

Abstract
The first total synthesis of aeruginosin 98B (1) was accomplished. The key step includes a highly diastereoselective Pd-catalyzed intramolecular asymmetric allylic alkylation (AAA) reaction of a diastereomeric mixture of allylic carbonates, which is enabled by the use of racemic phosphine ligand L1.
Aeruginosin 98B (1) is a tetrapeptide with an unnatural amino acid, isolated from a blue green algae Microcystis aeruginosa by Murakami and co-workers (Figure 1).1 Its structure was determined by 2D-NMR analysis, and the absolute configurations of the seven stereogenic centers were determined by X-ray crystallographic analysis of its cocrystal with trypsin.2 Aeruginosin 298A (2), another member of aeruginosin family, was synthesized by several groups.3 Oscillarin (3) was also isolated by Boehringer Mannheim GmbH.4 Recently, Hanessian and co-workers reported a total synthesis of aeruginosin 205B (4),5 which has a unique sulfonic sugar in the aeruginosin family.
Figure 1.

Structure of Aeruginosin 98B (1) and related compounds.
Aeruginosin family has gathered attention because of their promising biological activities. Aeruginosins exhibit inhibitory activities against serine proteases, especially thrombin and trypsin, and their activity profiles can be explained by a high degree of pharmacophoric and structural homology within the family.5 This array of structural and functional features is responsible for their high affinity to the catalytic binding pocket of trypsin, thrombin, and other serine proteases involved in the blood coagulation cascade. Aeruginosin 98B (1) was shown to inhibit the serine proteases thrombin, plasmin, and trypsin with IC50 values of 10.0, 7.0 and 0.6 μg/mL, respectively.1 The trypsin selectivity could be attributed to its unique 2-carboxy-6-hydroxyoctahydroindole sulfate (Choi sulfate) structure. The sulfonic acid group could interact with Tyr96 of trypsin through a H2O-mediated hydrogen bonding.2 However, other aeruginosins, aeruginosin 298A (2)6 and oscillarin (3)7 for example, are known to interact with Asp102 of thrombin. The negative interaction between the sulfonyl group and Asp102 may explain the IC50 value of aeruginosin 98B, which is 33 times smaller than that of aeruginosin 298. For these reasons, SAR studies of aeruginosin family were continued vigorously. 3c, 8 Herein, we describe the first total synthesis of aeruginosin 98B.
A major goal in developing a new strategy is to enable facile structural diversity to examine structure-function relationships. Aeruginosin 98B can be dissected into four, suitably protected building blocks: 4-hydroxyphenyl lactic acid derivative (Hpla, 5), bis-protected agmatine (6), 2-carboxyl-6-hydroxyoctahydroindole core (Choi, 7), and D-allo-isoleucine (8) (Figure 2). It was further reasoned that the octahydroindole subunit could be prepared in a highly diastereoselective fashion by employing an intramolecular Pd-catalyzed AAA reaction 9 on allylic carbonate 9a and 9b. Since the palladiumcatalyzed allylic alkylation reaction is a stereospecific double inversion process, it is only necessary to control the relative stereochemistry between the protected hydroxyl and carbonate functional groups during the preparation of the AAA precursors 9a and/or 9b. An important component of developing a route was to avoid the use of tyrosine as a precursor to both Hpla and Choi fragments as was commonly done in the synthesis of the other aeruginosins, in order to allow more facile modification of those subunits.
Figure 2.
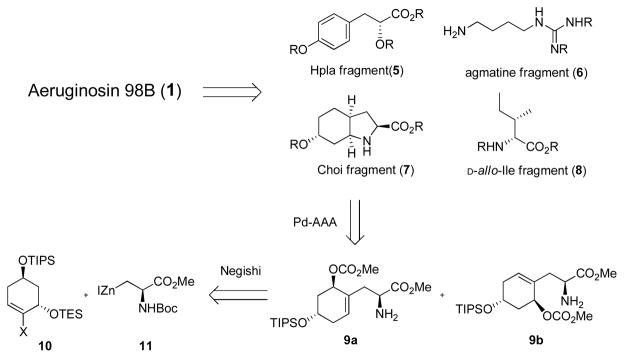
Retrosynthetic strategy of Aeruginosin 98B (1).
The allyl carbonates 9a and 9b were prepared from 4-methoxyphenol (12). Birch reduction11 followed by TIPS silylation of the resultant alcohol provided methyl enol ether in 97% overall yield. Dihydroxylation of methyl enol ether provided a 2:1 mixture of α-hydroxyketones 13a and 13b. The direct hydroxylation of the TIPS ether of 4-hydroxylcyclohexanone with MoOPH gave 13a selectively in 72% yield. The anti-diastereomer 13a was treated with TESCl under standard conditions to afford bis-silyl ether 14 in 97% yield. The enolate of ketone 14 was then allowed to react with Comins reagent12 to furnish vinyl triflate 15 in 88% yield. Gratifyingly, compound 15 could be efficiently coupled with alkylzinc reagent 1113 to give 16a and 16b as a 1:1 diastereomeric mixture. Amino esters 16a and 16b were treated with HF-pyridine complex to remove the TES protecting group selectively. The resulting allylic alcohols were easily separable by flash column chromatography and thus each diastereomer could be transformed separately toward the desired heterocycle. Treating 17a and 17b with methyl chloroformate and catalytic DMAP in CH2Cl2 furnished allylic carbonates 18a and 18b in 98% yield. Subsequent removal of the Boc protecting group under standard conditions, gave aminoesters 9a and 9b in 93% and 97% yield, respectively.
With the key intermediates 9a and 9b in hand, the diastereoselectivity of the Pd catalyzed allylic alkylation was examined. Using dppp as ligand with [(η3-C3H5)PdCl]2 in THF at 60°C, no reaction was obtained (Table 1, entry 1).14 On the other hand, using the (R, R)-chiral ligand L1, a 4.4:1 ratio of 19a:19b in a combined yield of 70% (Table 1, entry 2) was obtained. Surprisingly, switching to the (S, S) enantiomer of L1, a 3.8:1 ratio still favoring 19a was obtained in 63% yield (Table 1, entry 3). Gratifyingly, switching to the racemic mixture of L1 as ligand, this ratio favoring 19a jumped to 11:1 in a 96% combined yield (Table 1, entry 4). Thus, a new role has been established for this family of ligands.
Table 1.
Asymmetric allylic alkylation of diastereomeric mixture 9a and 9b.
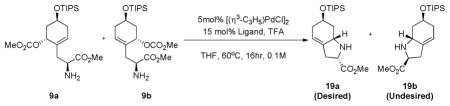
| ||||
|---|---|---|---|---|
| entry | Ligand | 19a (%) | 19b (%) | Recovery (%) |
| 1 | dppp | 0 | 0 | 71 |
| 2 | (R, R)-L1 | 57 | 13 | 0 |
| 3 | (S, S)-L1 | 50 | 13 | trace |
| 4 | racemic L1 | 88 | 8 | 0 |
Completion of the synthesis of the Choi subunit required diastereoselective reduction of the olefin. Since the simple amino group proved unstable to TBAF conditions, this amino group was benzylated and subsequently treated with TBAF in the same pot to give alcohol 20 in 89% yield whose hydrogenation then directly led to amino alcohol 21 in 92% yield. The relative stereochemistry of 21 was assigned on the basis of nOe analysis (see Supporting Information). More importantly, the spectroscopic properties of its TFA salt exactly matched those previously reported by Bonjoch and coworkers. 3a
The synthetic routes to Hpla and agmatine fragments are shown in Scheme 2. The cinnamic ester 22a was subjected to Sharpless asymmetric dihydroxylation to provide diol 23a.15 The product was shown to have >99%ee by chiral HPLC analysis. Removal of the benzylic hydroxyl group was effected using triethylsilane and TFA in CH2Cl2 to afford α-hydroxyester 24a. The hydroxyl group of 24a was protected by TIPS by treatment with TIPSCl and imidazole. The same route starting from cinnamate 22b (X=Cl) allows access to the hydroxylated ester 25b, the intermediate required for the synthesis of aeruginosin 98A. Agmatine fragment 2816 was prepared from N-Boc-1, 4-diaminobutane 26, according to Rich and co-workers.17 Treatment of 26 with N, N′-bis(benzyloxycarbonyl)-1H-pyrazole-1-carboxamidine in THF smoothly provided 27 in 65% yield. The Boc group was removed under standard conditions in 95% yield. D-allo-Ile methyl ester (29) was prepared by an esterification of D-allo-Ile, which was obtained by an optical resolution of DL-allo-Ile.18 Analytical data of 29 matched with that previously reported.19
Scheme 2. Synthesis of other fragmentsa.
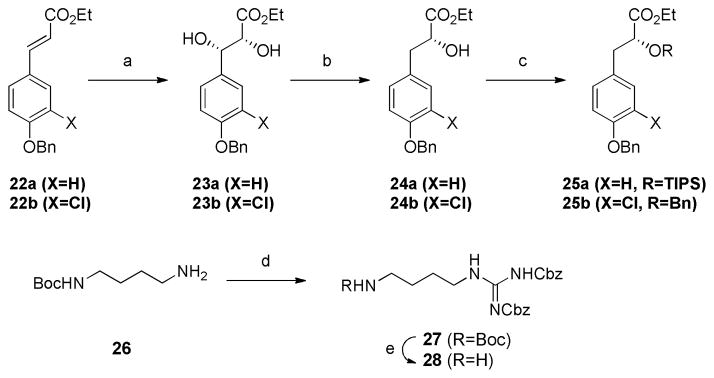
a(a) AD-mix α, MeSO2NH2, t-BuOH/H2O=1/1. 0°C, 23a; 86%, >99%ee, 23b; 91%, 98%ee. (b) Et3SiH, TFA, CH2Cl2, r.t., 24a; 80%, 24b; 82%. (c) 25a; TIPSCl, imidazole, DMAP, CH2Cl2, r.t., 79%, 25b; BnBr, Ag2O, TBAI, PhH, reflux, >63%. (d) N, N′-bis(benzyloxycarbonyl)-1H-pyrazole-1-carboxamidine, THF, r.t., 65%. (e) TFA, CH2Cl2, 0°C, 95%.
Ethyl ester 25a was hydrolyzed with 1N LiOH aq, and the resulting carboxylic acid was coupled with D-allo-Ile methyl ester (29) to afford dipeptide 30 in 73% yield (Scheme 3). Subsequent hydrolysis of the ester of 30 and the following peptide coupling with Choi fragment 21 provided tripeptide 31 in 69% yield. The tetrapeptide 32 was prepared in the same manner as the synthesis of tripeptide 31, using agmatine fragment 28. For the introduction of the sulfonic acid group, sulfur trioxide pyridine complex was used to afford sulfonic acid 33 in 75% yield. Removal of TIPS group was accomplished by treatment with HF-pyridine complex in acetonitrile to provide access to the precursor 34. The removal of Bn and Cbz groups provided aeruginosin 98B (1) in quantitive yield. The crude material was purified by reverse phase HPLC to afford pure aeruginosin 98B (1) in 98% yield as a colorless solid. The 1H and 13C-NMR spectra matched those of the natural product. Surprisingly, the optical rotation of synthetic 1 was higher (−11.24, c 0.25, H2O) than the natural product (−5.24, c 0.25, H2O).
Scheme 3. Total synthesis of Aeruginosin 98Ba.

a(a) 1N LiOHaq, THF/MeOH=3/2, r.t. (b) D-allo-Isoleucine methyl ester (29), TBTU, Et3N, DMF, r.t. 73% in 2 steps. (c) 1N LiOHaq, THF/MeOH=3/2, r.t. (d) 21, WSC, HOBt, Et3N, DMF, 0°C-r.t., 69% in 2 steps. (e) 1N LiOHaq, THF/MeOH=3/2, r.t. (f) 28, WSC, HOBt, Et3N, DMF, 0°C-r.t., 60% in 2 steps. (g) SO3-pyridine, pyridine, 40°C, 75%. (h) HF-pyridine, CH3CN, 0°C-r.t., 57%, 91%brsm. (i) Pd(OH)2, H2, MeOH, r.t., 98%.
In summary, we achieved the first total synthesis of aeruginosin 98B in eight steps from the four fragments (Choi, D-allo-Ile, Hpla and agmatine). After the construction of aeruginosin core, sulfonic acid was directly introduced by treatment with SO3-pyridine complex. In the preparation of Choi fragment, a Pd-catalyzed intramolecular AAA reaction of a diastereomeric mixture of allyl carbonates 9a and 9b using racemic ligand L1 provided hexahydroindole derivative 19a in high diastreo- and enantioselectivity wherein the asymmetry derived from 3-iodo-S-alanine. Curiously, higher reactivity of the racemic mixture of the chiral ligands suggests that each enantiomer of the racemic ligand might have recognized the matched diastereomer of 9a and 9b imparting better kinetics for ionization but the substrate controls the diastereoselectivity. We are currently exploring the precise reaction mechanism that explains the observed diastereo- and enantioselectivity.
Supplementary Material
Scheme 1. Synthesis of Choi fragmenta.

a(a) i) Li, NH3, EtOH, −78°C, ii) TIPSOTf, 2, 6-lutidine, CH2Cl2, 0°C, iii) OsO4, NMO, THF/H2O=3/1, 0°C, 96%, 13a/13b=2/1. (b) TESCl, imidazole, DMAP, CH2Cl2, r.t., 97%. (c) LHMDS, Comins reagent, −78°C to −40°C, 88%. (d) N-Boc-3-iodoalanine methyl ester, Zn, TMSCl, Pd(PPh3)4, LiCl, DMA, 60°C, 87%. (e) HF-pyridine, CH2Cl2, 0°C, 93%. (f) Methyl chloroformate, pyridine, DMAP, CH2Cl2, 0°C, 98%. (g) TFA, CH2Cl2, −10°C, 93–97%. (h) [(η3-C3H5)PdCl]2, racemic L1, TFA, THF, 60°C, 96%, 19a/19b=11/1. (i) BnBr, Et3N, CH3CN, r.t., then TBAF, 89%. (j) 5% Pd/C, H2, MeOH, r.t., 92%.
Acknowledgments
We thank the National Institutes of Health (GM 033049) and Teijin Pharma Ltd for their generous support of our program.
Footnotes
Experimental procedures and analytical data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Murakami M, Ishida K, Okino T, Okita Y, Matsuda H, Yamaguchi K. Tetrahedron Lett. 1995;36:2785.For a review see Ersmark K, Del Valle JR, Hanessian S. Angew Chem Int Ed. 2008;47:1202. doi: 10.1002/anie.200605219.
- 2.Sandler B, Murakami M, Clardy J. J Am Chem Soc. 1998;120:595. [Google Scholar]
- 3.(a) Valls N, Lopez-Canet M, Vallribera M, Bonjoch J. Chem Eur J. 2001;7:3446. doi: 10.1002/1521-3765(20010817)7:16<3446::aid-chem3446>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]; (b) Wipf P, Methot JL. Org Lett. 2000;2:4213. doi: 10.1021/ol006759x. [DOI] [PubMed] [Google Scholar]; (c) Ohshima T, Gnanadesikan V, Shibuguchi T, Fukuta Y, Nemoto T, Shibasaki M. J Am Chem Soc. 2003;125:11206. doi: 10.1021/ja037290e. [DOI] [PubMed] [Google Scholar]
- 4.(a) Konetschny-Rapp S, Krell H-W, Martin U. PCT WO96/11941. 1996; (b) Engh R, Konetschny-Rapp S, Krell H-W, Martin U, Tsaklakidis C. PCT WO97/21725. 1997
- 5.Hannesian S, Wang X, Ersmark K, Del Valle JR, Klegraf E. Org Lett. 2009;11:4232. doi: 10.1021/ol901702k. [DOI] [PubMed] [Google Scholar]
- 6.Steiner JLR, Murakami M, Tulinsky A. J Am Chem Soc. 1998;120:597. [Google Scholar]
- 7.Hanessian S, Tremblay M, Petersen JFW. J Am Chem Soc. 2004;126:6064. doi: 10.1021/ja030669g. [DOI] [PubMed] [Google Scholar]
- 8.(a) Radau G, Schermuly S, Fritsche A. Arch Pharm Pharm Med Chem. 2003;336:300. doi: 10.1002/ardp.200300765. [DOI] [PubMed] [Google Scholar]; (b) Hanessian S, Ersmark K, Wang X, Del Valle JR, Blomberg N, Xuec Y, FjellströM O. Bioorg Med Chem Lett. 2007;17:3480. doi: 10.1016/j.bmcl.2007.03.075. [DOI] [PubMed] [Google Scholar]; (c) Radau G, Rauh D. Bioorg Med Chem Lett. 2000;10:779. doi: 10.1016/s0960-894x(00)00101-3. [DOI] [PubMed] [Google Scholar]; (d) Radau G, Stürzebecher J. Pharmazie. 2002;57:729. [PubMed] [Google Scholar]; (e) Radau G, Gebel J, Rauh D. Arch Pharm Pharm Med Chem. 2003;336:372. doi: 10.1002/ardp.200300726. [DOI] [PubMed] [Google Scholar]; (f) Doi T, Hoshina Y, Mogi H, Yamada Y, Takahashi T. J Comb Chem. 2006;8:571. doi: 10.1021/cc0600270. [DOI] [PubMed] [Google Scholar]; (h) Nie X, Wang G. Tetrahedron. 2008;64:5784. [Google Scholar]
- 9.For reviews, see, Trost BM, Van Vranken DL. Chem Rev. 1996;96:395. doi: 10.1021/cr9409804.Trost BM, Crawley ML. Chem Rev. 2003;103:2921. doi: 10.1021/cr020027w.Lu Z, Ma S. Angew Chem Int Ed. 2008;47:258. doi: 10.1002/anie.200605113.Trost BM, Zhang T, Sieber JD. Chem Sci. 2010;1:427.
- 10.Negishi E, Owczarczyk ZR, Swanson DR. Tetrahedron Lett. 1991;32:4453. [Google Scholar]
- 11.(a) Marshall JA, Flynn GA. Synth Commun. 1979;9:123. [Google Scholar]; (b) Radlick P, Crawford HT. J Org Chem. 1972;37:1669. [Google Scholar]
- 12.Comins DL, Dehghani A. Tetrahedron Lett. 1992;33:6299. [Google Scholar]
- 13.Trost BM, Rudd MT. Org Lett. 2003;24:4599. doi: 10.1021/ol035752n. [DOI] [PubMed] [Google Scholar]
- 14.All of these results involved formation of the TFA salt since irreproducible results were obtained with pure 9a/9b in its absence.
- 15.Jagadeesh BY, Rao V. Tetrahedron Lett. 2011;52:6366. [Google Scholar]
- 16.Préville P, He JX, Tarazi M, Siddiqui MA, Cody WL, Doherty AM. Bioorg Med Chem Lett. 1997;7:1563. [Google Scholar]
- 17.West CW, Estiarte MA, Rich DH. Org Lett. 2001;3:1205. doi: 10.1021/ol015678d. [DOI] [PubMed] [Google Scholar]
- 18.Noda H, Sakai K, Murakami H. Tetrahedron: Asymmetry. 2002;13:2649. [Google Scholar]
- 19.Lloyd-Williams P, Monerris P, Gonzalez I, Jou G, Giralt E. J Chem Soc, Perkin Trans 1. 1994:1969. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.