ that is needed to stimulate cartilage and bone cell differentiation; these events might induce a stronger response of longer duration than is normal, owing to the mechanism of mutant receptor activation or an impaired negative feedback response, or both. In normal, non-FOP tissues, the set point is sufficiently low such that the equivalent activation of BMP signaling does not have sufficient activation energy to overcome the threshold that leads to cartilage and bone formation, and normal tissue repair occurs, for example, in response to injury. Abbreviation: ACVR1, activin receptor type-1; BMP, bone morphogenetic protein; FOP, fibrodysplasia ossificans progressiva.
FOP and fracture healing
Although normal skeletal formation is limited after the skeleton has been generated during early development, vertebrates retain the ability to regenerate skeletal elements during fracture repair. Fracture healing parallels the stages of endochondral bone formation during skeletal development, a process that is also induced by BMPs.23,62,63 BMPs and components of the BmP signaling pathway are expressed during the course of fracture healing and are required for this process.64–67
If the heterotopic bone in a patient with FOP sustains a fracture, this extraskeletal bone will undergo a qualitatively normal fracture repair process.68 The rate of fracture repair in the normal skeleton of an FOP patient is similar to that of healing of normal skeletal bone; however, anecdotal observations indicate that the rate of repair within the heterotopic bone seems to be accelerated. Of particular note is that fracture occurrence and subsequent healing in FOP patients have not been as sociated with inducing new heterotopic ossification.
FOP and embryonic development
The underlying mutation in ACVR1 in FOP alters bone formation during embryonic skeletal development in addition to inducing heterotopic bone formation postnatally.8,9,11,13,69 When a child with FOP is born, the only obvious physical finding that might provoke suspicion of the disease is congenital malformation of the big toes. However, other skeletal changes are commonly, but variably, present, including a short broad femoral neck, spine/vertebral malformations and tibial osteochondromas.70,71
Mutations in specific components of the BMP signaling pathway, including receptors, ligands, and BMP antagonists, cause various human skeletal disorders,72 providing valuable information about the temporal activity and the tissue-specific expression of specific components of this complex signaling pathway. The FOP Arg206His ACVR1 mutation has relatively subtle effects on the skeleton, which suggests that this mutation modulates embryonic skeletal development, perhaps by altering the level or duration of BMP signaling. In addition to the Arg206His ACVR1 mutation identified in patients with a classic clinical presentation of FOP, a small number of patients have been identified with increased or decreased severity of FOP-type ectopic ossification or developmental skeletal malformations, or both,73 and are also caused by a mutation in ACVR1. These mutations, however, do not affect residue 206 but fall within the GS domain or in the protein kinase domain. In at least some cases, genotype–phenotype correlations are observed. Of particular note are mutations within codon 328; specific mutations are associated with little or no toe malformation and late onset of heterotopic ossification, whereas other mutations in this codon correlate with extensive heterotopic ossification and severe malformation or absence of thumbs and big toes.73
Although the first identified function of BMPs was in promoting bone formation,23,74 BMPs are important in the development of a wide range of vertebrate tissues and organs.19,75–80 A gradient of BMP signaling establishes dorsal–ventral polarity in the developing embryo, thereby specifying the initial differentiation of ectoderm-derived and mesoderm-derived tissues and organs.17 Mouse models in which ACVR1 has been knocked out or activated have shown that roles for BMP signal transduction through ACVR1 include embryonic patterning during gastrulation, the migration of neural crest cells that contribute to cardiac and craniofacial development, eye development, and the formation of germ cells.81–86 Patient observations (F. S. Kaplan, unpublished observations) and preliminary data from mouse models (S. Chakkalakal and E. M. Shore, unpublished observations) support the notion that activating mutations in ACVR1 that are associated with FOP affect the development and function of a range of tissues and organs.73
Progressive osseous heteroplasia
Similar to patients with FOP, patients with POH experience extensive formation of bone within soft connective tissues (Figure 5).87 Like FOP, this bone formation is episodic and progressive. However, POH and FOP can be distinguished on the basis of several clinical cri teria.8,9,88,89 Unlike in FOP patients, ossification within the dermis typically occurs in patients with POH, often in association with adipose tissue (Figure 6).8,88 POH heterotopic ossification progresses from the dermis to the underlying deep connective tissues; bone forms within skeletal muscle, sometimes fusing with skeletal bone. Distinct from FOP, POH is not associated with inflammation, predictable regional patterns of heterotopic ossification, or FOP-like big toe malformations. Heterotopic bone formation in POH patients occurs in an asymmetric mosaic distribution and is mainly intramembranous (Figure 5).8,88,90,91
Figure 5.

Heterotopic ossification in POH. Radiographic appearance of heterotopic ossification. Lateral serial X-rays of the lower leg of a child, showing progression of heterotopic ossification from the ages of 18 months a | to 30 months b | to 8 years (c | amputation specimen). Extensive ossification of the soft tissues of the superficial and deep posterior compartments of the leg, disuse osteopenia and anterior bowing of the tibia can be seen. Abbreviation: POH, progressive osseous heteroplasia. Permission obtained from The Journal of Bone and Joint Surgery, Inc. © Kaplan, F. S. et al. J. Bone Joint Surg. 76, 425-436 (1994).
Figure 6.
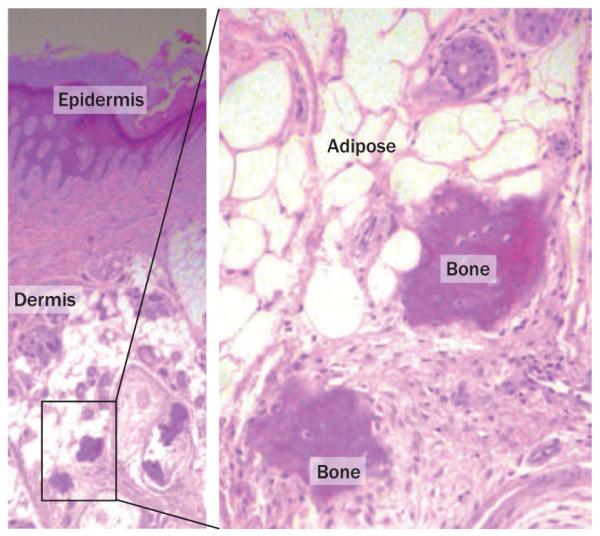
Histopathology of a POH lesion. Lower power image (left, magnification ×50) shows the epidermis and dermis. Irregular deposits of bone within the dermal tissue (shown at higher power on the right, magnification ×200) are often observed in proximity to subcutaneous adipose tissue. Both images were obtained following hematoxylin and eosin staining. Abbreviation: POH, progressive osseous heteroplasia.
The GNAS locus
POH is caused by inactivating mutations in GNAS,90 a transcriptionally complex locus that contains multiple promoters and first exons that are spliced to a common set of exons 2–13 (Figure 7).92–95 Genomic imprinting mechanisms, which differentially methylate the maternally inherited and paternally inherited copy of each gene, contribute to promoter selection and transcriptional regulation within the locus. The most abundant protein product of this gene is Gsα, a ubiquitously expressed heterotrimeric G-protein alpha subunit that activates adenylyl cyclases, thereby increasing levels of cyclic AMP. The Gsα transcript is expressed from both alleles in most tissues, but is expressed only from the maternally inherited allele in a subset of cells and tissues. The GNAS locus also encodes XLαs, a variant form of Gsα with a longer amino-terminal domain that seems to function similarly to Gsα but has a more restricted expression pattern and lower expression levels. Transcription of XLαs is restricted to the paternally inherited GNAS allele. A third transcript produces neuroendocrine secretory protein 55 (Nesp55); expression is limited to the maternally inherited allele and is highest in the nervous system and endocrine tissues, but little is known about the function of this protein. In addition, noncoding transcripts are generated from the locus and have been implicated in the transcriptional regulation of other transcripts within the GNAS locus. Exon A/B (exon 1A in mice) is only expressed from the paternal allele and, like the coding transcripts, splices to exons 2–13. Nespas is an antisense transcript that overlaps Nesp55 exon 1 and is only expressed from the paternal allele of the GNAS locus.
Figure 7.
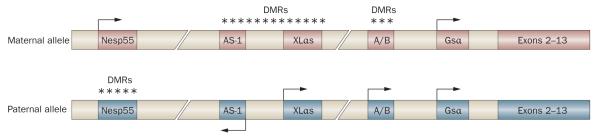
The GNAS gene locus. GNAS is a complex gene, encoding multiple transcripts that are expressed from several promoters within the locus. In the figure, exons are depicted by boxes, with arrows indicating transcriptional activity and direction. The first exons of Nesp55, XLαs, A/Band Gsα splice to a set of common downstream exons (exons 2–13). The antisense transcript (AS-1) is also indicated. Genomic imprinting differentially marks the maternally inherited and paternally inherited GNAS alleles by DNA methylation (indicated by asterisks) in a reciprocal pattern (differentially methylated regions, DMrs). Methylation is associated with transcriptional silencing.
Other GNAS inactivation disorders
POH is a rare disorder, with fewer than 60 clinically-confirmed cases worldwide. The rarity of the disease seems to be a function, at least in part, of the incomplete penetrance of inactivating GNAS mutations91 and the wide and variable range of clinical phenotypes that are associated with these mutations. POH is among a number of related genetic disorders that are associated with heterozygous inactivating mutations in GNAS, including Albright hereditary osteodystrophy (AHO), pseudohypoparathyroidism (PHP) and osteoma cutis (Box 1).91–97 This spectrum of GNAS inactivation disorders has the common feature of superficial/dermal ossification; however, POH is unique in that it causes heterotopic ossification that is not limited to the dermis and subcutaneous tissues.88,90,91
Box 1. A spectrum of human disorders with inactivating GNAS mutations.
Several human disorders share the common features of superficial (subcutaneous or dermal) ossification and association with inactivating mutations in the GNAS gene locus.91,94,96
Albright hereditary osteodystrophy (AHO)
A variable constellation of clinical features, including short adult stature, obesity, round face, brachydactlyly and subcutaneous ossification. Some cases are also associated with neurobehavioral problems or mental retardation, or both. AHO features are more frequently associated with maternal inheritance of mutations in GNAS, but can also be caused by paternal inheritance. The term ‘pseudopseudohypoparathyroidism’ (PPHP) has been used to describe some patients who have GNAS mutations on the paternally inherited allele and clinical features of AHO without endocrine abnormalities; these cases generally occur within family pedigrees that also show PHP1a with maternal inheritance of GNAS mutations (see below).
Pseudohypoparathyroidism type Ia (PhPIa)
End-organ resistance to parathyroid hormone and other hormones, including thyroidstimulating hormone and gonadotrophins. Associated with AHO features, particularly obesity. Only caused by GNAS mutations on the maternally inherited allele.
Osteoma cutis
Heterotopic ossification that is limited to superficial (subcutaneous) tissues without hormone resistance or AHO features.
Progressive osseous heteroplasia (POH)
Heterotopic ossification that initiates in early childhood then progresses within deeper connective tissues such as skeletal muscle and fascia. Some patients show limited AHO features, but never signs of obesity. POH is not associated with hormone resistance and mutations are found almost exclusively on the paternally inherited GNAS allele. However, in rare cases, extensive POH-like progressive bone formation occurs in patients with a maternal GNAS mutation and/or AHO or PHP1a features, indicating a range in clinical response to GNAS mutations.
POH–AHO and POH–PhP1a
A small number of patients with POH present with multiple features of AHO or hormone resistance, or both. A maternal or paternal inheritance pattern of GNAS mutations in these patients is undetermined.
Heterotopic ossification in POH
Heterotopic bone formation in POH is typically intramembranous, but evidence of ectopic cartilage has been observed.91 As heterotopic ossification in POH progresses into the deeper connective tissues, the appearance is diffuse and web-like; discrete skeletal-like elements do not form (Figure 5).8,88,90 Bone formation in POH originates spontaneously in the dermis; the initial clinical presentation is indistinguishable from the subcutaneous ossification that is a common feature of AHO. Subcutaneous ossification and other features of AHO, which include short stature, round face and brachydactyly, are common in patients with PHP type 1a (PHP1a).92–94,96 However, most POH patients show no AHO features, and POH patients also lack the hormone resistance and obesity that are characteristically associated with PHP1a.88,91
PHP1a is caused by mutation of the maternally inherited GNAS allele and is rarely associated with extensive progression of bone formation beyond initial dermal ossification. By contrast, paternal inheritance of inactivating GNAS mutations in patients with POH correlates highly with progressive formation of bone that extends from superficial sites in the dermis into deeper soft connective tissues such as skeletal muscle and fascia.90,91 These observations support the theory that induction of bone formation within the dermis results from GNAS haploinsufficiency, whereas the progression of bone to deeper tissues might be regulated by skewed expression levels from the two GNAS alleles or by loss of specific transcripts, such as XLαs, that are expressed from the paternally inherited GNAS allele. Consistent with the possibility that specific transcripts might be involved in the extensive progression of bone formation, no mutations in GNAS exon 1, which is specific for the Gsα transcript, have so far been identified in patients with POH.90,91 However, POH-like progressive bone formation has, in rare cases, been reported to occur in patients with maternal GNAS mutation and/or features of AHO or PHP1a92,94,98 (E. M. Shore and F. S. Kaplan, unpublished observations), suggesting a range of patient sensitivities and responses to GNAS mutations, which perhaps reflects variations in individual genetic backgrounds.
Lessons from mouse models
The association of features of AHO with those of both PHP1a and POH supports the concept that these features are a result of GNAS haploinsufficiency during early skeletal development, which can be caused by deficiency of either the maternally or paternally inherited GNAS allele. Further support has come from mouse models of Gnas haploinsufficiency; these models show brachydactyly and reduced adult length with either maternal or paternal null alleles.92,95 Mouse models in which the maternal or paternal Gnas allele has been knocked out also experience subcutaneous ossification, similar to clinical findings of AHO, although only at advanced adult ages (E. M. Shore and F. S. Kaplan, unpublished observations).90,97 However, these mouse models show no evidence of progression to deep tissues, as occurs in patients with POH.
Mouse models with a maternally inherited Gnas null allele90,93 show characteristics of AHO and PHP1a, with hormone resistance and increased adiposity. By contrast, mice with paternally inherited Gnas null alleles show a normal hormone response and lean body mass, similar to findings in POH patients. Results from Gnas-transcript-specific knockout models have shown that the lean body mass associated with paternal Gnas allele inactivation correlates with the loss of the paternally expressed Gnasxl (XLαs) transcript.100
Gnas mouse models have also shown that Gsα is important in osteoblast and chondrocyte differentiation.92,95 Studies of chimeric mice revealed that Gsα null or Gsα haploinsufficient chondrocytes undergo accelerated differentiation to hypertrophic chondrocytes, resulting in shorter growth plates and bones, probably as a result of defects in signaling through parathyroid hormone and parathyroid hormone-related protein.101,102 Osteoblast-specific Gsα-null mouse models showed reduced long bone size, reduced amounts of trabecular bone and thicker cortical bone, with an overall reduction in the rate of bone turnover,103 suggesting that an effect of Gsα haploinsufficiency on the normal skeleton is to reduce bone growth and maintenance. By contrast, Gsα haploinsufficiency enhances initiation of osteogenesis during ectopic bone formation.
GNAS is widely expressed in various cells and tissues, and mutation or insufficiency would be expected to have effects on multiple developing tissues and organs. Such consequences have been observed in patients and mouse models and include, in addition to effects on skeletal development and subcutaneous ossification, effects on energy metabolism, accumulation of adipose tissue, renal function, cognitive abnormalities, and bone marrow hematopoiesis.92,95,104
Therapeutic strategies for FOP and POH
No effective treatment options for FOP or POH are currently available. For FOP, anti-inflammatory agents such as steroids have, in some cases, been associated with the suppression of flare-ups if used at early stages of onset. However, current care regimens for FOP and POH patients are mainly palliative. Knowledge of the genetic causes of these disorders greatly increases the possibility that effective treatments can be developed.
The identification of an activating mutation in a cellsurface receptor, ACVR1, in FOP provides a target with good potential for therapeutic intervention. Most cases of FOP are caused by the identical ACVR1 mutation, suggesting that treatments could be directed against a highly specific region of the ACVR1 protein or specific signaling event, or both. The identification of this mutation together with a growing understanding of its mechanism of activity mean that at least four broad approaches for intervention are possible (reviewed elsewhere105). First, the enhanced activity of the ACVR1-induced signaling pathway could be inhibited, potentially by the use of signal transduction inhibitors to block receptor activity, RNA inhibition, monoclonal antibodies, and/or secreted antagonists. This approach is likely to be the most promising, at least in the short term. Second, the chondrogenic or osteogenic progenitor cell that gives rise to heterotopic bone and/or a cell that stimulates differentiation of the progenitor cell could be targeted. Suppressing the inflammatory stimulus that accompanies the initiation of FOP lesions is an additional possibility. Finally, it might be feasible to alter the connective tissue microenvironment that supports heterotopic ossification.
Compared to identifying plausible therapeutic strategies for FOP, determining an appropriate target for treating POH is a much greater challenge. Unlike approaches for FOP that involve inhibition (or partial inhibition) of an activating mutation, developing agents or strategies to compensate for the inactivating GNAS mutations in POH is more difficult, partly owing to the complexity of the GNAS gene and our dearth of knowledge. The development of a therapy for POH is confounded by several factors. First, several products are expressed from the GNAS gene. Moreover, genetic imprinting occurs at the GNAS locus. Furthermore, several signaling pathways are initiated downstream of GNAS products. GNAS expression is ubiquitous, as are the activities of its products, which would make it difficult to limit any desired effect to a specific cell or tissue. Related to this point, there is minimal understanding of the roles of GNAS in osteogenesis and a lack of knowledge of the progenitor cells in the target tissue.
Our group, and other researchers, are investigating the functions of GNAS products, with a particular interest in paternal allele-specific transcripts (such as XLαs) that are relevant to osteogenesis and the progression of heterotopic ossification to extensively infiltrate connective tissues.106–110 Such investigations have identified cyclic AMP signaling, the major pathway downstream of Gsα and XLαs, as being important in regulating cellular differentiation. Crosstalk between BMP signaling and GNAS–cyclic AMP signaling pathways is thought to be relevant to osteogenesis (S. Zhang and E. M. Shore, unpublished observations),110 suggesting the possibility that common treatment strategies might be applicable to POH and FOP. Further investigations to understand the regulation of cell-fate decisions and osteogenic differentiation will help to identify the best targets for POH and FOP, as well as strategies that are likely to be applicable to a range of other disorders that affect cell-fate determination.
Conclusions
FOP and POH are two rare human genetic disorders of heterotopic ossification that have broad implications for understanding and manipulating physiologic bone formation. In FOP, activating mutations in the gene encoding the BMP type I receptor ACVR1 induce endochondral heterotopic ossification that results in a functional bone organ system. In POH, the heterotopic ossification forms mainly intramembranous bone tissue in response to inactivating mutations in GNAS.
Identification of the gene mutations that cause these diseases provides opportunities to discover cellular pathways and mechanisms that regulate bone and cartilage cell differentiation and that direct the formation of the skeleton. Although the clinical presentations of heterotopic ossification in FOP and POH are distinct, the two underlying pathways regulate overlapping cellular processes, which plausibly function in part through cellular crosstalk but also carry out distinct roles to regulate where, when, and how bone forms.
Investigations into FOP and POH provide new information that is relevant to understanding the basic biology of cell-fate decisions during embryogenesis and in adult connective tissues. Although these are rare conditions, the application of knowledge gained from the study of FOP and POH is potentially great, with relevance to therapeutic development for more common conditions of nonhereditary heterotopic ossification and to strategies for tissue engineering.
MedspaceCME Continuing Medical Education online.
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education through the joint sponsorship of Medscape, LLC and Nature Publishing Group. Medscape, LLC is accredited by the ACCME to provide continuing medical education for physicians.
Medscape, LLC designates this educational activity for a maximum of 0.75 AMA PRA Category 1 Credits™. Physicians should only claim credit commensurate with the extent of their participation in the activity. All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test and/or complete the evaluation at http://www.medscapecme.com/journal/nrrheum; and (4) view/print certificate.
Learning objectives
Upon completion of this activity, participants should be able to:
Describe the genetics and pathologic characteristics of fibrodysplasia ossificans progressiva (FOP).
Apply knowledge of the clinical course of FOP in counseling patients and families.
Describe progressive osseous heteroplasia and its etiology.
Key points.
▪ Heterotopic ossification is the formation of extraskeletal bone in soft connective tissues
▪ The bone tissue that forms during heterotopic ossification is qualitatively normal
▪ Two rare inherited forms of heterotopic ossification are fibrodysplasia ossificans progressiva (FOP) and progressive osseous heteroplasia (POH)
▪ FOP is caused by a mutation in ACVR1, which encodes a bone morphogenetic protein type I receptor; POH is caused by a mutation in the GNAS locus
▪ The genes and signaling pathways that are altered in these genetic disorders are key regulators of skeletal development and cell differentiation
▪ Understanding the cellular mechanisms responsible for these rare disorders might lead to the development of therapeutic approaches relevant to common conditions of excessive and insufficient bone formation
Acknowledgments
We thank the members of our research laboratories and our many collaborators and colleagues for their contributions, notably J. Haupt and R. Mauck who generously provided figures (Figure 2 and Figure 4, respectively) that were adapted for this manuscript. We also thank the NIH/NIAMS-supported Penn Center for Musculoskeletal Disorders (AR050950). This work was supported in part by the Center for Research in FOP and Related Disorders, the International FOP Association (IFOPA), the Ian Cali Endowment, the Weldon Family Endowment, the Isaac and Rose Nassau Professorship of Orthopedic Molecular Medicine, the Rita Allen Foundation, and by grants from the National Institutes of Health (R01- AR41916 and R01-AR046831).
Charles P. Vega, University of California, Irvine, CA, is the author of and is solely responsible for the content of the learning objectives, questions and answers of the MedscapeCME-accredited continuing medical education activity associated with this article.
Footnotes
Competing interests The authors, the Journal Editor J. Buckland and the CME questions author C. P. Vega declare no competing interests.
Review criteria English-language, full-text research papers and review articles published between 1990 and 2009 were sourced for inclusion in this review from a PubMed search using the following terms: “heterotopic ossification”, “osteogenic differentiation”, “endochondral ossification”, “intramembranous ossification”, “bone morphogenetic protein (BMP) signaling pathway”, “fibrodysplasia ossificans progressiva”, and “progressive osseous heteroplasia”. The OMIM database was also searched using the terms “heterotopic ossification” and “extra-skeletal bone”.
Author contributions E. M. Shore researched the data for the article and wrote the article. Both E. M. Shore and F. S. Kaplan contributed equally to discussion of content and to reviewing and editing the manuscript before submission.
References
- 1.Yang Y. In: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Rosen CJ, editor. American Society of Bone and Mineral Research; Washington, DC: 2008. pp. 2–10. [Google Scholar]
- 2.McCarthy EF, Sundaram M. Heterotopic ossification: a review. Skeletal Radiol. 2005;34:609–619. doi: 10.1007/s00256-005-0958-z. [DOI] [PubMed] [Google Scholar]
- 3.Pignolo RJ, Foley KL. Nonhereditary heterotopic ossification. Implications for injury, arthropy, and aging. Clin. Rev. Bone Miner. Metab. 2005;3:261–266. [Google Scholar]
- 4.Forsberg JA, et al. Heterotopic ossification in high-energy wartime extremity injuries: prevalence and risk factors. J. Bone Joint Surg. Am. 2009;91:1084–1091. doi: 10.2106/JBJS.H.00792. [DOI] [PubMed] [Google Scholar]
- 5.Neal B, Gray H, MacMahon S, Dunn L. Incidence of heterotopic bone formation after major hip surgery. ANZ J. Surg. 2002;72:808–821. doi: 10.1046/j.1445-2197.2002.02549.x. [DOI] [PubMed] [Google Scholar]
- 6.Potter BK, Burns TC, Lacap AP, Granville RR, Gajewski D. Heterotopic ossification in the residual limbs of traumatic and combat-related amputees. J. Am. Acad. Orthop. Surg. 2006;14(10 Spec. No):S191–S197. doi: 10.5435/00124635-200600001-00042. [DOI] [PubMed] [Google Scholar]
- 7.van Kuijk AA, Geurts ACH, van Kuppevelt HJM. Neurogenic heterotopic ossification in spinal cord injury. Spinal Cord. 2002;40:313–326. doi: 10.1038/sj.sc.3101309. [DOI] [PubMed] [Google Scholar]
- 8.Kaplan FS, Shore EM. Progressive osseous heteroplasia. J. Bone Miner. Res. 2000;15:2084–2094. doi: 10.1359/jbmr.2000.15.11.2084. [DOI] [PubMed] [Google Scholar]
- 9.Shore EM, Kaplan FS. Insights from a rare genetic disorder of extra-skeletal bone formation, fibrodysplasia ossificans progressiva (FOP) Bone. 2008;43:427–433. doi: 10.1016/j.bone.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.National Center for Biotechnology Information. 2010 MIM #135100. [online], http://www.ncbi.nlm.nih.gov/omim/135100.
- 11.Kaplan FS, et al. The phenotype of fibrodysplasia ossificans progressiva. Clin. Rev. Bone Miner. Metab. 2005;3:183–188. [Google Scholar]
- 12.Kaplan FS, et al. Fibrodysplasia ossificans progressiva. Best Pract. Res. Clin. Rheumatol. 2008;22:191–205. doi: 10.1016/j.berh.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaplan FS, Shore EM. In: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Rosen CJ, editor. American Society for Bone and Mineral Research; Washington, DC: 2008. pp. 442–446. [Google Scholar]
- 14.Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva and progressive osseous heteroplasia: two genetic disorders of heterotopic ossification. Clin. Rev. Bone Miner. Metab. 2005;3:257–259. [Google Scholar]
- 15.Shore EM, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- 16.Derynck R, Akhurst RJ. Differentiation plasticity regulated by TGF-β family proteins in development and disease. Nat. Cell Biol. 2007;9:1000–1004. doi: 10.1038/ncb434. [DOI] [PubMed] [Google Scholar]
- 17.Eivers E, Fuentealba LC, De Robertis EM. Integrating positional information at the level of Smad1/5/8. Curr. Opin. Genet. Dev. 2008;18:304–310. doi: 10.1016/j.gde.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heisenberg CP, Solnica-Krezel L. Back and forth between cell fate specification and movement during vertebrate gastrulation. Curr. Opin. Genet. Dev. 2008;18:311–316. doi: 10.1016/j.gde.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hogan BLM. Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev. 1996;10:1580–1594. doi: 10.1101/gad.10.13.1580. [DOI] [PubMed] [Google Scholar]
- 20.Watabe T, Miyazono K. Roles of TGF-β family signaling in stem cell renewal and differentiation. Cell Res. 2009;19:103–115. doi: 10.1038/cr.2008.323. [DOI] [PubMed] [Google Scholar]
- 21.Wu MY, Hill CS. TGF-β superfamily signaling in embryonic development and homeostasis. Dev. Cell. 2009;16:329–343. doi: 10.1016/j.devcel.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 22.Xu J, Lamouille S, Derynck R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Urist MR. Bone: formation by autoinduction. Science. 1965;150:893–899. doi: 10.1126/science.150.3698.893. [DOI] [PubMed] [Google Scholar]
- 24.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 25.Huse M, et al. The TGFβ receptor activation process: an inhibitor- to substrate-binding switch. Mol. Cell. 2001;8:671–682. doi: 10.1016/s1097-2765(01)00332-x. [DOI] [PubMed] [Google Scholar]
- 26.Krause C, de Gorter DJJ, Karperien M, ten Dijke P. In: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Rosen CJ, editor. American Society of Bone and Mineral Research; Washington, DC: 2008. pp. 10–16. [Google Scholar]
- 27.Schmierer B, Hill CS. TGF-β–SMAD signal transduction: molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 28.Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 29.ten Dijke P, Hill CS. New insights into TGF-β–Smad signalling. Trends Biochem. Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 30.Guo X, Wang X-F. Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. 2009;19:71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moustakas A, Heldin CH. Non-Smad TGF-β signals. J. Cell Sci. 2005;118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 32.Nohe A, et al. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J. Biol. Chem. 2002;277:5330–5338. doi: 10.1074/jbc.M102750200. [DOI] [PubMed] [Google Scholar]
- 33.Chen YG, et al. Determinants of specificity in TGF-β signal transduction. Genes Dev. 1998;12:2144–2152. doi: 10.1101/gad.12.14.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Little SC, Mullins MC. Bone morphogenetic protein heterodimers assemble heteromeric type I receptor complexes that pattern the dorsoventral axis. Nat. Cell Biol. 2009;11:637–643. doi: 10.1038/ncb1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Massague J. How cells read TGF-β signals. Nat. Rev. Mol. Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 36.Gazzerro E, Canalis E. Bone morphogenetic proteins and their antagonists. Rev. Endocr. Metab. Disord. 2006;7:51–65. doi: 10.1007/s11154-006-9000-6. [DOI] [PubMed] [Google Scholar]
- 37.Yu PB, et al. BMP type I receptor inhibition reduces heterotopic ossification. Nat. Med. 2008;14:1363–1369. doi: 10.1038/nm.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang D, et al. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. J. Bone Miner. Res. 2003;18:1593–1604. doi: 10.1359/jbmr.2003.18.9.1593. [DOI] [PubMed] [Google Scholar]
- 39.Billings PC, et al. Dysregulated BMP signaling and enhanced osteogenic differentiation of connective tissue progenitor cells from patients with fibrodysplasia ossificans progressiva (FOP) J. Bone Miner. Res. 2008;23:305–313. doi: 10.1359/JBMR.071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fiori JL, Billings PC, Serrano de la Pena LS, Kaplan FS, Shore EM. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP) J. Bone Miner. Res. 2006;21:902–909. doi: 10.1359/jbmr.060215. [DOI] [PubMed] [Google Scholar]
- 41.Serrano de la Pena LS, et al. Fibrodysplasia ossificans progressiva (FOP), a disorder of ectopic osteogenesis, misregulates cell surface expression and trafficking of BMPRIA. J. Bone Miner. Res. 2005;20:1168–1176. doi: 10.1359/JBMR.050305. [DOI] [PubMed] [Google Scholar]
- 42.Shafritz AB, et al. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N. Engl. J. Med. 1996;335:555–561. doi: 10.1056/NEJM199608223350804. [DOI] [PubMed] [Google Scholar]
- 43.Fukuda T, et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J. Biol. Chem. 2009;284:7149–7156. doi: 10.1074/jbc.M801681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen Q, et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J. Clin. Invest. 2009;119:3462–3472. doi: 10.1172/JCI37412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gu Z, et al. The type I serine/threonine kinase receptor ActRIA (ALK2) is required for gastrulation of the mouse embryo. Development. 1999;126:2551–2561. doi: 10.1242/dev.126.11.2551. [DOI] [PubMed] [Google Scholar]
- 46.Mishina Y, Crombie R, Bradley A, Behringer RR. Multiple roles for activin-like kinase-2 signaling during mouse embryogenesis. Dev. Biol. 1999;213:314–326. doi: 10.1006/dbio.1999.9378. [DOI] [PubMed] [Google Scholar]
- 47.Pignolo RJ, Suda RK, Kaplan FS. The fibrodysplasia ossificans progressiva lesion. Clin. Rev. Bone Miner. Metab. 2005;3:195–200. [Google Scholar]
- 48.Cohen RB, et al. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. J. Bone Joint Surg. Am. 1993;75:215–219. doi: 10.2106/00004623-199302000-00008. [DOI] [PubMed] [Google Scholar]
- 49.Rocke DM, Zasloff M, Peeper J, Cohen RB, Kaplan FS. Age- and joint-specific risk of initial heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. Clin. Orthop. Relat. Res. 1994;301:243–248. [PubMed] [Google Scholar]
- 50.Lanchoney TF, Cohen RB, Rocke DM, Zasloff MA, Kaplan FS. Permanent heterotopic ossification at the injection site after diphtheria-tetanus-pertussis immunizations in children who have fibrodysplasia ossificans progressiva. J. Pediatr. 1995;126:762–764. doi: 10.1016/s0022-3476(95)70408-6. [DOI] [PubMed] [Google Scholar]
- 51.Scarlett RF, et al. Influenza-like viral illnesses and flare-ups of fibrodysplasia ossificans progressiva. Clin. Orthop. Rel. Res. 2004;423:275–279. doi: 10.1097/01.blo.0000129557.38803.26. [DOI] [PubMed] [Google Scholar]
- 52.Gannon FH, Valentine BA, Shore EM, Zasloff MA, Kaplan FS. Acute lymphocytic infiltration in an extremely early lesion of fibrodysplasia ossificans progressiva. Clin. Orthop. Rel. Res. 1998;346:19–25. [PubMed] [Google Scholar]
- 53.Glaser DL, et al. In vivo somatic cell gene transfer of an engineered noggin mutein prevents BMP4-induced heterotopic ossification. J. Bone Joint Surg. Am. 2003;85A:2332–2342. doi: 10.2106/00004623-200312000-00010. [DOI] [PubMed] [Google Scholar]
- 54.Hegyi L, et al. Stromal cells of fibrodysplasia ossificans progressiva lesions express smooth muscle lineage markers and the osteogenic transcription factor Runx2/Cbfa-1: clues to a vascular origin of heterotopic ossification? J. Pathol. 2003;201:141–148. doi: 10.1002/path.1413. [DOI] [PubMed] [Google Scholar]
- 55.Kaplan FS, et al. Immunological features of fibrodysplasia ossificans progessiva and the dysregulated BMP4 pathway. Clin. Rev. Bone Miner. Metab. 2005;3:189–193. [Google Scholar]
- 56.Kaplan FS, et al. The histopathology of fibrodysplasia ossificans progressiva. An endochondral process. J. Bone Joint Surg. Am. 1993;75:220–230. doi: 10.2106/00004623-199302000-00009. [DOI] [PubMed] [Google Scholar]
- 57.Lounev VY, et al. Identification of progenitor cells that contribute to heterotopic skeletogenesis. J. Bone Joint Surg. Am. 2009;91:652–663. doi: 10.2106/JBJS.H.01177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaplan FS, et al. Hematopoietic stem-cell contribution to ectopic skeletogenesis. J. Bone Joint Surg. Am. 2007;89A:347–357. doi: 10.2106/JBJS.F.00472. [DOI] [PubMed] [Google Scholar]
- 59.Kaplan FS, Strear CM, Zasloff MA. Radiographic and scintigraphic features of modeling and remodeling in the heterotopic skeleton of patients who have fibrodysplasia ossificans progressiva. Clin. Orthop. Rel. Res. 1994;304:238–247. [PubMed] [Google Scholar]
- 60.Kaplan F, et al. Urinary basic fibroblast growth factor. A biochemical marker for preosseous fibroproliferative lesions in patients with fibrodysplasia ossificans progressiva. Clin. Orthop. Rel. Res. 1998;346:59–65. [PubMed] [Google Scholar]
- 61.Lutwak L. Myositis ossificans progressiva. Mineral, metabolic and radioactive calcium studies of the effects of hormones. Am. J. Med. 1964;37:269–293. doi: 10.1016/0002-9343(64)90011-7. [DOI] [PubMed] [Google Scholar]
- 62.Ferguson C, Alpern E, Miclau T, Helms JA. Does adult fracture repair recapitulate embryonic skeletal formation? Mech. Dev. 1999;87:57–66. doi: 10.1016/s0925-4773(99)00142-2. [DOI] [PubMed] [Google Scholar]
- 63.Vortkamp A, et al. Recapitulation of signals regulating embryonic bone formation during postnatal growth and in fracture repair. Mech. Dev. 1998;71:65–76. doi: 10.1016/s0925-4773(97)00203-7. [DOI] [PubMed] [Google Scholar]
- 64.Dean DB, Watson JT, Moed BR, Zhang Z. Role of bone morphogenetic proteins and their antagonists in healing of bone fracture. Front. Biosci. 2009;14:2878–2888. doi: 10.2741/3419. [DOI] [PubMed] [Google Scholar]
- 65.Gerstenfeld LC, Cullinane DM, Barnes GL, Graves DT, Einhorn TA. Fracture healing as a post-natal developmental process: molecular, spatial, and temporal aspects of its regulation. J. Cell. Biochem. 2003;88:873–884. doi: 10.1002/jcb.10435. [DOI] [PubMed] [Google Scholar]
- 66.Kloen P, et al. BMP signaling components are expressed in human fracture callus. Bone. 2003;33:362–371. doi: 10.1016/s8756-3282(03)00191-1. [DOI] [PubMed] [Google Scholar]
- 67.Tsuji K, et al. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat. Genet. 2006;38:1424–1429. doi: 10.1038/ng1916. [DOI] [PubMed] [Google Scholar]
- 68.Einhorn TA, Kaplan FS. Traumatic fractures of heterotopic bone in patients who have fibrodysplasia ossificans progressiva. A report of 2 cases. Clin. Orthop. Rel. Res. 1994;308:173–177. [PubMed] [Google Scholar]
- 69.Connor JM, Evans DA. Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. J. Bone Joint Surg. Br. 1982;64:76–83. doi: 10.1302/0301-620X.64B1.7068725. [DOI] [PubMed] [Google Scholar]
- 70.Deirmengian GK, et al. Proximal tibial osteochondromas in patients with fibrodysplasia ossificans progressiva. J. Bone Joint Surg. Am. 2008;90:366–374. doi: 10.2106/JBJS.G.00774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schaffer AA, et al. Developmental anomalies of the cervical spine in patients with fibrodysplasia ossificans progressiva are distinctly different from those in patients with Klippel-Feil syndrome —Clues from the BMP signaling pathway. Spine. 2005;30:1379–1385. doi: 10.1097/01.brs.0000166619.22832.2c. [DOI] [PubMed] [Google Scholar]
- 72.Seemann P, Mundlos S, Lehmann K. In: Bone Morphogenetic Proteins: From Local to System Therapeutics. Vukicevic S, Sampath KT, editors. Birkhäuser; Basel: 2008. pp. 141–159. [Google Scholar]
- 73.Kaplan FS, et al. Classic and atypical FOP phenotypes are caused by mutations in the BMP type I receptor ACVR1. Hum. Mutat. 2009;30:379–390. doi: 10.1002/humu.20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wozney JM, et al. Novel regulators of bone formation: molecular clones and activities. Science. 1988;242:1528–1534. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- 75.Deng ZL, et al. Regulation of osteogenic differentiation during skeletal development. Front. Biosci. 2008;13:2001–2021. doi: 10.2741/2819. [DOI] [PubMed] [Google Scholar]
- 76.Furuta Y, Hogan BL. BMP4 is essential for lens induction in the mouse embryo. Genes Dev. 1998;12:3764–3775. doi: 10.1101/gad.12.23.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lawson KA, et al. BMP4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 1999;13:424–436. doi: 10.1101/gad.13.4.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mitu GM, Wang S, Hirschberg R. BMP7 is a podocyte survival factor and rescues podocytes from diabetic injury. Am. J. Physiol. Renal Physiol. 2007;293:F1641–F1648. doi: 10.1152/ajprenal.00179.2007. [DOI] [PubMed] [Google Scholar]
- 79.Morty RE, et al. Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arterioscler. Thromb. Vasc. Biol. 2007;27:1072–1078. doi: 10.1161/ATVBAHA.107.141200. [DOI] [PubMed] [Google Scholar]
- 80.Valdimarsdottir G, et al. Stimulation of Id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein-induced activation of endothelial cells. Circulation. 2002;106:2263–2270. doi: 10.1161/01.cir.0000033830.36431.46. [DOI] [PubMed] [Google Scholar]
- 81.de Sousa Lopes SM, et al. BMP signaling mediated by ALK2 in the visceral endoderm is necessary for the generation of primordial germ cells in the mouse embryo. Genes Dev. 2004;18:1838–1849. doi: 10.1101/gad.294004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dudas M, Sridurongrit S, Nagy A, Okazaki K, Kaartinen V. Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech. Dev. 2004;121:173–182. doi: 10.1016/j.mod.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 83.Kaartinen V, et al. Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development. 2004;131:3481–3490. doi: 10.1242/dev.01214. [DOI] [PubMed] [Google Scholar]
- 84.Kevenaar ME, et al. Variants in the ACVR1 gene are associated with AMH levels in women with polycystic ovary syndrome. Hum. Reprod. 2009;24:241–249. doi: 10.1093/humrep/den353. [DOI] [PubMed] [Google Scholar]
- 85.Komatsu Y, Scott G, Nagy A, Kaartinen V, Mishina Y. BMP type I receptor ALK2 is essential for proper patterning at late gastrulation during mouse embryogenesis. Dev. Dyn. 2007;236:512–517. doi: 10.1002/dvdy.21021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rajagopal R, et al. Functions of the type 1 BMP receptor Acvr1 (Alk2) in lens development: cell proliferation, terminal differentiation, and survival. Invest. Ophthal. Vis. Sci. 2008;49:4953–4960. doi: 10.1167/iovs.08-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.National Center for Biotechnological Information. 2010 MIM #166350. [online], http://www.ncbi.nlm.nih.gov/omim/166350.
- 88.Kaplan FS, et al. Progressive osseous heteroplasia: a distinct developmental disorder of heterotopic ossification. Two new case reports and follow-up of three previously reported cases. J. Bone Joint Surg. Am. 1994;76:425–436. [PubMed] [Google Scholar]
- 89.Shore EM, Feldman GJ, Xu M, Kaplan FS. The genetics of fibrodysplasia ossificans progressiva. Clin. Rev. Bone Miner. Metab. 2005;3:201–204. [Google Scholar]
- 90.Shore EM, et al. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N. Engl. J. Med. 2002;346:99–106. doi: 10.1056/NEJMoa011262. [DOI] [PubMed] [Google Scholar]
- 91.Adegbite NS, Xu M, Kaplan FS, Shore EM, Pignolo RJ. Diagnostic and mutational spectrum of progressive osseous heteroplasia (POH) and other forms of GNAS-based heterotopic ossification. Am. J. Med. Genet. A. 2008;146A:1788–1796. doi: 10.1002/ajmg.a.32346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Plagge A, Kelsey G, Germain-Lee EL. Physiological functions of the imprinted Gnas locus and its protein variants Gαs and XLαs in human and mouse. J. Endocrinol. 2008;196:193–214. doi: 10.1677/JOE-07-0544. [DOI] [PubMed] [Google Scholar]
- 93.Rubin MR, Levine MA. In: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Rosen CJ, editor. American Society for Bone and Mineral Research; Washington, DC: 2008. pp. 354–361. [Google Scholar]
- 94.Weinstein LS, Chen M, Xie T, Liu J. Genetic diseases associated with heterotrimeric G proteins. Trends Pharmacol. Sci. 2006;27:260–266. doi: 10.1016/j.tips.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 95.Weinstein LS, Xie T, Zhang QH, Chen M. Studies of the regulation and function of the Gsα gene Gnas using gene targeting technology. Pharmacol. Ther. 2007;115:271–291. doi: 10.1016/j.pharmthera.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bastepe M, Jüppner H. GNAS locus and pseudohypoparathyroidism. Horm. Res. 2005;63:65–74. doi: 10.1159/000083895. [DOI] [PubMed] [Google Scholar]
- 97.Yeh GL, et al. GNAS1 mutation and Cbfa1 misexpression in a child with severe congenital platelike osteoma cutis. J. Bone Miner. Res. 2000;15:2063–2073. doi: 10.1359/jbmr.2000.15.11.2063. [DOI] [PubMed] [Google Scholar]
- 98.Eddy MC, et al. Deficiency of the α-subunit of the stimulatory G protein and severe extraskeletal ossification. J. Bone Miner. Res. 2000;15:2074–2083. doi: 10.1359/jbmr.2000.15.11.2074. [DOI] [PubMed] [Google Scholar]
- 99.Huso DL, McGuire S, Germaine-Lee EL. Heterotopic subcutaneous ossifications in a mouse model of Albright hereditary osteodystophy. Presented at The Endocrine Society 89th Annual Meeting; Toronto, Canada. 2007. [Google Scholar]
- 100.Plagge A, et al. The imprinted signaling protein XLαs is required for postnatal adaptation to feeding. Nat. Genet. 2004;36:818–826. doi: 10.1038/ng1397. [DOI] [PubMed] [Google Scholar]
- 101.Bastepe M, et al. A form of Jansen’s metaphyseal chondrodysplasia with limited metabolic and skeletal abnormalities is caused by a novel activating parathyroid hormone (PTH)/PTH-related peptide receptor mutation. J. Clin. Endocrinol. Metab. 2004;89:3595–3600. doi: 10.1210/jc.2004-0036. [DOI] [PubMed] [Google Scholar]
- 102.Sakamoto A, Chen M, Kobayashi T, Kronenberg HM, Weinstein LS. Chondrocyte-specific knockout of the G protein Gsα leads to epiphyseal and growth plate abnormalities and ectopic chondrocyte formation. J. Bone Miner. Res. 2005;20:663–671. doi: 10.1359/JBMR.041210. [DOI] [PubMed] [Google Scholar]
- 103.Sakamoto A, et al. Deficiency of the G-protein α-subunit Gsα in osteoblasts leads to differential effects on trabecular and cortical bone. J. Biol. Chem. 2005;289:21369–21375. doi: 10.1074/jbc.M500346200. [DOI] [PubMed] [Google Scholar]
- 104.Wu JY, Scadden DT, Kronenberg HM. Role of the osteoblast lineage in the bone marrow hematopoietic niches. J. Bone Miner. Res. 2009;24:759–764. doi: 10.1359/jbmr.090225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kaplan FS, Groppe JC, Shore EM. When one skeleton is enough: approaches and strategies for the treatment of fibrodysplasia ossificans progressiva (FOP) Drug Discov. Today Ther. Strateg. 2008;5:255–262. doi: 10.1016/j.ddstr.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Aydin C, et al. Extralarge XLαs (XXLαs), a variant of stimulatory G protein α-subunit (Gsα), is a distinct, membrane-anchored GNAS product that can mimic Gsα. Endocrinology. 2009;150:3567–3575. doi: 10.1210/en.2009-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bastepe M, et al. Stimulatory G protein directly regulates hypertrophic differentiation of growth plate cartilage in vivo. Proc. Natl Acad. Sci. USA. 2004;101:14794–14799. doi: 10.1073/pnas.0405091101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Michienzi S, et al. GNAS transcripts in skeletal progenitors: evidence for random asymmetric allelic expression of Gsα. Hum. Mol. Genet. 2007;16:1921–1930. doi: 10.1093/hmg/ddm139. [DOI] [PubMed] [Google Scholar]
- 109.Yang DC, et al. cAMP/PKA regulates osteogenesis, adipogenesis and ratio of RANKL/OPG mRNA expression in mesenchymal stem cells by suppressing leptin. PLoS ONE. 2008;3:e1540. doi: 10.1371/journal.pone.0001540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang S, Xu M, Kaplan FS, Pignolo FS, Shore EM. G protein–cAMP pathway regulates early stage embryonic stem cell-derived osteoblast differentiation. J. Bone Miner. Res. 2009;24:S115. [Google Scholar]