

Abstract
One of the most important considerations in designing clinical trials is the choice of outcome measures. These outcome measures could be clinically meaningful endpoints that are direct measures of how patients feel, function and survive. Alternatively, indirect measures, such as biomarkers that include physical signs of disease, laboratory measures and radiological tests, often are considered as replacement endpoints or “surrogates” for clinically meaningful endpoints. We discuss the definitions of clinically meaningful endpoints and surrogate endpoints, and provide examples from recent clinical trials. We provide insight into why indirect measures such as biomarkers may fail to provide reliable evidence about the benefit-to-risk profile of interventions. We also discuss the nature of evidence that is important in assessing whether treatment effects on a biomarker reliably predict effects on a clinically meaningful endpoint, and provide insights into why this reliability is specific to the context of use of the biomarker. .
Keywords: validation, accelerated approval, correlate, indirect measure, clinically meaningful endpoint, replacement endpoint, clinical efficacy measure, effect modifiers
1. Introduction: Important Characteristics of Study Endpoints
The selection of the primary ‘endpoint’ or ‘outcome measure’ has considerable influence on the reliability and interpretability of clinical trials intended to evaluate the benefit-to-risk profile of an intervention. This influence can be better understood by considering several important characteristics of these outcome measures.
To enhance the informativeness of clinical trials regarding benefits and risks of treatments, primary endpoints should have the characteristics of being well defined and reliable [1] measures that assess important aspects of patient health status. A key step in assessing these properties is to evaluate content validity, which is “the extent to which an instrument measures the important aspects of concepts most significant and relevant to the patient’s condition and its treatment” [2,3]. Effects on many important aspects of patient health status can best be assessed by using Patient-Reported Outcomes (PROs), defined to be “any report of the status of a patient’s health condition that comes directly from the patient, without interpretation of the patient’s response by a clinician or anyone else” [4]. The 2009 FDA Guidance on PROs summarizes good measurement principles that are applicable to any assessment based on a PRO. These principles also apply for endpoints based on obtaining information through a clinician (i.e., a ClinRO) or an observer (i.e., an ObsRO) assessment.
Another characteristic is that the primary endpoint should be sensitive to the effects of the intervention. For example, if an analgesic is being evaluated in a pre-terminal patient, while overall survival would be particularly relevant to the patient, a validated PRO measuring pain relief would be more sensitive to the expected effects of the intervention. This characteristic for sensitivity usually plays a dominant role in endpoint selection, in order to reduce the size and duration of clinical trials and to increase the likelihood of achieving positive results. However, when an endpoint is chosen based on sensitivity, evidence regarding effects on more clinically important outcomes should be particularly influential, such as data suggesting adverse effects on survival for an agent achieving relief of pain.
Another consideration is that the primary endpoint should be readily measurable and interpretable. If invasive procedures such as frequent biopsies or intubation are required to assess effects on histological measures, then the challenges in measuring these outcomes induce a risk for high levels of missing data that could cause substantial bias and a meaningful reduction in interpretability of study results.
Interpretability also might be reduced if composite endpoints are used. Composites are often considered to increase the trial’s sensitivity or statistical power by increasing the number of patients experiencing the primary endpoint in time-to-event settings. However, the interpretability of such endpoints is greatly influenced by whether each component of the composite has similar clinical relevance to the other components. The Major Cardiovascular Event (MACE) composite endpoint, i.e., the composite of “cardiovascular death, stroke or myocardial infarction”, is interpretable in clinical trials in patients with acute coronary syndrome because each component of the composite is a measure of irreversible morbidity or mortality. However, the interpretability of such a measure was substantially reduced when the components “acute coronary syndrome, cardiac intervention including coronary artery bypass graft or percutaneous coronary intervention, major leg amputation, or revascularization in the leg” were added to the MACE components, “death, stroke or myocardial infarction” in the PROactive clinical trial evaluating pioglitazone in patients with type-2 diabetes, [5]. Interpretability of the MACE endpoint also was meaningfully compromised when “asymptomatic distal deep venous thrombosis” was added to the composite in the EXULT-A and -B clinical trials evaluating ximelagatran in patients having total knee replacement surgery [6,7].
The most important characteristic in guiding the selection of the primary endpoint in definitive trials is that effects on such an endpoint should provide reliable evidence about whether the intervention provides clinically meaningful benefit. Thus, the primary outcome measure in definitive trials should be “a clinical event relevant to the patient” [8], or an endpoint that “measures directly how a patient feels, functions or survives” [9], where function refers to patients’ ability to perform activities in their daily lives. Such outcome measures are hereafter referred to as ‘clinically meaningful endpoints’ or ‘clinical efficacy measures’. Alternatively, an endpoint can be a validated surrogate for such an outcome measure. A surrogate endpoint is an outcome measure “used as a substitute for a clinically meaningful endpoint…changes induced by a therapy on a surrogate endpoint are expected to reflect changes in a clinically meaningful endpoint” [9]. Many outcome measures used in clinical research are not clinically meaningful endpoints, but are indirect measures that are used as surrogate endpoints. Validating a surrogate endpoint requires providing an evidence based justification, often from randomized controlled clinical trials, that achievement of substantial effects on the surrogate endpoint reliably predicts achievement of clinically important effects on a clinically meaningful endpoint.
Some indirect measures that are considered as potential surrogate endpoints, such as 6 minute walk test, limb spasticity, pulmonary function tests, or rescue medications for pain, may be dependent on patient motivation or clinical judgment. However, most indirect measures to be considered in this article do not have such dependence. Rather, they are measurements of biological processes. They will be called biomarkers, and “include physiological measurements, blood tests and other chemical analyses of tissue or bodily fluids, genetic or metabolic data, and measurements from images” [10].
In the remainder of the manuscript, we discuss the challenges in validation of a surrogate endpoint. Prior to that discussion, we provide examples of clinically meaningful endpoints, i.e., true clinical efficacy measures, examples of validated surrogate endpoints, and examples of outcome measures that are neither. Table 1 provides a categorization of many outcome measures commonly used as primary endpoints in Phase 3 clinical trials designed to provide reliable or definitive assessments of interventions’ benefit-to-risk profiles. A 4-level hierarchy for endpoints is used in this categorization, as given in Fleming [11]:
“Level 1 is a true clinical efficacy measure; Level 2 is a validated surrogate (for a specific disease setting and class of interventions); Level 3 is a non-validated surrogate, yet one established to be ‘reasonably likely to predict clinical benefit’ (for a specific disease setting and class of interventions); and Level 4 is a correlate that is a measure of biological activity but that has not been established to be at a higher level.”
Table 1.
Categorization of Outcome Measures, according to Level of Evidence regarding Efficacy. Composite Endpoints will be denoted by {brackets}.
| Level 1 A true clinical efficacy measure (When evidence establishing risk is acceptable in the context of evidence of benefit)
|
| Level 2 A validated surrogate (for a specific disease setting and class of interventions.) (When interventions are safe, with strong evidence that risks from off target effects are acceptable)
|
| Level 3 A non-validated surrogate, yet one established to be ‘reasonably likely to predict clinical benefit’ (for a specific disease setting and class of interventions) (When interventions are safe, with evidence that risks from off target effects are acceptable)
|
Level 4 A correlate that is a measure of biological activity, but not established to be at a higher level.
|
In Table 1, EDSS is ‘expanded disability status scale score’; T2DM is ‘type 2 diabetes mellitus’; VRE is ‘vancomycin resistant enterococci’; PSA is ‘prostate specific antigen’; FEV-1 is ‘forced expiratory volume in 1 second’; FVC is ‘forced vital capacity’; MPS is ‘mucopolysaccharidosis’
In this categorization, all endpoints at Levels 2, 3, and 4 are indirect outcome measures. Measures at Level 1 or 2 likely would be appropriate primary endpoints in definitive or registration clinical trials. Level 3 measures might be considered as primary endpoints in clinical trials using the subpart E or subpart H “Accelerated Approval” regulatory approach [12] for applications to the U.S. Food and Drug Administration (FDA). Level 4 measures may be strongly correlated in natural history with direct measures of the patient’s clinical status, and as such might be useful for disease diagnosis, for assessment of prognosis, or by caregivers to guide patient management decisions in the absence of more reliable evidence to guide clinical practice. However, it is problematic to propose use of these Level 4 measures as primary endpoints in definitive clinical trials.
2. “A Correlate does not a Surrogate Make”
Suppose it is of interest to use a biomarker as a replacement endpoint in a Phase 3 clinical trial intended to provide reliable evidence about efficacy and safety of an intervention. When querying clinical researchers about the available evidence to justify their acceptance that effects on their favorite biomarker measure should reliably predict effects on a clinical efficacy measure, a frequent response is that there is a strong correlation between these measures in natural history observations. For example, in oncology, since responders (i.e., patients who experience substantial tumor shrinkage following therapy) live longer than non-responders, many have believed that increases in the response rate should predict improvement in overall survival. However, such evidence about correlations does not allow one to understand the true nature of causality. Was the longer survival duration in responders causally induced by the anti-tumor effects of the intervention, or did the treatment-induced tumor response simply allow identification of the immunologically or inherently stronger patients who both responded and lived longer because of their inherently better status?
As indicated by Fleming and DeMets [8], “A correlate does not a surrogate make.” While the effect of an intervention on a biomarker does provide direct evidence regarding biological activity, such evidence could be unreliable regarding effects on true clinical efficacy measures even when the biomarker is strongly correlated with these clinical efficacy measures in natural history observations. Clarification about this paradox is provided by Figures 1–3 that are variations of previously published illustrations, [8, 10, 11].
Figure 1.

Illustrations where the biomarker is not in a causal pathway of the disease process, reducing the likelihood it could be shown to be a valid surrogate endpoint. “CEA” is carcinoembryonic antigen; “PSA” is the prostate specific antigen; “Ca.” is cancer.
Figure 3.
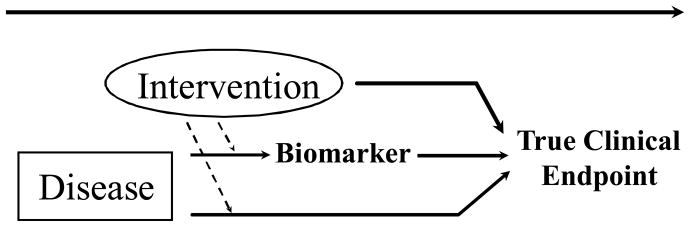
An illustration where the biomarker lies in a causal pathway of the disease process that is impacted by the intervention, yet off target effects of the intervention reduce the likelihood the biomarker would be a valid surrogate endpoint.
Biomarkers that are strongly correlated with clinical efficacy measures in natural history observations, yet are not in the causal pathway of the disease process, are likely to provide misleading information about clinical efficacy. Figure 1 provides illustrations of this setting. While the risk that HIV-infected pregnant women will transmit the infection to their infants is strongly correlated with maternal CD4 counts, an intervention such as interleukin-2 given late in pregnancy to spike maternal CD4 counts would not impact this transmission risk. This correlation between maternal CD4 and risk of mother-to-child transmission of HIV exists because both measures are influenced by maternal viral load. More reliable insights about potential effects on mother-to-child transmission of HIV would be obtained by assessing whether an anti-retroviral intervention provides large reductions in maternal viral load, where these reductions are sustained during pregnancy, labor and delivery, and during breastfeeding. Of course, the preferred approach would be to assess the effect of the intervention directly on the outcome of the proportion of infants infected with HIV.
In oncology, tumor markers such as prostate specific antigen (PSA) and carcinoembryonic antigen (CEA) are correlated in natural history observations with clinical efficacy measures, such as cancer symptoms and death. These correlations are sufficient to allow these measures to be useful for assessing prognosis in patients receiving treatment for their disease, or for disease diagnosis. However, effects on CEA and PSA likely would provide unreliable information about clinical efficacy since it is the tumor burden process, rather than levels of CEA or PSA, that is a true causal mechanism for risk of cancer induced symptoms and mortality.
A second factor complicating the reliability of an evaluation of efficacy based on biomarkers is the multidimensionality of the causal mechanisms of the disease process, as illustrated in Figure 2. There is risk of false negative conclusions about clinical efficacy if the biomarker does not lie in the disease process causal pathway that is meaningfully impacted by the intervention. For example, in a registration trial in chronic granulomatous disease, interferon-γ provided a statistically and clinically significant 70% reduction in rate of recurrent serious infections [13]. However, the agent did not have a detectable effect on the biomarkers of bacterial killing and superoxide production. These biomarkers had been seriously considered during trial design as possible primary endpoints due to interests in reducing the size and duration of this trial conducted in children.
Figure 2.

Illustrations where the disease process has multiple causal pathways, and the biomarker lies in just one of those pathways. “MI” is myocardial infarction; “CGD” is chronic granulomatous disease.
False positive conclusions about clinical efficacy could arise if a biomarker captures the substantial effects of an intervention on one causal pathway of the disease process, while the intervention has an inadequate impact on other principal causal pathways. Consider, for example, the three arm Sweden I Acellular Pertussis trial, where all children received vaccines having diphtheria and tetanus components, along with the addition of a Smith-Kline Beecham or an Aventis Pasteur acellular pertussis component or a placebo, [14]. Relative to the diphtheria +tetanus+ placebo control arm, the Aventis Pasteur vaccine provided an 85% reduction, (95% CI, 81% to 89%), in the rate of pertussis cases, while the Smith-Kline Beecham vaccine provided only a 58% reduction, (95% CI, 51% to 66%). When comparing these two vaccines having active acellular pertussis components, even though the Aventis Pasteur vaccine had strongly superior vaccine efficacy, the Smith-Kline Beecham vaccine had superior effect on two leading biomarkers of Filamentous Haemagglutinin and Pertussis Toxoid antibody responses. The misleading information provided by these two antibody biomarkers regarding relative efficacy of these acellular pertussis vaccines might be explained by differences between vaccines in durability of their antibody responses, yet more likely is explained by additional immune responses generated by the Pertactin and Fimbrae (types 2 and 3) antigens in the Aventis Pasteur vaccine.
Even when the biomarker does capture effects on the principal causal pathway of the disease process, it often is unclear what magnitude and duration of effect on that pathway is required to meaningfully affect the clinical efficacy measure, (see Figure 2). For example, consider the evaluation of coronary thrombolysis to speed reperfusion of infarct-related coronary arteries, and in turn to decrease 30-day mortality post myocardial infarction. In this setting, the Phase 2b RAPID II trial provided evidence that the experimental agent Reteplase, (Recombinant Plasminogen Activator, r-PA), provided better effects than Alteplase (Recombinant Tissue Plasminogen Activator, t-PA), in achieving “patency”, i.e., TIMI-III blood flow rates at 60 minutes (51% versus 37%) and at 90 minutes (60% versus 45%) post randomization [15]. Based on these positive biomarker results for r-PA, it was somewhat surprising that 30-day mortality was numerically higher on r-PA than t-PA (i.e., 7.43% versus 7.22%) in the 15,000-patient GUSTO-III confirmatory trial [16]. However, re-inspection of the RAPID-II trial revealed that TIMI-III blood flow rates at 30 minutes were lower on r-PA than on t-PA (i.e., 27% versus 39%). The lack of knowledge about the magnitude and duration of effect on a pathway of the disease process that is required to achieve a given effect size on a clinically meaningful endpoint compromises the reliability and interpretability of any trial designed to use biomarkers as surrogate endpoints, and is particularly problematic in the setting of non-inferiority trials.
Another factor complicating the reliability of an evaluation of efficacy based on biomarkers, as illustrated in Figure 3, is the likelihood that these measures do not capture important off target effects of the intervention, even though such effects could meaningfully alter the true clinical efficacy of the intervention. There are numerous examples where biomarkers have failed for this reason (8,10,11). A classic example arose when over a quarter million U.S. patients annually were provided encainide and flecanide to suppress their arrhythmias post myocardial infarction, due to the increased risk of sudden death in patients with arrhythmia. Eventually, the two thousand-patient placebo-controlled Cardiac Arrhythmia Suppression Trial was completed. Many were stunned by the trial results that revealed the two anti-arrhythmia agents actually tripled the death rate, likely due to off-target effects not captured by the suppression of arrhythmia biomarker, [17,18].
Among more recent experiences of failed biomarkers due to off-target effects of interventions, in end stage renal disease, the TREAT, CHOIR, CREATE and Normal Hematocrit trials [19–22] revealed that regimens involving more aggressive use of erythropoiesis stimulating agents provided better normalization of hematocrit. However, in these trials, these aggressive regimens had unfavorable effects on overall survival, at least in part due to off target effects on thrombosis, on stroke risk, and potentially on malignancy. In type 2 diabetes mellitus, rosiglitazone effectively lowered levels of glycosylated hemoglobin, (HbA1c), yet subsequent trials provided evidence that its use increased the risk of cardiovascular morbidity and mortality[23–24]. The ACCORD trial in type 2 diabetes mellitus revealed that a therapeutic strategy providing an additional absolute 1% reduction in HbA1c resulted in an increase in mortalitythrough off target effects that might include inducing risk of hypoglycemia [25]. In coronary heart disease, the ILLUMINATE trial confirmed that the addition of torcetrapid to atorvastatin enhanced the effects on lipids, providing a substantial increase in high-density lipoprotein cholesterol in addition to enhancing atorvastatin’s effect on lowering low-density lipoprotein cholesterol [26]. Nevertheless, the trial was terminated early due to the combination regimen’s unfavorable effects on mortality, potentially due to unintended off target effects of torcetrapid on the renin angiotensin system that caused increases in blood pressure. The torcetripid experience illustrates the hazards of a development strategy where evidence that a biomarker (e.g., lipids) is a valid surrogate endpoint for clinical efficacy measures (e.g., cardiovascular morbidity and mortality) when evaluating an original class of agents (e.g., statins), then is used to justify using that biomarker as a surrogate endpoint in the evaluation of a new class of agents (e.g., cholesteryl ester transfer protein inhibitors that impact the renin angiotensin system). Such “bridging” may not be justified if the original and new class of agents have different profiles regarding meaningful off target effects.
3. Validation of Biomarkers as Surrogate Endpoints
Given the substantial risk that effects on biomarkers can provide misleading information about the true effect of an intervention on clinical efficacy measures, it is important to consider the nature of scientific evidence that would allow one to use biomarkers in place of clinically meaningful endpoints in definitive clinical trials.
The Normal Hematocrit Trial [22] in Figure 4, conducted in 1233 patients with end stage renal disease, illustrates that even if a strong correlation between a biomarker (i.e., hematocrit) and clinical efficacy measures (i.e., death and myocardial infarction-free survival), apparent on the “standard of care” control regimen (i.e., standard dose Epogen), is maintained on the experimental regimen (i.e., high dose Epogen), a favorable effect on the biomarker still can be misleading about the net effect of the intervention on the clinical efficacy measure. In this trial, through off-target effects including increased risk of thrombosis not captured by the biomarker, use of the experimental high dose Epogen regimen resulted in a net 30% increase in the rate of death or myocardial infarction with a net increase of 38 patients who either died or experienced a myocardial infarction.
Figure 4.

Results from the Normal Hematocrit Trial (18). The x-axis is the achieved level of hematocrit %, after treatment. The dark bars represent percent deaths and the light bars represent the percent of patients, for each level of hematocrit %. “RR” is relative risk. “Hem” is hematocrit.
The Normal Hematocrit trial illustrates that a strong correlation between the biomarker and the clinical efficacy measure, even if apparent on the experimental as well as on the control regimen, is not sufficient to establish a biomarker to be a valid surrogate endpoint. However, derivations by Prentice [27] show it would be sufficient if a second very restrictive condition is also satisfied: namely, the biomarker fully captures the net effect of the intervention on the clinical efficacy measure.
A rational argument about the validity of Prentice’s second condition could be developed for a specific setting if one could have a comprehensive understanding about (i) the principal pathways through which the disease process affects how a patient feels, functions or survives, (ii) the extent to which effects on the biomarker capture the meaningful “on-target” effects of the intervention on those causal pathways of the disease process, and (iii) any “off-target” effects of the intervention that would meaningfully affect the clinical efficacy measures and yet would not be captured by the biomarker.
While in theory such a rational argument could be developed, in reality a sufficiently comprehensive depth of understanding about the causal pathways of the disease process and about the unintended as well as intended mechanisms of action of the intervention rarely could be achieved. In recognition of these limitations, the most reliable evidence regarding the validity of a biomarker as a surrogate endpoint for a clinical efficacy measure might be provided by an extensive overview of trials that give reliable estimates of the net effects of the intervention on the clinically meaningful endpoint as well as on the biomarker.
For illustration, such an overview was presented to the FDA Cardiovascular and Renal Drugs Advisory Committee on June 15, 2005 to address the validity of using blood pressure measures as surrogate endpoints for cardiovascular outcomes in anti-hypertension clinical trials [28]. As an example of the evidence presented to the Advisory Committee, Figure 5 provides a graph that shows the relationship between the odds ratio (experimental to reference) for cardiovascular clinical events when plotted against the intervention effects (reference minus experimental) on systolic blood pressure. In this figure, where the results from each large scale clinical trial are represented by a single point on the graph, effects on systolic blood pressure do allow one to make reliable predictions about effects on the cardiovascular clinical efficacy measure.
Figure 5.

Data presented at the 15 June 2005 FDA Cardiovascular and Renal Drugs Advisory Committee. For controlled trials evaluating antihypertensive agents, the treatment effect on systolic blood pressure (x-axis) is plotted against the odds ratio for major cardiovascular events (y-axis).
There are other important insights from this experience in the anti-hypertensive setting. The validity of blood pressure measures as surrogates was addressed independently for several classes of agents, including low-dose diuretics, β-blockers, angiotensin-converting enzyme inhibitors, calcium channel blockers, and angiotension II receptor blockers. This was important due to the potential that off-target effects not captured by the blood pressure biomarker could differ across these classes. A consequence of this need to consider the surrogacy issue separately across drug classes is the very large size of the data set used in this validation process. In total, these analyses were based on studies collectively involving 500,000 patients. Further, these studies were randomized clinical trials since the use of data from patient registries would not allow an unbiased assessment of treatment effects on either the biomarker or the clinical outcome measures. Another important insight is that the validity of blood pressure measures as surrogates depended quite strongly on the definition of the clinical efficacy measures. Specifically, effects on blood pressure measures were very predictive of effects on stroke, less predictive of effects on myocardial infarction, cardiovascular death and overall mortality, and poorly predictive of effects on heart failure [29]. This reinforces the point that when using a biomarker as a substitute for a clinically meaningful endpoint, one must first be clear about the clinically meaningful endpoint for which the biomarker is a proposed surrogate.
4. Conclusions
There are many potential roles for biomarkers in clinical research [10]. For some of these roles, the biomarker can be used to effectively achieve the intended objective even if it is not on a pathway through which the disease process causally induces risk of symptoms or mortality. For example, when assessing the prognosis in a patient receiving treatment for their disease or when diagnosing a disease, it is sufficient that a biomarker used for these purposes simply be correlated with clinical efficacy measures in evidence obtained from observational studies and patient registries. Biomarkers might also be useful in providing insights about whether an intervention has a detectable effect on a biological pathway, and thus might serve as endpoints in a proof-of-concept trial or as supportive measures in definitive Phase 3 clinical trials.
The greatest clinical utility of biomarkers might be in the two clinical settings where it can be most challenging to justify their validity and reliability. These two settings are use as surrogate endpoints in place of clinical efficacy measures in definitive trials, or use to achieve enrichment when one expects greater effects with interventions in specific groups of subjects (i.e., effect modification). Regarding enrichment, there is considerable interest in identifying the subset of the patient population for whom an intervention would have a clinically meaningfully favorable benefit-to-risk profile due to greater benefits or fewer adverse outcomes. Since the key mechanisms of treatment effect on the causal factor(s) of the disease process might be specific to a targeted population (e.g., those patients having a specific genetic characterization), being able to define this targeted population can avoid diluting the benefit-to-risk profile of an intervention, both in clinical research as well as in clinical practice. For example, the level of effect of trastuzumab in breast cancer patients appears to be specific to the level of her-2-neu over-expression [30], and the level of effect of epidermal growth factor receptor-inhibiting drugs in colorectal cancer patients appears to depend upon whether tumors express the wild type or the mutated version of the KRAS gene [31]. When pursuing biomarkers as effect modifiers, it is insufficient to simply establish the biomarker to be prognostic, which can be done by using data from observational studies and patient registries to show the biomarker is associated with outcome risk. Simply stated, a prognostic factor does not an effect modifier make. To determine whether biomarkers are useful in identifying those patients most likely to receive clinically important benefits from an intervention, considerable biological insights as well as data intensive approaches based on evidence from randomized controlled trials are needed.
The second clinical setting where considerable biological insights as well as data intensive approaches are needed to justify effective implementation of biomarkers is their use as replacement or surrogate endpoints for clinical efficacy measures in clinical trials intended to provide a definitive assessment of the benefit-to-risk profile of an intervention. As discussed in this article, simply establishing a biomarker to be correlated with clinical efficacy measures in natural history observations does not provide reliable evidence that the effects on that biomarker will predict the intervention’s effects on those clinical efficacy measures. Simply stated, a correlate does not a surrogate make, (8). The most reliable evidence regarding the validity of a biomarker as a surrogate endpoint for a clinical efficacy measure might be provided by an extensive overview of trials that give reliable estimates of the net effects of the intervention on the clinically meaningful endpoint as well as on the biomarker.
We should carefully consider the consequences of relying on biomarkers as surrogate endpoints and thus as the primary source of efficacy information when determining whether interventions should be used in clinical practice. Such reliance has the benefit of allowing clinical trials for regulatory approval to be smaller in size and shorter in duration. However, an unfortunate consequence is that this leads not only to more limited insights about efficacy but also to less reliable assurances about safety given the smaller safety dataset upon which to base assessments. It should not be surprising, then, that agents receiving regulatory approval using efficacy assessments based on surrogate endpoints are more vulnerable to having clinically unacceptable safety issues discovered during the post-marketing period. In type-2 diabetes mellitus, rosiglitazone was approved based on reducing levels of HbA1c, yet clinical trials results that were evaluated in the post-marketing setting provided substantial evidence that the agent increases risks of cardiovascular morbidity and mortality. The simvastatin/ezetimibe combination (Vytorin) was approved based on lowering low-density lipoprotein cholesterol, but data from 3 large post-marketing trials suggest it has harmful effects on risk of cancer-related mortality [32–34]. Erythropoiesis stimulating agents (ESAs) received regulatory approval for use in the clinical settings of end stage renal disease and chemotherapy induced anemia, based on short term effects on increasing the levels of the biomarker, hematocrit, and reducing the need for blood transfusions. However, as discussed earlier, subsequent trials provided strong evidence of harmful effects of ESAs on thrombosis, stroke, mortality and possibly malignancy.
The concerns about a biomarker based approach for evaluating agents become greater when recognizing that the determination of the threshold for acceptable safety risks depends on the strength of evidence regarding efficacy. Thus, not only does the biomarker based approach provide an increased likelihood that safety signals will not be discovered until post-marketing, there is much greater risk that such signals, when discovered, cannot readily be justified to be acceptable within the context of the strength of evidence regarding efficacy. The assessment of the balance of benefit and risk is particularly challenging when benefit measures are based on biomarkers or on short-term intermediate endpoints but evidence for risk is based on clinically meaningful measures of major morbidity. For illustration, natalizumab was granted an accelerated approval under the FDA’s sub-part E provision for biologics, based on evidence from short term trials in multiple sclerosis patients that evaluated effects on short term relapse rates but did not provide direct evidence about effects of the agent on the clinically much more important endpoint of time to irreversible loss of mobility [35]. As a consequence, not only was the discovery of natalizumab’s effects on progressive multifocal leukoencephalopathy delayed until after marketing approval had been granted, but also the ability to judge the acceptability of such a highly morbid and often fatal safety risk, when evaluated in a benefit-to-risk manner, was hampered by uncertainties about whether natalizumab truly provided benefit on measures of irreversible morbidity for multiple sclerosis patients, such as a delay in time to walking with a cane (i.e., Expanded Disability Status Scale Score = 6) or being wheelchair bound (i.e., Expanded Disability Status Scale Score = 7). Since reliance on biomarkers or on short term intermediate endpoints leads to having less reliable insights about risks of rare but clinically important safety events or about longer term safety and efficacy, their use as replacement endpoints should be considered only when there is substantial evidence to establish their reliability in predicting effects on clinical efficacy measures and where there is interest in interventions that could offer added benefits over existing therapies.
The Institute of Medicine of the National Academies of Science released a major report discussing an array of useful roles for biomarkers and why rigor is important regarding their proper use [10]. In particular, this report recommends the evaluation process for using biomarkers as surrogate endpoints consists of 3 steps: (i) Analytical Validation, which includes an analysis of the analytical performance of an assay used in formulating the biomarker; (ii) Qualification, which includes assessing available information regarding the relationship of effects on biomarkers and effects on clinical efficacy measures; and (iii) Utilization, which includes determining whether the validation and qualification provide sufficient support for use of a biomarker in the context proposed. The Institute of Medicine recognized the need for this third step on a per use basis because “the status of a biomarker as a surrogate endpoint is context specific, and a biomarker cannot be assumed to be a general surrogate endpoint”. For a specific intervention in a given clinical setting, suppose the effect on a biomarker reliably predicts the effect on a clinical efficacy endpoint. This “validity of surrogacy” for evaluating clinical efficacy cannot be extrapolated to another intervention in that clinical setting if the interventions differ (i) in the magnitude and duration of their effects on the causal pathway of the disease process that is captured by the biomarker, or (ii) in how they affect causal pathways of the disease process not captured by the biomarker, or (iii) in their off-target effects. Furthermore, the “validity of surrogacy” for evaluating the effect of a specific intervention in one clinical setting cannot be assumed to hold in another clinical setting if there are differences across settings in either the on-target or the off-target effects ofthe intervention.
Using biomarkers as surrogate endpoints often is motivated by interests to reduce the size and duration of definitive clinical trials, with the hope that this will allow more timely evaluation of the benefit-to-risk profile of experimental interventions, and an improved ability to offer patients another choice in their clinical care. However, a rigorous evidence based justification should be provided in any setting where use of biomarkers as surrogate endpoints is proposed because the scientific evaluation of benefit and risk needs to be valid and reliable as well as timely. More important than offering patients a choice is offering them an informed choice.
Acknowledgments
For T. Fleming, the source of financial support for research described in this article is an NIH/NIAID grant entitled “Statistical Issues in AIDS Research” (R37 AI 29168).
For JH Powers, this project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported in part by the National Institute of Allergy and Infectious Disease. Collaborative Clinical Research Branch, John H Powers, MD, SAIC-Frederick, Inc., NCI-Frederick, Frederick, Maryland 21702
The authors acknowledge Journal of Hypertension as the source for Figure 5.
Contributor Information
Thomas R. Fleming, Department of Biostatistics, University of Washington, Seattle, Washington
John H Powers, Senior Medical Scientist, SAIC in support of the Collaborative Clinical Research Branch, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), And Assistant Clinical Professor of Medicine, George Washington University School of Medicine, And University of Maryland School of Medicine.
Reference List
- 1.US Government Printing Office. Applications for FDA approval to market a new drug: adequate and well-controlled studies. [Accessed November 29, 2011.];US Code title 21, section 314.126. Available at: http://edocket.access.gpo.gov/cfr_2009/aprqtr/21cfr314.126.htm.
- 2.Patrick DL, Burke LB, Gwaltney CJ, Leidy NK, Martin ML, Molsen E, Ring L. [Accessed November 29, 2011.];Content Validity—Establishing and Reporting the Evidence in Newly Developed Patient-Reported Outcomes (PRO) Instruments for Medical Product Evaluation: ISPOR PRO Good Research Practices Task Force Report: Part 1—Eliciting Concepts for a New PRO Instrument. doi: 10.1016/j.jval.2011.06.014. Available at: http://www.valueinhealthjournal.com/article/S1098-3015(11)03323-7/abstract. [DOI] [PubMed]
- 3.Patrick DL, Burke LB, Gwaltney CJ, Leidy NK, Martin ML, Molsen E, Ring L. [Accessed November 29, 2011.];Content Validity—Establishing and Reporting the Evidence in Newly Developed Patient-Reported Outcomes (PRO) Instruments for Medical Product Evaluation: ISPOR PRO Good Research Practices Task Force Report: Part 2—Assessing Respondent Understanding. doi: 10.1016/j.jval.2011.06.013. Available at: http://www.valueinhealthjournal.com/article/S1098-3015(11)03321-3/abstract. [DOI] [PubMed]
- 4.Guidance for Industry Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims. U.S. Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Center for Devices and Radiological Health (CDRH); Dec, 2009. [Accessed November 29, 2011.]. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM193282.pdf. [Google Scholar]
- 5.Dormandy JA, Charbonnel B, Eckland EJA, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes: a randomized trial of pioglitazone. The PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events) Lancet. 2005;366:1279–1289. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- 6.Francis CW, Berkowitz SD, Comp PC, Lieberman JR, Ginsberg JS, Paiement G, Peters GR, Roth AW, McElhattan J, Colwell CW, Jr EXULT A Study Group. Comparison of ximelagatran with warfarin for the prevention of venous thromboembolism after total knee replacement. N Engl J Med. 2003;349:1703–1712. doi: 10.1056/NEJMoa035162. [DOI] [PubMed] [Google Scholar]
- 7.Colwell CW, Jr, Berkowitz SD, Lieberman JR, Comp PC, Ginsberg JS, Paiement G, McElhattan J, Roth AW, Francis CW The EXULT-B Study Group. Oral direct thrombin inhibitor ximelagatran compared with warfarin for the prevention of venous thromboembolism after total knee arthroplasty. J Bone Joint Surg Am. 2005;87:2169–2177. doi: 10.2106/JBJS.D.02184. [DOI] [PubMed] [Google Scholar]
- 8.Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled? Ann Intern Med. 1996;125(7):605–13. doi: 10.7326/0003-4819-125-7-199610010-00011. [DOI] [PubMed] [Google Scholar]
- 9.Temple RJ. A regulatory authority’s opinion about surrogate endpoints. In: Nimmo WS, Tucker GT, editors. Clinical Measurement in Drug Evaluation. New York: J. Wiley; 1995. [Google Scholar]
- 10.IOM. Evaluation of Biomarkers and Surrogate Endpoints in Chronic Disease. Washington DC: National Academies Press; 2010. http://www.iom.edu/Reports/2010/Evaluation-of-Biomarkers-and-Surrogate-Endpoints-in-Chronic-Disease.aspx. [PubMed] [Google Scholar]
- 11.Fleming TR. Surrogate endpoints and FDA’s accelerated approval process. Health Affairs. 2005 Jan;24(1):67–78. doi: 10.1377/hlthaff.24.1.67. [DOI] [PubMed] [Google Scholar]
- 12.US Code of Federal Regulations FDA Subpart H 21 CFR, Secs. 314.500–314.560
- 13.The International Chronic Granulomatous Disease Cooperative Group Study. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. N Engl J Med. 1991;324:509–516. doi: 10.1056/NEJM199102213240801. [DOI] [PubMed] [Google Scholar]
- 14.Gustafsson L, Hallander HO, Olin P, Reizenstein E, Storsaeter J. A controlled trial of a two-component acellular, a five-component acellular, and a whole-cell pertussis vaccine. N Engl J Med. 1996;334:349–355. doi: 10.1056/NEJM199602083340602. [DOI] [PubMed] [Google Scholar]
- 15.Smalling RW, Bode C, Kalbfleisch J, et al. More rapid, complete, and stable coronary thrombolysis with bolus administration of reteplase compared with alteplace infusion in acute myocardial infarction. Circulation. 195;91:2725–2732. doi: 10.1161/01.cir.91.11.2725. [DOI] [PubMed] [Google Scholar]
- 16.GUSTO Investigators. A comparison of reteplase with alteplase for acute myocardial infarction. N Engl J Med. 1997;337:1118–1123. doi: 10.1056/NEJM199710163371603. [DOI] [PubMed] [Google Scholar]
- 17.Preliminary Report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST) investigators. N Engl J Med. 1989;321:406–412. doi: 10.1056/NEJM198908103210629. [DOI] [PubMed] [Google Scholar]
- 18.Echt DS, Liebson PR, Mitchell LB, Peters JW, Oblas-Manno D, Barker AH, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med. 1991;324:781–788. doi: 10.1056/NEJM199103213241201. [DOI] [PubMed] [Google Scholar]
- 19.Pfeffer MA, Burdmann EA, Chen CY, Cooper ME, de Zeeuw D, Eckardt KU, Feyzi JM, Ivanovich P, Kewalramani R, Levey AS, Lewis EF, McGill JB, McMurray JJ, Parfrey P, Parving HH, Remuzzi G, Singh AK, Solomon SD, Toto R for the TREAT Investigators. A Trial of Darbepoetin Alfa in Type II Diabetes and Chronic Kidney Disease. N Engl J Med. 2009;361:2019–32. doi: 10.1056/NEJMoa0907845. [DOI] [PubMed] [Google Scholar]
- 20.Singh AK, MBBS, Szczech L, MD, Tang KL, PhD, Barnhart H, PhD, Sapp S, MS, Wolfson M, MD, Reddan D, MBBS for the CHOIR Investigators. Correction of Anemia with Epoetin Alfa in Chronic Kidney Disease. N Engl J Med. 2006;355:2085–2098. doi: 10.1056/NEJMoa065485. November 16, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Drüeke TB, MD, Locatelli F, MD, Clyne N, MD, Eckardt K-U, MD, Macdougall IC, MD, Tsakiris D, MD, Burger H-U, PhD, Scherhag A, MD for the CREATE Investigators. Normalization of Hemoglobin Level in Patients with Chronic Kidney Disease and Anemia. N Engl J Med. 2006 Nov 16;355:2071–2084. doi: 10.1056/NEJMoa062276. [DOI] [PubMed] [Google Scholar]
- 22.Besarab A, MD, Kline Bolton W, MD, Browne JK, PhD, Egrie JC, PhD, Nissenson AR, MD, Okamoto DM, PhD, Schwab SJ, MD, Goodkin DA., MD The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med. 1998;339:584–90. doi: 10.1056/NEJM199808273390903. [DOI] [PubMed] [Google Scholar]
- 23.Nissen SE, Wolski K. Effect of Rosiglitazone in the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 24.Meeting Transcript of the FDA Endocrinologic and Metabolic Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee; July 13–14, 2010; www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/EndocrinologicandMetabolicDrugsAdvisoryCommittee/UCM222628.pdf and UCM222629.pdf. [Google Scholar]
- 25.The Action to Control Cardiovascular Risk in Diabetes Study Group. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJP, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall TR, Brewer B for the ILLUMINATE Investigators. Effects of Torcetrapid in patients at high risk of coronary events. N Engl J Med. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 27.Prentice RL. Surrogate endpoints in clinical trials: Definition and operational criteria. Statistics in Medicine. 1989;8:431–440. doi: 10.1002/sim.4780080407. [DOI] [PubMed] [Google Scholar]
- 28.Meeting Transcript of the FDA Cardiovascular and Renal Drugs Advisory Committee. 2005 Jun 15; www.fda.gov/ohrms/dockets/ac/05/transcripts/2005-4145T1.pdf.
- 29.Reboldi G, Gentile G, Angeli F, Ambrosio G, Mancia G, Verdecchia P. Effects of intensive blood pressure reduction on myocardial infarction and stroke in diabetes: a meta-analysis in 73,913 patients. J Hypertens. 2011;29:1253–1269. doi: 10.1097/HJH.0b013e3283469976. [DOI] [PubMed] [Google Scholar]
- 30.Herceptin product labeling. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/103792s5175lbl.pdf.
- 31.Karapetis Christos S, Khambata-Ford Shirin, Jonker Derek J, O’Callaghan Chris J, Tu Dongsheng, Tebbutt Niall C, John Simes R, Chalchal Haji, Shapiro Jeremy D, Robitaille Sonia, Timothy, Zalcberg John R, et al. K-ras Mutations and Benefit from Cetuximab in Advanced Colorectal Cancer. N Engl J Med. 2008;359:1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 32.Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Bärwolf C, Holme I, Kesäniemi YA, Malbecq W, Nienaber CA, Ray S, Skjærpe T, Wachtell K, Willenheimer R for the SEAS Investigators. Intensive lipid lowering with Simvastatin and Ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- 33.Cannon CP, Guigliano RP, Blaxing MA, et al. Rationale and design of IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial): comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes in patients with acute coronary syndrome. Am Heart Journal. 2005;149:464–473. doi: 10.1016/j.ahj.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 34.Baigent C, Landry M. Study of heart and renal protection (SHARP) Kidney Int. 2003;63(Suppl 84):S207–S210. doi: 10.1046/j.1523-1755.63.s84.4.x. [DOI] [PubMed] [Google Scholar]
- 35.Assche GV, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, Verbeeck J, Geboes K, Robberecht W, Rutgeerts P. Progressive multifocal leukoencephalopathy after Natalizumab therapy for Crohn’s Disease. N Engl J Med. 2005 Jul 28;353:362–368. doi: 10.1056/NEJMoa051586. [DOI] [PubMed] [Google Scholar]