

Abstract
Congenital heart disease (CHD) has been reported to occur in 14–70% of individuals with Cornelia de Lange syndrome (CdLS, OMIM 122470) and accounts for significant morbidity and mortality when present. Charts from a cohort of 479 patients with CdLS were reviewed for cardiac evaluations, gene testing and information to determine phenotypic severity. Two hundred fifty-nine individuals had either documented structural defects or minor cardiac findings. The presence of CHD was then quantified as a function of mutation status and severity of CdLS: mild, moderate, or severe. Different types of CHD were also evaluated by mutation status to assess for any genotype –phenotype correlation. NIPBL, SMC1A, and SMC3 mutation-positive patients were equally likely to have CHD, although the number of SMC1A and SMC3 mutation-positive patients were small in comparison. Structural CHDs were more likely to be present in individuals with moderate and severe CdLS than in the mild phenotype. This study evaluates the trends of CHD seen in the CdLS population and correlates these findings with genotype.
Keywords: Cornelia de Lange syndrome (CdLS), congenital heart disease (CHD), mutation, phenotype, cohesin, NIPBL, SMC1A, SMC3
INTRODUCTION
The diagnosis of Cornelia de Lange syndrome (CdLS) can be made clinically based on distinctive craniofacial appearance, limb deformities, hirsutism, and growth failure with significant developmental delays and intellectual disability. To date, three genes, NIPBL, SMC1A, and SMC3, all involved in the regulation or structure of the cohesin complex, have been identified to cause CdLS when mutated [Krantz et al., 2004; Tonkin et al., 2004; Musio et al., 2006; Deardorff et al., 2007]. Mutations are detected in approximately 60% of patients. Although the diagnosis of CdLS is made primarily based on the aforementioned features, congenital heart disease (CHD) is a commonly recognized feature of CdLS. The estimated prevalence of CHD in CdLS varies between 14% and 70% in previously studied patient cohorts of between 10 and 310 individuals, with several studies reporting a prevalence near 25% [Hawley et al., 1985; Robinson et al., 1985; Jackson et al., 1993; Kousseff et al., 1994; Mehta and Ambalavanan, 1997; Tsukahara et al., 1998; Barisic et al., 2008; Selicorni et al., 2009]. The largest group published to date by Jackson et al. included 310 patients, 25% of whom had a heart condition as indicated on a parent questionnaire, with 14% having a confirmed congenital heart defect. A systematic evaluation of 87 patients for CHD reported by Selicorni et al. [2009] revealed a prevalence of 33%. The estimated rates of CHD may vary, but nonetheless account for a significant proportion of the morbidity and mortality associated with CdLS [Beck and Fenger, 1985]. A recent review of causes of death in CdLS revealed congenital anomalies, including CHD, to account for 15% of CdLS-related deaths [Schrier et al., 2011]. A variety of defects have been described, with ventricular and atrial septal defects being the most common; complex heart defects such as tetralogy of Fallot and single ventricle anomalies have also been described.
An NIPBL+/− transgenic mouse with haploinsufficiency of NIPBL has been reported by Kawauchi et al. which showed that an estimated 75–80% of all NIPBL+/− mice die prior to 4 weeks of life, when genotyping is performed [Kawauchi et al., 2009]. Mutants were found to constitute 41% of all progeny genotyped just prior to birth (E17.5–E18.5) indicating a nearly normal 50% Mendelian distribution. Atrial septal defects, typically large, were found in half of NIPBL+/− embryos examined at E17.5–E18.5. Abnormalities in ventricular and interventricular myocardium were also observed. No cardiac structural defects were found in NIPBL+/− mice that survived the perinatal period, suggesting that CHDs resulted in the demise of embryos that did not survive.
CdLS can be phenotypically classified as mild, moderate, or severe based on three primary features including degree of growth retardation, severity of developmental delays/intellectual disability, and presence or absence of structural upper limb defects [Gillis et al., 2004]. The wide range of phenotypic variability in CdLS is becoming more appreciated and genotype–phenotype correlations have been described in regard to the primary diagnostic features. Missense mutations in the NIPBL gene typically confer a milder phenotype than nonsense, frameshift, or splice-site mutations that result in a truncated protein, and mutations in SMC1A/3 have been associated with a milder form of CdLS [Gillis et al., 2004; Deardorff et al., 2007]. To date, no genotype–phenotype correlation has been described to associate congenital heart defects with CdLS mutation status. In this study the prevalence of CHD in CdLS was assessed and compared to patient genotype and correlated with phenotypic severity of disease.
PATIENTS AND METHODS
Clinical Evaluation
Charts for 479 patients with a diagnosis of CdLS based on clinical criteria evaluated at The Children’s Hospital of Philadelphia (CHOP) between 1999 and 2009 were reviewed for this study. All patients were evaluated under an IRB-approved protocol of informed consent at CHOP. For all subjects a clinical diagnosis of CdLS was determined by one or more dysmorphologists experienced in the diagnosis of CdLS (I.D.K, A.D.K, L.G.J, M.A.D). In addition to documentation of characteristic dysmorphology used for clinical diagnosis, records for each patient were reviewed for description of anomalies used to determine severity of disease (growth, limb reduction, and development), in addition to documentation of other associated differences including hearing impairment, ophthalmologic findings, cleft palate, gastrointestinal, genitourinary, and renal anomalies used to support the clinical diagnosis. This cohort has previously been included in prior studies of genotype–phenotype correlation in CdLS [Gillis et al., 2004; Deardorff et al., 2007]. Ages of the individuals in this cohort ranged from aborted fetuses to 57 years. Charts were reviewed to document a formal cardiac evaluation with or without echocardiogram reports identifying the presence or absence of CHD. Information was available regarding the presence or absence of CHDs for 337 (70%) of the 479 probands. Of these individuals, 97 had obtainable echocardiogram reports. Only these 337 patient records were included for further analysis.
CHD Classification
The types of CHDs were collected from chart review, and categorized by hemodynamic consequences as has been used in other similar studies, as opposed to using an anatomic or embryologic classification system [Selicorni et al., 2007; Barisic et al., 2008]. This categorization was more useful in determining the clinical severity of the CHD to evaluate for genotype/phenotype correlation. Due to the nature of a retrospective chart review, the variability of particular information, including specific anatomic descriptions of congenital heart disease, which would be necessary for a more thorough anatomic analysis of cardiac defects in CdLS, was not available in every case.
Phenotype Severity
CdLS was categorized based on severity of phenotype. A mild phenotype is defined by no limb reduction defect, growth >75th centile and milder developmental delays including motor milestones <2 years delayed with speech and communication skills present. The moderate phenotype is defined by the presence of a partial limb defect, oligodactyly (>2 digits on each hand), growth between the 25th and 75th centile, and motor milestones delayed more than 2 years with limited speech and communication. A severe phenotype is defined by severe limb defects (<2 digits in either hand), growth at <25th centile, and profound developmental delay with absence of meaningful communication skills. Two hundred thirty-one of the 259 patients with documented structural or minor CHD had adequate phenotypic information to classify by severity. Only these patient records were included in further analysis.
Mutation Analysis
Mutational analysis of the three genes known to be associated with CdLS (NIPBL, SMC1A, and SMC3) was performed as reported previously [Gillis et al., 2004; Deardorff et al., 2007]. Individuals were screened for mutations in the 46 coding exons of NIPBL (inclusive of splice donor and acceptor sites), by conformation-sensitive gel electrophoresis and/or direct sequencing. SMC1A and SMC3 were analyzed by PCR amplification and direct sequencing of exons 1–29 and 2–25, respectively. The presence or absence of an identified mutation in NIPBL, SMC1A, or SMC3, was known for 319 (95%) of individuals with cardiac documentation. Of these 319 individuals, 279 had adequate phenotypic information to classify by severity. Overall, 231 probands had both known mutation status and phenotype severity. Only these probands were included in further analysis.
RESULTS
A total of 479 patients were evaluated at CHOP over a 10-year period and given a diagnosis of CdLS based on clinical features. Information regarding presence or absence of congenital heart disease was known for 337 (70% of the total cohort; Table I). Of the individuals for whom there was information about the presence of a CHD, phenotypic classification into mild, moderate and severe categories was possible for 279 (83%). There was a 50/50 distribution of males versus females in the group.
TABLE I.
Cohort Demographics
| CHD information | 337 (70%) |
| With known mutation status | 319 (96%) |
| With phenotype classification | 279 (83%) |
| With documented structural CHD or minor findings | 259 (77%) |
| With known mutation status AND phenotype | 231 (68%) |
| Male | 168 (50%) |
| Female | 169 (50%) |
| Mutation detection rate in those tested | 146/319 (46%) |
CHD, congenital heart disease.
Mutation Status
Mutation status was known for 319 of the 337 probands with a documented cardiac evaluation. Mutations were detected in 46% of the 319 patients, the distribution of which is shown in Table II. The spectrum of NIPBL mutations included missense, nonsense, in-frame deletion, frameshift, and splice site mutations. Among these a total of 53 individuals carried missense or in-frame deletion mutations (41% of NIPBL mutation-positive individuals), and 77 (59% of NIPBL mutation-positive individuals) had truncating mutations. All SMC1A mutations were missense changes or in-frame deletions, and the one patient with an SMC3 mutation had an in-frame deletion.
TABLE II.
Number of Patients With CHD as a Function of CdLS Severity and Mutation Status
| Total number (% of individuals in category) | |||||
|---|---|---|---|---|---|
| Mutation | Total with CHDb (% of total) | Mild | Moderate | Severe | Unknown |
| NIPBL missense | 18/53 (34) | 3 (13) | 8 (42) | 5 (82) | 2 |
| NIPBL other | 22/77 (29) | 2 (16) | 5 (33) | 15 (36) | 0 |
| SMC1A or 3 | 5/16 (31) | 5 (42) | 0 | 0 | 0 |
| Mutation negative | 51/173 (29) | 20 (26) | 21 (42) | 3 (27) | 7 |
| Mutation unknowna | 4/18 (22) | 1 | 1 | 1 | 1 |
| Total | 98/337 (30) | 29 | 35 | 24 | 10 |
CHD, congenital heart disease; CdLS, Cornelia de Lange syndrome.
These 18 individuals were CdLS patients with a CHD who were not screened for mutations in the CdLS genes.
This includes both the structural cardiac defects and the minor findings of PFO, PDA, innocent murmur.
The relatively high proportion of mildly affected individuals (126 of the 279 patients with a classifiable severity score) in this cohort is likely the reason for an overall mutation detection rate of only 46%, which is less than the generally quoted mutation detection rate amongst CdLS probands of 60% [Deardorff et al., 2007] as mutation detection is highest amongst individuals with the more severe or “classic” form of CdLS and less in the moderate and mild forms.
Mutation Status as a Function of Severity
In this cohort, as has been reported previously in patients with NIPBL mutations, missense mutations correlated with a mild phenotype whereas NIPBL non-missense mutations correlated with a more severe phenotype [Gillis et al., 2004]. Phenotypic classification by severity was possible for 279 of the 319 patients with known mutation status. All phenotyped individuals in this study with SMC1A or SMC3 mutations had a mild CdLS classification as has previously been reported [Deardorff et al., 2007]. Among 59 individuals categorized as severe with known mutation status, 42 (71%) had NIPBL truncating mutations, 6 (10%) had NIPBL missense mutations and 11 (19%) were mutation negative.
Cardiac Findings
Of 337 patients in whom the presence or absence of CHD was documented, a structural heart defect was found in 98 (29%). Minor findings also occurred including innocent murmurs, and persistent fetal circulatory findings (PDA or PFO), seen in an additional 161 (48%) of patients. Therefore, 259 probands had a structural congenital heart defect or minor findings. It was not clear in most cases if PDA and PFO findings were persistent beyond the neonatal period. It could not be assumed whether or not these were truly abnormal cardiac findings, and while they were important to include in the total, they were included in a separate category of “minor” findings so as not to convey definitive pathogenicity (Fig. 1).
FIG. 1.
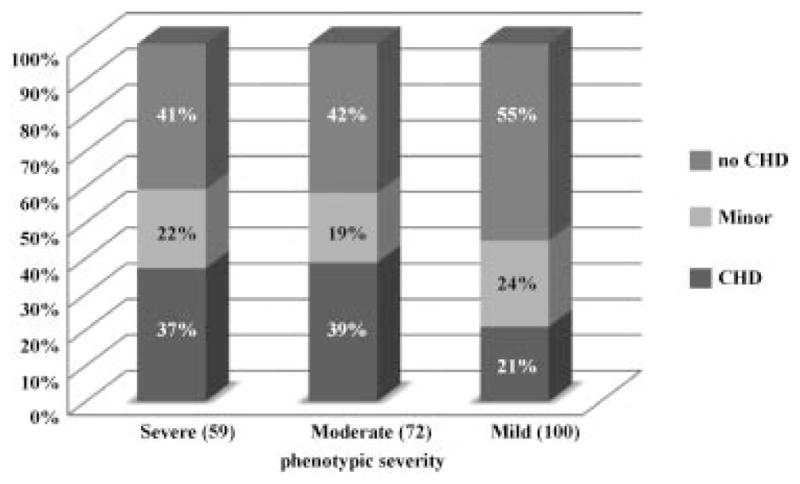
Structural and minor CHDs as related to phenotypic severity. CHD: congenital heart disease. Note: Only 231 out of 259 probands with known CHD status had adequate phenotypic data available to include in analysis.
Cardiac Findings as a Function of Severity
The types of cardiac findings in all patients with CHD were tabulated and then categorized by severity of CdLS. Of the 259 probands with known CHD, 231 (89%) had sufficient phenotypic information to classify by severity. Percentages of types of cardiac defects as correlated with phenotypic severity are outlined in Table IV.
TABLE IV.
Types of Congenital Heart Defects and CdLS Severity
| Defect (# of probands) | Total number (279) | Mild (130) | Moderate (89) | Severe (60) |
|---|---|---|---|---|
| Septal defects | ||||
| VSD | 20 | 8 | 5 | 5 |
| ASD | 19 | 6 | 5 | 3 |
| AVSD | 6 | 1 | 2 | 3 |
| Other (unspecified) | 5 | — | 2 | 2 |
| Overall prevalence | 18% | 12% | 16% | 22% |
| Outflow obstruction | ||||
| Pulmonic valve | 23 | 4 | 8 | 8 |
| Aortic | 11 | 3 | 6 | 1 |
| Mitral | 3 | 1 | 2 | 0 |
| Overall prevalence | 13% | 6% | 18% | 15% |
| Vascular anomalies | ||||
| Coarctation/hypoplastic aorta | 15 | 5 | 5 | 4 |
| PS/PPS | 15 | 4 | 5 | 5 |
| Enlarged aortic root | 1 | 1 | — | — |
| Overall prevalence | 11% | 8% | 11% | 15% |
| Complex defect | ||||
| TOF | 3 | — | 2 | 1 |
| TOF/DORV | 2 | — | — | 2 |
| DORV | 1 | — | — | 1 |
| AV canal defect | 1 | 1 | — | 1 |
| TAPVR | 1 | — | — | 1 |
| Truncus art | — | — | 1 | — |
| Overall prevalence | 5% | 1% | 3% | 10% |
| Dextrocardia | 2 | 1 | — | 1 |
| Fetal persistent | ||||
| PFO | 28 | 8 | 9 | 8 |
| PDA | 27 | 6 | 4 | 11 |
| Innocent murmur | 42 | 23 | 10 | 8 |
VSD, ventricular septal defect; ASD, atrial septal defect; AVSD, atrioventricular septal defect; PS, pulmonic stenosis; PPS, peripheral pulmonic stenosis; TOF, tetralogy of Fallot; DORV, double-outlet right ventricle; AV, atrioventricular; TAPVR, total anomalous pulmonary venous return; PFO, patent foramen ovale; PDA, patent ductus arteriosus.
Although more than one anomaly was identified in many CdLS patients, the frequency of individual types of findings was tallied independently. As reported in the literature, pulmonic stenosis, peripheral pulmonic stenosis, and atrial and ventricular septal defects were the most common findings (Table III) [Jackson et al., 1993; Tsukahara et al., 1998; Selicorni et al., 2009]. Other common defects included aortic valve anomalies (bicuspid valve, stenotic, and dysplastic), and coarctation of the aorta. All of these more common defects were observed in patients with mild, moderate and severe CdLS (Table IV).
TABLE III.
Most Common Congenital Heart Defects
| Defect | Total number of patients |
|---|---|
| PS/PPS | 30 |
| VSD | 20 |
| ASD | 19 |
| ASD/VSD | 6 |
| Coarctation/hypoplastic arch | 16 |
| Aortic valve anomaly | 14 |
| Pulmonary valve stenosis | 9 |
| Severe/cyanotic defects | |
| TOF | 5 |
| DORV | 3 |
| AV canal defect | 3 |
| TAPVR | 1 |
| Truncus arteriosus | 1 |
PS, pulmonic stenosis; PPS, peripheral pulmonic stenosis; VSD, ventricular septal defect; ASD, atrial septal defect; TOF, tetralogy of Fallot; DORV, double-outlet right ventricle; AV, atrioventricular; TAPVR, total anomalous pulmonary venous return.
Cardiac Findings as a Function of Mutation Status
The presence of CHD as a function of mutation status was analyzed and the results are presented in Tables II and V. The overall prevalence of a congenital heart defect was similar between the two subsets of NIPBL mutation positive patients: 34% among those with a missense mutation or in-frame deletion and 29% among those with all other types of mutations. Among those with missense mutations or in-frame deletions, a majority had a mild phenotype and of these only 13% had any structural cardiac defect compared with 42% of those with a moderate phenotype, and 82% of those with a severe phenotype (five out of six total individuals). Similarly, in patients with a truncating mutation, CHD was found to be more frequent in the severe phenotype (36%) when compared with the moderate or mild form, which had a 33% and 16% prevalence of CHD, respectively.
TABLE V.
Types of Congenital Heart Defects by Genotype
| Defect | Missense/in-frame deletion (53) | Truncating (77) | SMC1A or 3 (16) | No Mutation |
|---|---|---|---|---|
| Combined defects | ||||
| ASD +VSD +AS +PDA +arch hypoplasia | 1 | |||
| BAV +VSD +coarct | 1 | |||
| VSD +PDA +coarct | 1 | |||
| AV canal +ASD +coarct | 1 | |||
| ASD + hypoplastic arch | 1 | |||
| Vascular ring +R aortic arch | 1 | |||
| Epsteins anomaly +aberrant R subclavian | 1 | |||
| PS +PFO | 1 | |||
| PS +ASD | 1 | 1 | 1 | |
| PS +VSD | 1 | |||
| PS +AVSD | 1 | |||
| PS +PDA +coarctation | 1 | |||
| PS + aberrant subclavian | 1 | |||
| PPS +ASD | 1 | |||
| PPS +VSD | 1 | |||
| PPS +PFO (+PDA) | 3 | 1 | ||
| PPS +PS +AVSD +PAPVR | 1 | |||
| PPS +hypoplastic arch | 1 | |||
| PV stenosis +PFO/ASD | 1 | |||
| PV stenosis +PPS | 1 | |||
| Aortic valve abnormality | ||||
| BAV | 1 | |||
| +VSD | 2 | 2 | ||
| BAV +PV stenosis | 1 | |||
| +PS | 1 | 1 | ||
| +coarctation | ||||
| Isolated Septal defects | ||||
| VSD | 4 | 0 | 6 | |
| ASD | 1 | 1 | 4 | |
| AVSD | 1 | 1 | ||
| Other (unspecified) | 0 | 1 | ||
| Isolated Valve defects | ||||
| Pulmonary stenosis | 1 | 1 | ||
| Mitral | 1 | |||
| MR/TR | 1 | |||
| Cyanotic defect | ||||
| TOF | 2 | 3(2DORV) | ||
| Truncus arteriosus | 1 | |||
| TAPVR | 1 | |||
| Outflow obstruction | ||||
| Coarctation | 2 | 1 | 2 | |
| Pulmonary artery (PS/PPS) | 2 | 2 | ||
| Other isolated defects | ||||
| Dextrocardia | 1 | 1 | ||
| LVH | 1 | 1 | ||
| Small RV | 1 | |||
| Tortuous vessel | 1 | |||
| Fatal cardiac defect NOS | 1 | |||
| Minor findings | ||||
| PFO/PDA | 1 | 3 | ||
| PFO | 2 | 3 | 2 | |
| PDA | 3 | 3 | 3 | |
| Isolated murmur | 6 | 13 | 1 | 16 |
| Total prevalence of CHD | 18/53 = 34% | 29/77 = 35% | 5/16 = 31% | 49/173 = 28% |
ASD, atrial septal defect; VSD, ventricular septal defect; AS, aortic stenosis; PDA, patent ductus arteriosus; BAV, bicuspid aortic valve; PV, pulmonic valve; PS pulmonic stenosis; AV, atrioventricular; PFO, patent foramen ovale; AVSD, atrio-ventricular septal defect; MR, mitral regurgitation; TR, tricuspid regurgitation; TOF, tetralogy of Fallot; TAPVR, total anomalous pulmonary venous return; LVH, left ventricular hypertrophy; RV, right ventricle; NOS, not otherwise specified.
Of those patients with a clinical diagnosis of CdLS who were mutation negative, 24% had a structural heart defect (Table II). Septal defects, ASD and VSD, were also common in this group, as was coarctation of the aorta, and pulmonary artery stenosis (Table V). There were four mutation-positive individuals with other cyanotic and complex defects including total anomalous pulmonary venous return, truncus arteriosus, vascular ring (right aortic arch with ligamentum arteriosum), and Ebstein anomaly with aberrant subclavian artery.
DISCUSSION
There is a large burden of CHD in patients with CdLS. This study corroborates others, which estimate an approximate incidence of structural heart defects in CdLS at 30% [Selicorni et al., 2009]. The overall prevalence of CHD may be underrepresented in this study in which an additional 19% of patients had a minor cardiac finding, either a murmur or a persistent PDA or PFO, and not all patients had a formal cardiac evaluation. This study has a degree of ascertainment bias, as it is more likely for a child with complex CHD and CdLS to be referred to our center. As a result, it was more valuable to look at trends of CHD in CdLS rather than perform statistical analyses.
In this cohort the most common CHD was pulmonic and peripheral pulmonic stenosis, followed by septal defects (VSD and ASD), aortic valve defects, and aortic hypoplasia/coarctation. There were a number of cyanotic and complex heart defects as well, with tetralogy of Fallot being most common.
The overall prevalence of CHD was increased in the severe and moderate compared with a mild CdLS phenotype, and more complex and cyanotic defects were seen almost exclusively in patients with a moderate or severe phenotype. A cardiac finding was as likely to be diagnosed in patients with NIPBL as those with SMC1A mutations, although the types of defects in SMC1A-positive patients were generally milder and the number of patients with SMC1A and SMC3 mutations was fewer in comparison to those with NIPBL mutations. Given that the phenotype is universally milder in SMC1A and SMC3 mutation positive individuals [Rohatgi et al., 2010], it is likely that these individuals are under-diagnosed and CHD may be over-represented in patients that come to medical attention.
In 54% of this cohort no detectable mutation was identified in any of the three known genes and 29% of this mutation-negative cohort had a CHD. Although the proportion of patients with a mild phenotype was greater in this group compared to the NIPBL-positive group (45% compared with 28%, Table II), there was still a frequent incidence of CHD (30%), and severe structural abnormalities were found in some of these patients.
Missense and frameshift changes that preserve the open reading frame are presumed to lead to the production of a NIPBL protein with attenuated function, while nonsense, frameshift, and splice-site mutations are likely to result in a truncated gene product and haploinsufficiency [Deardorff et al., 2010]. There was no difference observed in the overall prevalence of congenital heart disease in patients with NIPBL missense mutations or in-frame deletions of NIPBL compared to those with other types of NIPBL mutations. A clear association of prevalence of CHD with severity of CdLS phenotype was more evident when cardiac defects were broken down by type. There is also a correlation between severity of CdLS phenotype and NIPBL genotype, with truncating mutations being associated with a more severe phenotype [Gillis et al., 2004; Deardorff et al., 2010]. Therefore, patients with NIPBL truncating mutations are likely to be at higher risk of severe CHD.
CdLS remains a clinical diagnosis with roughly half of all patients testing negative for mutations in the three known genes. There is also wide phenotypic variability making a definitive diagnosis more difficult when no mutation is identified. In this study phenotypic severity was correlated with risk of CHD, although cardiac defects were still identified in patients with a mild phenotype. Overall these data underscore the importance of screening for heart defects regardless of an individual’s clinical severity or genotype. Given that some heart defects commonly seen in this cohort can initially be asymptomatic and not detected on auscultation alone, cardiac evaluation including imaging such as echocardiography would be prudent in all children with a diagnosis of CdLS, in order to effectively diagnose cardiac involvement. The role of genetic testing for all patients with a new or possible diagnosis of CdLS, while valuable for counseling, family planning and in predicting severity, does not appear to be completely predictive of the presence or absence of CHD, but may correlate with the severity of CHD.
Acknowledgments
The authors are exceptionally grateful to the families and the patients who participated in this study as well as the Cornelia De Lange Foundation of the United States. This work was supported by the following NIH grants: NIH/NICHD PO1HD052860 (IDK), NIH/NICHD R21HD050538 (IDK), NIH/NICHD KO8HD055488 (MAD), T32GM008638 (SAS), as well as CHOP Institutional Development Funds (IDK) and The Center for Cornelia de Lange Syndrome and Related Diagnoses at CHOP.
References
- Barisic I, Tokic V, Loane M, Bianchi F, Calzolari E, Garne E, Wellesley D, Dolk H. Descriptive epidemiology of Cornelia de Lange syndrome in Europe. Am J Med Genet Part A. 2008;146A:51–59. doi: 10.1002/ajmg.a.32016. [DOI] [PubMed] [Google Scholar]
- Beck B, Fenger K. Mortality, pathological findings and causes of death in the de Lange syndrome. Acta Paediatr Scand. 1985;74:765–769. doi: 10.1111/j.1651-2227.1985.tb10028.x. [DOI] [PubMed] [Google Scholar]
- Deardorff MA, Kaur M, Yaeger D, Rampuria A, Korolev S, Pie J, Gil-Rodríguez C, Arnedo M, Loeys B, Kline AD, Wilson M, Lillquist K, Ramos FJ, Musio A, Jackson LS, Dorsett D, Krantz ID. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of Cornelia de Lange syndrome with predominant mental retardation. Am J Hum Genet. 2007;80:485–494. doi: 10.1086/511888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis LA, McCallum J, Kaur M, DeScipio C, Yaeger D, Mariani A, Kline AD, Li H, Devoto M, Jackson LG, Krantz ID. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype–phenotype correlations. Am J Hum Genet. 2004;75:610–623. doi: 10.1086/424698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley PP, Jackson LG, Kurnit DM. Sixty-four patients with Brachmann–de Lange syndrome: A survey. Am J Med Genet. 1985;20:453–459. doi: 10.1002/ajmg.1320200306. [DOI] [PubMed] [Google Scholar]
- Jackson L, Kline AD, Barr MA, Koch S. de Lange syndrome: A clinical review of 310 individuals. Am J Med Genet. 1993;47:940–946. doi: 10.1002/ajmg.1320470703. [DOI] [PubMed] [Google Scholar]
- Kawauchi S, Calof AL, Santos R, Lopez-Burks ME, Young CM, Hoang MP, Chua A, Lao T, Lechner MS, Daniel JA, Nussenzweig A, Kitzes L, Yokomori K, Hallgrimsson B, Lander AD. Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/−) mouse, a model of Cornelia de Lange syndrome. PLoS Genet. 2009;5:e1000650. doi: 10.1371/journal.pgen.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousseff BG, Newkirk P, Root AW. Brachmann–de Lange syndrome. 1994 update. Arch Pediatr Adolesc Med. 1994;148:749–755. doi: 10.1001/archpedi.1994.02170070087016. [DOI] [PubMed] [Google Scholar]
- Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D, Jukofsky L, Wasserman N, Bottani A, Morris CA, Nowaczyk MJ, Toriello H, Bamshad MJ, Carey JC, Rappaport E, Kawauchi S, Lander AD, Calof AL, Li HH, Devoto M, Jackson LG. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet. 2004;36:631–635. doi: 10.1038/ng1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta AV, Ambalavanan SK. Occurrence of congenital heart disease in children with Brachmann–de Lange syndrome. Am J Med Genet. 1997;71:434–435. doi: 10.1002/(sici)1096-8628(19970905)71:4<434::aid-ajmg12>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Musio A, Selicorni A, Focarelli ML, Gervasini C, Milani D, Russo S, Vezzoni P, Larizza L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet. 2006;38:528–530. doi: 10.1038/ng1779. [DOI] [PubMed] [Google Scholar]
- Rohatgi S, Clark D, Kline AD, Jackson LG, Pie J, Siu V, Ramos FJ, Krantz ID, Deardorff MA. Facial Diagnosis of mild and variant CdLS: Insights from a dysmorphoogist survey. Am J Med Genet Part A. 2010;152A:1641–1653. doi: 10.1002/ajmg.a.33441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson LK, Wolfsberg E, Jones KL. Brachmann–de Lange syndrome: Evidence for autosomal dominant inheritance. Am J Med Genet. 1985;22:109–115. doi: 10.1002/ajmg.1320220112. [DOI] [PubMed] [Google Scholar]
- Schrier SA, Sherer I, Deardorff MA, Clark D, Audette L, Gillis L, Kline AD, Ernst L, Loomes K, Krantz ID, Jackson LG. Causes of death and autopsy findings in a large study cohort of individuals with Cornelia de Lange syndrome and review of the literature. Am J Med Genet Part A. 2011;155A:3007–3024. doi: 10.1002/ajmg.a.34329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selicorni A, Russo S, Gervasini C, Castronovo P, Milani D, Cavalleri F, Bentivegna A, Masciadri M, Domi A, Divizia MT, Sforzini C, Tarantino E, Memo L, Scarano G, Larizza L. Clinical score of 62 Italian patients with Cornelia de Lange syndrome and correlations with the presence and type of NIPBL mutation. Clin Genet. 2007;72:98–108. doi: 10.1111/j.1399-0004.2007.00832.x. [DOI] [PubMed] [Google Scholar]
- Selicorni A, Colli AM, Passarini A, Milani D, Cereda A, Cerutti M, Maitz S, Alloni V, Salvini L, Galli MA, Ghiglia S, Salice P, Danzi GB. Analysis of congenital heart defects in 87 consecutive patients with Brachmann–de Lange syndrome. Am J Med Genet Part A. 2009;149A:1268–1272. doi: 10.1002/ajmg.a.32838. [DOI] [PubMed] [Google Scholar]
- Tonkin ET, Wang T, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet. 2004;36:636–641. doi: 10.1038/ng1363. [DOI] [PubMed] [Google Scholar]
- Tsukahara M, Okamoto N, Ohashi H, Kuwajima K, Kondo I, Sugie H, Nagai T, Naritomi K, Hasegawa T, Fukushima Y, Masuno M, Kuroki Y. Brachmann–de Lange syndrome and congenital heart disease. Am J Med Genet. 1998;75:441–442. doi: 10.1002/(sici)1096-8628(19980203)75:4<441::aid-ajmg20>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]