

Abstract
The PDZ domains of the trimeric DegS protease bind unassembled outer-membrane proteins (OMPs) that accumulate in the E. coli periplasm. This cooperative binding reaction triggers a proteolytic cascade that activates a transcriptional stress response. To dissect the mechanism of allosteric activation, we generated hybrid DegS trimers with different numbers of PDZ domains and/or protease-domain mutations. By studying the chemical reactivity and enzymatic properties of these hybrids, we show that all subunits experience a strongly coupled energetic landscape. For example, OMP-peptide binding to a single PDZ domain stimulates active-site chemical modification and proteolytic cleavage in the attached and neighboring protease domains. OMP-peptide binding relieves inhibitory PDZ interactions, whereas the interfaces between protease domains in the trimeric DegS core mediate positively cooperative activation driven both by substrate binding and inhibition relief.
HtrA-family proteases consist of a protease domain, which employs a classical Ser-His-Asp catalytic triad and assembles into a stable trimer, and one or two PDZ domains, which regulate activity by binding the C-termini of regulatory or substrate molecules1. Human HtrA enzymes play roles in apoptosis, growth, and differentiation, and have been linked to arthritis, cancer, vascular disease, and macular and neural degeneration2–8, but much of our understanding of these proteases is based on studies of DegS, a bacterial ortholog. E. coli DegS is a single PDZ-domain protease that senses environmental stress in the periplasm by binding the C-termini of outer-membrane proteins (OMPs) that fail to assemble9. This binding event activates DegS proteolysis of a transmembrane anti-sigma factor, initiating an intramembrane proteolytic cascade that ultimately results in transcription of envelope-stress genes in the cytoplasm10.
DegS trimers are anchored to the periplasmic face of the inner membrane, with each subunit containing a trypsin-like protease domain and a PDZ domain1,11. Heat shock and other stresses slow insertion of OMPs into the outer membrane, and unassembled OMPs accumulate in the periplasm, where their C-terminal Tyr-Xxx-Phe peptides bind to the DegS PDZ domains and activate cleavage of the RseA periplasmic domain (RseAP)9. RseA spans the inner membrane and its cytoplasmic domain binds and inhibits the σE transcription factor12–14. DegS cleavage of RseAP creates a new C-terminal residue that recruits RseP, an integral-membrane protease, resulting in a second cleavage of RseA and release of its cytoplasmic domain plus bound σE from the membrane15–18. AAA+ proteases then degrade the RseA portion of this complex19,20, liberating σE to activate transcription.
The core of the DegS enzyme is formed by symmetric packing between its three protease domains, with peripheral PDZ domains contacting only the protease domain in the same subunit21,22 (Fig. 1a). The ligand-free PDZ domains inhibit proteolysis, as RseA is cleaved more than 100-fold faster by a DegSΔPDZ variant than by DegS in the absence of OMP peptides9,23,24. The site for OMP-peptide binding in each PDZ domain is ~25 Å from the nearest proteolytic active site in the trimer21, indicating that the mechanism of activation must be indirect. Many features of OMP activation of DegS cleavage of RseA can be explained by a Monod-Wyman-Changeux (MWC) model of allostery in which DegS trimers exist either in an inactive/tense conformation or in an active/relaxed conformation, with the binding of specific OMP peptides and/or the RseA substrate altering the equilibrium between these states24–27. Indeed, the trimeric protease domain displays two basic conformations, which correspond to active and inactive structures, in a large number of crystal structures21–24,27,28. The free energies and percentage of active enzymes for the different unbound or ligand-bound states of DegS and DegSΔPDZ (Fig. 1b) have been calculated based on MWC modeling of experimental data24,26. However, other models of DegS activation have been proposed in which OMP binding to a specific PDZ domain selectively tunes the cleavage activity of only one DegS subunit rather than the entire trimer21,28.
Figure 1.

DegS and hybrid trimers containing mixtures of full length (S) and ΔPDZ (Δ) subunits. (a) Each subunit of the DegS trimer contains a trypsin-like protease domain (darker color; active site shown in red) and a peripheral PDZ domain (lighter color). Contacts between protease domains stabilize the trimer. OMP peptides (CPK representation) bind to the PDZ domains. The structure shown (3gcn)27 is the proteolytically active conformation. (b) Free-energy differences for DegS and DegSΔPDZ with and without saturating RseAP substrate and/or OMP peptide (Tyr-Tyr-Phe) were calculated (ΔG = − RT ln ([active]/[inactive]) from values taken from references 24 and 26. The right axis shows the percentage of active trimers for each molecular species. DegSΔPDZ has no binding site for OMP peptide. (c) The denaturation-renaturation protocol used to generate hybrid trimers is shown in the upper portion of the panel. The lower portion shows separation of hybrid trimers by ion-exchange chromatography. The inset shows SDS-PAGE of the four major peaks (see Supplementary Fig. 2 for complete gel). (d) The steady-state rate of RseAP (200 μM) cleavage by the SSΔ or SΔΔ variants showed a linear dependence (R2 > 0.98) on enzyme concentration. Reactions contained 150 μM Tyr-Tyr-Phe tripeptide.
Allostery is widely used to control biological reactions, and the molecular mechanisms that mediate and control the underlying cooperative conformational changes are therefore of substantial interest.29,30 To test the mechanism of allosteric activation by OMP peptides, here we generate and study the enzymatic properties of asymmetric DegS trimers that lack one or two PDZ domains. By using active-site chemical profiling31 and/or placing mutations that inactivate specific protease domains in these trimers, we show that OMP-peptide binding to a single PDZ domain affects the activity of the protease domain in the same subunit and neighboring subunits. Similarly, mutations that stabilize or destabilize the active conformation of one protease domain affect the activities of neighboring protease domains. Our results support a model in which the allosteric conformations and thus the proteolytic activities of each DegS subunit are cooperatively coupled through a shared free-energy landscape of the trimeric core. OMP-peptide binding relieves inhibitory PDZ interactions, allowing positively cooperative proteolytic activation driven in part by the intrinsic energetic landscape of the trimeric core and in part by substrate binding.
Results
DegS trimers with one or two PDZ domains
E. coli DegS variants that lack the N-terminal membrane anchor or that lack this anchor and the PDZ domain have been used for previous biochemical and structural studies9,21,23,24,26–28. Both variants form stable trimers in solution. Here, we used two types of DegS subunits to create mixed trimers. Subunits called “S” contain the protease and PDZ domains (residues 27–355) with an N-terminal His6 tag replacing the membrane anchor. Subunits called “Δ” contain the protease domain alone (residues 27–256) with an N-terminal Flag tag in place of the anchor sequence. We expressed and purified the SSS trimers and ΔΔΔ trimers separately.
To obtain hybrid trimers, we used a strategy (Fig. 1c) employed to obtain mixed oligomers of other proteins32,33. Purified SSS and ΔΔΔ trimers were mixed, incubated under denaturing conditions, and denaturant was removed by dialysis. The resulting mixture was first run on a gel-filtration column to remove aggregated material and then chromatographed on an ion-exchange column, allowing purification of individual species corresponding to SΔΔ, SSΔ, and SSS DegS trimers (Fig. 1c). The ΔΔΔ enzyme was recovered in the flow-through fraction but was contaminated with a small amount of at least one species containing S subunits (Fig. 1c). Thus, for experiments using the ΔΔΔ enzyme, we denatured and renatured ΔΔΔ by itself and then removed aggregated protein by gel filtration. Following denaturation, renaturation, and purification, ΔΔΔ cleaved 35S-RseAP with kinetic parameters similar to those of native DegSΔPDZ (Table 1), which differs only in the tag sequence. In addition, the activities of SSS and native DegS were similar (Table 1). Trimers are very stable under non-denaturing conditions, and thus subunit exchange or rearrangement should be insignificant during the short times (~10 min) used for activity assays. In the presence of saturating concentrations of an OMP tripeptide (Tyr-Tyr-Phe), both the SΔΔ and SSΔ enzymes cleaved 35S-RseAP at rates that varied linearly with enzyme concentration (Fig. 1d), establishing that active trimers are the major species populated over this concentration range. These assays and those presented below used sub-KM concentrations of RseAP to ensure sensitivity to small changes in enzyme activity.
Table 1.
Cleavage activities of DegS trimers containing different numbers of PDZ domains and/or mutations in specific subunits
| Michaelis-Menten parameters | ||||
|---|---|---|---|---|
| trimer | activating peptide | Vmax/[Etotal] (s−1 trimer−1) | KM (μM) | substrate Hill constant |
| SSS | Tyr-Tyr-Phe | 2.7 ± 0.1 | 300 ± 90 | 1.7 ± 0.1 |
| (a) DegS | Tyr-Tyr-Phe | 2.6 ± 0.2 | 370 ± 40 | 1.4 ± 0.2 |
| SSΔ | Tyr-Tyr-Phe | 3.7 ± 0.5 | 360 ± 70 | 1.9 ± 0.3 |
| SΔΔ | Tyr-Tyr-Phe | 2.6 ± 0.2 | 490 ± 100 | 1.8 ± 0.4 |
| ΔΔΔ | none | 1.0 ± 0.3 | 400 ± 110 | 1.5 ± 0.1 |
| (b) DegSΔPDZ | none | 0.8 ± 0.1 | 570 ± 50 | 1.7 ± 0.1 |
| Tyr-Tyr-Phe stimulated cleavage of 200 μM RseAP | ||||
| basal activity (M−1 s−1) | maximal activity (M−1 s−1) | Kact (μM) | OMP peptide Hill constant | |
| SHP/SAΔΔ | 590 ± 130 | 1700 ± 130 | 5 ± 2 | 1 (c) |
| SSAΔΔ | 110 ± 17 | 790 ± 50 | 18 ± 4 | 1 (c) |
| SHP/SASHP/SAΔ | 420 ± 140 | 1600 ± 410 | 7 ± 2 | 1.3 ± 0.3 |
| SSASSAΔ | 4.5 ± 2 | 350 ± 32 | 38 ± 2 | 1.3 ± 0.1 |
| SSΔYA | 27 ± 4 | 400 ± 80 | 13 ± 1 | 1.4 ± 0.4 |
| SSΔ | 87 ± 7 | 3300 ± 480 | 14 ± 2 | 1.3 ± 0.1 |
| SΔYAΔYA | 17 ± 1 | 220 ± 39 | 25 ± 7 | 1 (c) |
| SΔΔ | 360 ± 20 | 2900 ± 380 | 18 ± 6 | 1 (c) |
| SSΔSA | 30 ± 9 | 2200 ± 300 | 19 ± 9 | 1.4 ± 0.3 |
| SΔSAΔSA | 95 ± 27 | 1300 ± 430 | 15 ± 5 | 1 (b) |
| SSS | < 5 | 3180 ± 80 | 16 | 1.7 |
| (a) DegS | 2.9 ± 0.5 | 2500 ± 550 | 29 ± 3 | 1.7 ± 0.1 |
| ΔΔΔ | 575 ± 60 | 575 ± 60 | n.a. | n.a. |
| (b) DegSΔPDZ | 590 ± 60 | 590 ± 60 | n.a. | n.a. |
Data taken from ref. 26.
Data taken from ref. 23.
Data were fitted to a hyperbolic binding isotherm. Errors were calculated as , where n is the number of independent trials (typically 2 or 3). Maximal activities for enzymes containing PDZ domains were determined in the presence of 150 μM Tyr-Tyr-Phe tripeptide; basal activity was determined in the absence of OMP peptide. Kact is the concentration of Tyr-Tyr-Phe tripeptide required for half-maximal stimulation of RseAP cleavage. (n.a.) not applicable.
Cleavage activity and the number of PDZ domains
We initially measured the basal rate of cleavage of a sub-KM concentration of 35S-RseAP (200 μM) by the ΔΔΔ, SΔΔ, SSΔ, and SSS enzymes and observed an approximately exponential decrease in activity for each additional PDZ domain (Fig. 2a). This result supports a model in which each ligand-free PDZ domain stabilizes the inactive trimer relative to the active trimer by roughly 1.5 ± 0.5 kcal/mol.
Figure 2.
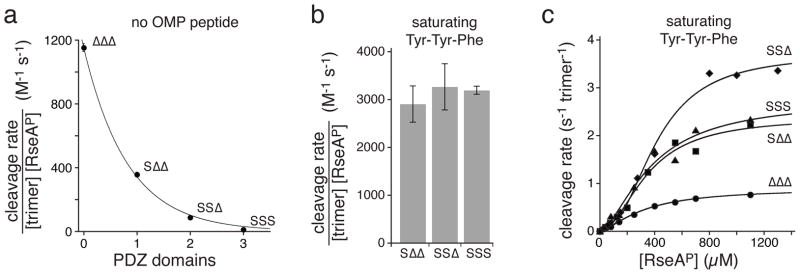
Basal and OMP-activated RseAP cleavage by the ΔΔΔ, SΔΔ, SSΔ, and SSS (0.5 μM trimer). (a) Basal cleavage rates divided by the enzyme and substrate (200 μM) concentration decreased with the number of PDZ domains in a trimer. The line is a fit to a single-exponential equation. (b) Cleavage rates divided by the enzyme and substrate concentration are plotted for the SΔΔ, SSΔ, and SSS trimers in the presence of 150 μM Tyr-Tyr-Phe tripeptide. Values are averages of 3 independent experiments ± 1 SD. (c) Dependence of the steady-state rate of cleavage on RseAP concentration for the ΔΔΔ, SΔΔ, SSΔ, and SSS trimers. Experiments were performed in the presence of 150 μM Tyr-Tyr-Phe tripeptide. The lines are fits to the Hill form of the Michaelis-Menten equation, .
Next, we measured SΔΔ, SSΔ, and SSS cleavage of 200 μM 35S-RseAP in the presence of saturating Tyr-Tyr-Phe tripeptide (Fig. 2b). Strikingly, despite having fewer PDZ domains, the SΔΔ and SSΔ variants had peptide-activated cleavage activities that were very similar to the SSS trimer.
We also assayed initial rates of cleavage of different concentrations of 35S-RseAP by the SSS, SSΔ, and SΔΔ enzymes in the presence of saturating Tyr-Tyr-Phe, and by the ΔΔΔ enzyme, which has no PDZ domains and thus does not bind this tripeptide (Fig. 2c; Table 1). Each cleavage reaction was positively cooperative, with a Hill constant for substrate between 1.5 and 1.9 and an apparent KM between 300 and 490 μM (Table 1). Thus, removing PDZ domains causes only minor changes in the positive cooperativity and apparent strength of substrate binding. In terms of Vmax, SSΔ had the highest value, SΔΔ and SSS were similar, and ΔΔΔ was lowest. Previous studies showed that DegSΔPDZ, which is almost identical to ΔΔΔ in sequence, had a somewhat low Vmax, at least in part, because the relative stabilities of the active and inactive conformations result in only ~50% of trimers assuming the functional conformation at saturating RseAP substrate concentrations (ref. 24). The Vmax values of the ΔΔΔ, SΔΔ, SSΔ, and SSS trimers are probably dictated by a combination of direct and indirect effects, in which the contribution of any subunit depends on the identities of its neighbors (see Discussion).
PDZ domains regulate cis and trans subunits
To determine if PDZ domains in hybrid trimers affect the activities of the attached (cis) protease domain and/or neighboring (trans) protease domains, we used chemical profiling to probe active-site reactivity in the SSΔ and SΔΔ hybrids with TAMRA-fluorophosphonate (TAMRA-FP). This fluorescent reagent covalently modifies the side-chain hydroxyl of the active-site Ser201 residue when the oxyanion hole and catalytic triad are properly formed26,31. Because S subunits are ~10 kDa larger than Δ subunits, we used SDS-PAGE followed by fluorescence imaging to determine the extent of TAMRA-FP labeling of each type of subunit.
For the SSΔ hybrid, neither S nor Δ subunits were labeled efficiently by TAMRA-FP without activating peptide, but both subunit types were labeled when Tyr-Tyr-Phe was present (Fig. 3a). Thus, the active-site reactivity of the Δ subunit is repressed by one or both of the unliganded PDZ domains in the neighboring S subunits and is stimulated by one or both of the peptide-bound PDZ domains in the neighboring subunits. For the SΔΔ hybrid, substantial TAMRA-FP modification of S and Δ subunits was observed without OMP peptide, consistent with the higher basal activity of this enzyme, and OMP peptide increased the reactivity of both types of subunits ~3-fold (Fig. 3b, c). In combination, these results provide strong evidence that unliganded PDZ domains repress activity and OMP-peptide binding activates proteolysis in both the cis and trans protease domains. OMP-bound S subunits were ~2-fold more reactive than Δ subunits after normalizing the rate of TAMRA-FP modification for subunit number, suggesting some degree of asymmetry in intrinsic subunit activity and/or OMP-peptide activation of cis versus trans subunits.
Figure 3.

OMP-peptide binding activates cis and trans subunits. (a) SSΔ (1 μM) was reacted with TAMRA-FP (20 μM) for different times, subjected to SDS-PAGE, and active-site modification of the S and Δ subunits was assayed by quantitative fluorescence imaging. Experiments were performed with Tyr-Tyr-Phe tripeptide (left panel) or no peptide (right panel). The fluorescence scan of the complete gel scan is shown in Supplementary Fig. 3. (b) Reaction of SΔΔ with TAMRA-FP. Same conditions as panel a. The S-subunit doublet probably results from cleavage of the tag sequence in some trimers. We do not know if both Δ subunits are labeled or are equally reactive. The fluorescence scan of the complete gel scan is shown in Supplementary Fig. 4. (c) Quantification of TAMRA-FP active-site modification from the panel-b experiment. Values were not adjusted for different numbers of S and Δ subunits. (d) Hybrid trimers (0.6 μM) containing different numbers of catalytically active S or Δ subunits and/or catalytically inactive SSA or ΔSA subunits were assayed for cleavage of RseAP (200 μM) in the presence or absence of 150 μM Tyr-Tyr-Phe tripeptide. Values are averages of 3 independent experiments ± 1 SD.
As a second method to test how the cleavage activities of S and Δ subunits in hybrid trimers are regulated by OMP-peptide binding in cis or trans subunits, we inactivated the catalytic triad by replacing the Ser201 nucleophile21 with alanine (henceforth called SA) in just S subunits or just Δ subunits to generate SSASSAΔ, SSAΔΔ, SSΔSA, and SΔSAΔSA variants. For each of these enzymes, addition of Tyr-Tyr-Phe enhanced the rate of cleavage of a sub-KM concentration of RseAP (Fig. 3d). Importantly, peptide binding to the single PDZ domain in SΔSAΔSA activated cleavage in the cis subunit, whereas peptide binding to the single PDZ domain in SSAΔΔ activated cleavage in trans subunits (Fig. 3d). Conversely, a single unliganded PDZ domain in a hybrid trimer suppressed the proteolytic activity of the attached protease domain or the protease domains in adjacent subunits. The ratio of stimulated/basal activities was 14 ± 6 for SΔSAΔSA and 7 ± 1 for SSAΔΔ. Thus, cis activation may be somewhat more efficient than trans activation, although the difference is close to the error and may not be significant.
Mutating inactive subunits can increase trimer activity
The His198 side chain is close to the oxyanion hole of DegS, and the His198-Pro mutation (henceforth called HP) increases basal activity and OMP-activated cleavage of sub-KM concentrations of RseAP (ref. 26). In crystal structures of the active HP enzyme, the Pro198 side chain makes favorable energetic contacts that would not be expected to be made in the inactive enzyme24,27, suggesting that the HP substitution increases activity indirectly by narrowing the free-energy gap between the inactive and active conformations. If HP does act indirectly by changing conformational preferences and the conformations of all protease domains in a trimer are tightly coupled, then HP mutations in catalytically inactive subunits should enhance proteolytic activity in neighboring subunits. By contrast, if the HP mutation acts directly by improving catalysis or only affects the conformational preference of the subunit in which it resides, then it should only increase activity in the context of a catalytically active subunit. To distinguish between these models, we generated hybrid trimers containing mixtures of S or Δ subunits with different combinations of the HP and active-site SA mutations.
We initially determined cleavage activities of the SSAΔΔ and SHP/SAΔΔ variants. At low substrate concentrations, Tyr-Tyr-Phe activated cleavage by SHP/SAΔΔ was faster than by SSAΔΔ (Supplementary Results, Supplementary Fig. 1a), suggesting that less substrate-binding energy is required to drive the conformational change from the inactive to the active SHP/SAΔΔ structure. Similarly, when we generated the SHP/SASHP/SAΔ and SSASSAΔ hybrid trimers, the HP mutations in the catalytically inactive subunits resulted in stronger apparent substrate binding as assayed by cleavage by the single active Δ subunit (Supplementary Fig. 1b). These results support a model in which HP mutations in some subunits reduce the free-energy gap between the inactive and active conformations of all subunits in hybrid trimers.
We also assayed the Tyr-Tyr-Phe dependence of cleavage of a sub- KM concentration of RseAP by the SHP/SAΔΔ and SSAΔΔ enzymes (Supplementary Fig. 1c) and by the SHP/SASHP/SAΔ and SSASSAΔ enzymes (Supplementary Fig. 1d). For the first pair, the HP mutation in the inactive subunit increased the maximal peptide-stimulated activity ~2-fold and decreased the peptide concentration required for half-stimulation ~3-fold compared to the otherwise identical enzyme without this mutation (Table 1). Basal activity was also ~5-fold higher for SHP/SAΔΔ than SSAΔΔ. These results support a model in which the HP mutation in the inactive SHP/SA subunit stabilizes the active conformations of neighboring Δ subunits. For the second pair, the HP mutations in both inactive subunits increased basal activity ~100-fold, increased maximal peptide-stimulated activity ~4-fold, and decreased the peptide concentration required for half-stimulation ~5-fold (Supplementary Fig. 1d; Table 1). Again, these results indicate that HP mutations in the inactive subunits of SHP/SASHP/SAΔ stabilize the active conformation of the single catalytically competent Δ subunit. These stabilizing effects allow the one functional Δ subunit of SHP/SASHP/SAΔ to have OMP-peptide stimulated cleavage activity similar to the combined activities of both functional Δ subunits of SHP/SAΔΔ (Table 1).
Additional evidence for a common subunit energy landscape
Contacts made by the side chain of Tyr162 in the protease domain appear to be important for stabilizing the active conformation of DegS21. Indeed, the Tyr162-Ala mutation (henceforth called YA) eliminates RseAP cleavage in DegS and DegS ΔPDZ backgrounds21,23,24. We generated and purified SSΔYA and SΔYAΔYA variants and measured the dependence of cleavage of a sub-KM concentration of RseAP on Tyr-Tyr-Phe concentration (Fig. 4a, b). Compared to the same enzymes without the YA substitutions, the maximum activated levels of cleavage were suppressed to extents far greater than would be expected if the ΔYA subunits did not reduce the proteolytic activities of the neighboring S subunits. For example, at saturating Tyr-Tyr-Phe tripeptide, the cleavage activity of SSΔ was ~8-fold higher than that of SSΔYA, and the cleavage activity of SΔΔ was ~13-fold higher than that of SΔYAΔYA (Fig 4a, b). By contrast, a model in which the ΔYA subunits themselves are inactive but do not affect the activities of neighboring subunits predicts much smaller differences.
Figure 4.
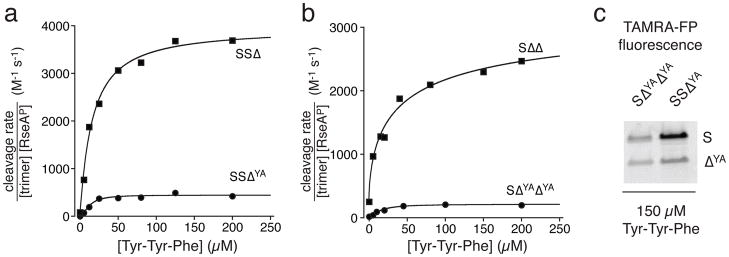
Trimers containing Δ subunits with the Tyr162-Ala mutation have reduced activity compared to otherwise isogenic trimers without this destabilizing mutation. (a, b) Tyr-Tyr-Phe activation of RseAP (200 μM) cleavage by 0.8 μM concentrations of mixed trimers. Panel-a data were fitted to a hyperbolic equation, ; panel-b data were fitted to a Hill equation, . Parameters are listed in Table 1. (c) Fluorescence imaging of SDS-PAGE showed that the S and ΔYA subunits in hybrid trimers reacted with TAMRA-FP (the fluorescence image of the complete gel lanes is shown in Supplementary Fig. 5). Reactions contained 1 μM trimer, 20 μM fluorophore, and were quenched after 12 min.
TAMRA-FP did not modify the YA variant of DegSΔPDZ, consistent with the active sites of this mutant trimer adopting an inactive conformation. Similarly, without OMP peptide, we found that neither the S nor ΔYA subunits of the SSΔYA and SΔYAΔYA variantswere appreciably modified by TAMRA-FP. Importantly, however, saturating Tyr-Tyr-Phe supported TAMRA-FP labeling of both the S and ΔYA subunits of the SSΔYA and SΔYAΔYA enzymes (Fig. 4c), although the normalized extent of modification was ~2-fold greater for the S subunits than the ΔYA subunits.
In combination, these results support a model in which all subunits of hybrid trimers experience a common energy landscape in which mutations that affect the conformational preferences of one subunit to adopt the active or inactive conformation also affect these preferences in neighboring subunits.
Discussion
How does OMP-peptide binding to the PDZ domains of trimeric DegS regulate proteolytic activity? Others had proposed that the penultimate side chain of a bound OMP peptide initiates a network of interactions that remodels the oxyanion hole in the active site of just the neighboring counterclockwise subunit21,28,34. By contrast, we find that OMP-peptide binding to a single PDZ domain in a hybrid trimer activates both the same subunit and neighboring subunits. Additional studies show that the penultimate OMP-peptide residue, which is poorly conserved, is not an important activation determinant9,23,26. Rather, the activation trigger is likely to involve the highly conserved C-terminal phenylalanine of OMPs, as the binding of this side chain requires movement of Met319 in the PDZ docking site from its position in inactive structures to one that should clash with and destabilize the inactive conformation of the protease domain. Indeed, the inactive conformation is stabilized by salt bridges between PDZ residues in the same helix as Met319 (e.g., Asp320) and residues in the protease domain (e.g., Arg178) (Fig. 5a). Deletion of the PDZ domain removes these inhibitory salt bridges, resulting in peptide-independent activation9,23,24. However, only ~50% of otherwise wild-type DegSΔPDZ trimers assume the active conformation in the presence of saturating substrate24, suggesting that structural features within the protease domain also contribute to the relative stabilities of the inactive and active conformations of this domain.
Figure 5.

Structural determinants of stabilization of the inactive and active DegS conformations. (a) Inactive DegS (1te0)22 contains salt bridges between acidic residues in the PDZ domain (colored cyan) and basic residues in the protease domain (colored green) of the same subunit. In the active conformation, OMP peptide binds slightly below and behind the 317–324 helix23 in the view shown. (b) The network shown appears to stabilize the active conformation of the protease domain trimer. Most residues are shown in stick representation, with the carbons of each subunit in a different color. The DFP-modified Ser201 residue is shown in CPK representation. The isopropyl group of the modified side chain mimics the P1 side chain of a substrate. Hydrogen bonds are shown as dotted lines. The structure shown (3gdv)27 contains the His198-Pro mutation. (c) A different view of a part of the network shown in panel b in which the packing interaction between the side chains of Tyr162 and Pro198 is more evident.
The principal result of the present work is that different protease domains in a DegS trimer share a cooperatively coupled energy landscape. This coupling allows OMP-peptide binding to a single PDZ domain to affect the conformations of cis and trans subunits in a trimer and allows sequence substitutions in one subunit of a trimer to affect the conformational preferences and thus the activities of neighboring subunits. The active conformation of the DegS protease-domain trimer contains a network of interactions involving side-chain and/or main-chain atoms of ~15 residues from each subunit (Fig. 5b, c)21,23,24,27,28. Many interactions within this network are absent in the inactive structures, including hydrogen bonds between the side chains of Thr167, Arg178, and Gln19. Indeed, in the inactive structure, Arg178 makes an inhibitory salt bridge with the PDZ domain (Fig. 5a). We propose that the cooperatively coupled energy landscape arises because the entire interaction network is substantially more stable than the sum of its parts, as expected in any highly coupled system in which each additional interaction reduces the entropic cost of successive interactions35. Supporting this model, the Thr167-Val, Arg178-Lys, and Gln191-Ala mutants of DegSΔPDZ are each missing only a small fraction of the network but have no detectable activity and crystallize in the inactive conformation24.
Two of the mutations studied here, Tyr162-Ala and His198-Pro, affect the interaction network in opposite ways. Tyr162-Ala removes a packing interaction with residue 198 and thus destabilizes the active network (Fig. 5c). We find that a Tyr162-Ala substitution in one subunit reduces the activity of neighboring wild-type subunits, as expected if fewer trimers adopt the active conformation. By contrast, the His198-Pro substitution adds additional contacts with the Tyr162 side chain and stabilizes the active conformation24,26. Introduction of His198-Pro into DegS ΔPDZ increases Vmax and almost completely eliminates the positive cooperativity of substrate binding, as expected if this variant predominantly assumes the active conformation, even in the absence of substrate24. Our current results show that the His198-Pro mutation in a catalytically inactive subunit increases the activity of a neighboring ΔPDZ subunit, as expected for cooperative stabilization of the functional trimer via formation of additional network contacts.
How does substrate binding stabilize the active conformation? There are no structures of the substrate-bound protease domain, but structures of diisopropylfluorophosphate-modified enzymes provide an important clue, as the isopropyl group of the modified Ser201 side chain mimics a valine side chain at the substrate P1 position24,27. This substrate mimic adds an additional network contact, helping tie together the active sites of each subunit of the active trimer (Fig. 5b, c).
OMP activation of DegS and mutant variants is fit well by the MWC model of allostery in which substrates and OMP-peptide effectors simply alter the equilibrium populations of inactive trimers versus active trimers, with the unliganded PDZ domain stabilizing inactive trimers25,26. Our present finding that basal enzyme activity is inversely proportional to the number of PDZ domains in the trimer supports this inhibitory role. We also find that hybrid DegS trimers with one or two PDZ domains have OMP-stimulated activities equal to or greater than trimers with three PDZ domains. These results suggest that the activities of these hybrid trimers are dictated, at least in part, by the effects of each type of subunit on the energy difference between the active and inactive conformational states. Because there is no simple trend between OMP-stimulated activities and the number of S or Δ subunits in different hybrids, the contribution of each subunit must depend, at least in part, on the identities of both neighboring subunits.
Within the context of an individual hybrid trimer, we find that the active sites of PDZ-containing subunits react with the chemical-profiling reagent TAMRA-FP ~2-fold faster than ΔPDZ subunits. Because altered reactivity suggests altered structure, this result is not expected by the MWC model25, which posits that all protease domains in a trimer should be symmetric and have the same structure irrespective of the presence of the PDZ domain. DegS might be better described by a general allosteric model that allows mixtures of active and inactive subunits within the same trimer36,37. Because the general model contains all species allowed by the MWC model, both models make similar predictions as long as DegS trimers with mixtures of active and inactive subunits are poorly populated relative to symmetric species. Alternatively, a variation of the MWC model in which minor structural asymmetry is allowed in terms of active-subunit conformations could also explain the small differences that we observe between S and Δ subunits. Indeed, in crystal structures of DegSΔPDZ, some catalytic triads in a given trimer are well formed, whereas others are not23,24,27.
OMPs represent one of the major macromolecular components of the outer membrane of Gram-negative bacteria and high rates of synthesis, secretion, and assembly are required to maintain exponential growth38. Thus, the concentration of unassembled OMPs passing through the periplasm is probably always significant. From a biological perspective, the energy landscape of DegS allows positively cooperative switch-like behavior in which modest increases in periplasmic OMPs, resulting from assembly defects caused by stress, cause substantial activation of the envelope-stress response.
Evolution produced a highly regulated DegS proteolytic machine with two important allosteric interfaces, one between the PDZ domain and the attached protease domain (Fig. 5a) and one between neighboring protease domains in the trimeric core (Fig. 5b, c). The unliganded PDZ domain inhibits proteolysis by stabilizing the inactive conformation, whereas OMP peptide binding to the PDZ domain antagonizes this inhibition. The design of the trimeric proteolytic core favors the inactive conformation in the absence of substrate and the active conformation in the substrate-bound state. This allosteric hierarchy allows robust control of RseA proteolysis linked to the concentrations of both substrate and OMP activator molecules. Trimers are the basic structural unit of all members of the HtrA family, and it will be important to determine if the allosteric and functional features of these enzymes resemble those of DegS.
Online Methods
Genes encoding E. coli DegS residues 27–355 with an N-terminal Met-Arg-Gly-Ser-His6-Gly-tag (called S subunits) or residues 27–256 with an N-terminal Flag Met-Asp-Tyr-Lys-Asp-Asp-Asp-Lys-Gly- tag (called Δ subunits) were cloned into a modified pet21b vector, and mutants were generated using the QuikChange PCR protocol. E. coli strain ER2566 harboring specific expression plasmids was grown at 37 °C to an OD600 of ~0.6, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM, and growth was continued for 4 h. Cells were harvested by centrifugation, resuspended in 50 mM NaPO4, 300 mM NaCl, and 10 mM imidazole (pH 8.0), and frozen at −80 °C. The cell suspension was thawed, lysed by sonication, and the lysate was centrifuged at 14,000×g for 30 min to remove insoluble debris. To purify His6-tagged SSS trimers, the supernatant was loaded onto a Ni++-NTA column (Pierce) equilibrated with 50 mM NaPO4, 300 mM NaCl, and 10 mM imidazole, pH 8.0. The column was washed extensively and the protein was eluted by the addition of 50 mM NaPO4, 300 mM NaCl, 300 mM imidazole, pH 8.0. To purify Flag-tagged ΔΔΔ trimers, the supernatant was brought to 20% saturation with ammonium sulfate, incubated at 4 °C for 1 h, centrifuged at 14000 ×g for 20 min, and the supernatant was discarded. The pellet was resuspended in 10 mM NaPO4, 50 mM NaCl, pH 8.0 (buffer A) and dialyzed extensively against the same buffer to remove residual ammonium sulfate. This solution was loaded onto a Q-Sepharose fast-flow column (GE Healthcare), washed extensively with buffer A, washed with buffer A plus 150 mM NaCl, and the Flag-tagged ΔΔΔ trimer was eluted with buffer A plus 300 mM NaCl. Both the His6-tagged SSS and Flag-tagged ΔΔΔ proteins were 95% pure as judged by SDS-PAGE. Each was concentrated to ~0.5 mM using an Ultracel-50k centrifugal filter (Millipore), 10% v/v glycerol was added, and the proteins were stored at −80 °C until use.
Generating and purifying hybrid trimers
To generate hybrid trimers, equimolar concentrations of His6-tagged SSS trimers and FLAG-tagged ΔΔΔ trimers were combined and diluted 15-fold into 7 M guanidinium chloride, 50 mM NaPO4, 300 mM NaCl (pH 8.0). After incubation at 50 °C for 2–3 h to promote disassembly of SSS and ΔΔΔ trimers, the solution was dialyzed extensively against 50 mM NaPO4, 300 mM NaCl, 5 mM EDTA (pH 8.0) at 4 °C. This mixture was concentrated by ultrafiltration and chromatographed on a Superdex-200 column (GE Healthcare) equilibrated with 10 mM NaPO4, 50 mM NaCl, 1 mM EDTA (pH 6.7) to remove aggregated protein. Appropriate fractions were combined, concentrated, and loaded onto a 1 mL ReSource 15S cation-exchange column (GE Healthcare) equilibrated with 10 mM NaPO4, 50 mM NaCl, 1 mM EDTA (pH 6.7). ΔΔΔ trimers were found in the flow-through fraction. The NaCl concentration was increased to 375 mM using a linear gradient of 62.5 mM/mL at which point the SΔΔ variant eluted. The gradient was then adjusted to 0.6 mM/mL and the SSΔ and SSS variants eluted over the course of 20 column volumes. A shallow gradient was necessary to obtain adequate separation between the SSΔ and SSS proteins.
Enzyme Assays
The periplasmic domain of E. coli RseA (residues 121–216) with an N-terminal Met- Gly-Ser2-His6-Ser2-Gly-Leu-Val-Pro-Arg-Gly-Ser-His-Met- tag was purified from E. coli X90DE3 cells grown in a defined rich medium lacking methionine (TekNova) supplemented with 35S-methionine (Perkin-Elmer). A cell lysate in 6 M GuHCl was applied to a Ni++-NTA column (Qiagen), which was subsequent washed with 50 mM NaPO4 (pH 8.0), 300 mM NaCl, and the RseAP protein was eluted with this buffer supplemented with 300 mM imidazole7. The synthetic Tyr-Tyr-Phe tripeptide was purified by reverse-phase chromatography on a C18 HPLC column. RseAP cleavage assays were performed in 150 mM NaHPO4 (pH 8.3), 380 mM NaCl, 10% glycerol, and 4 mM EDTA at room temperature23. Different concentrations of 35S-RseAP were incubated with DegS variants (0.5–0.8 μM trimer) in the presence or absence of Tyr-Tyr-Phe (150 μM) for different times, reactions were quenched by rapid dilution into 10% (w/v) trichloroacetic acid, samples were incubated on ice and centrifuged to remove insoluble material, and the extent of cleavage was detected by scintillation counting of the acid-soluble fraction. Data were fit using the nonlinear least squares algorithm implemented in SigmaPlot v12.0.
Active-site modification
Enzyme-modification assays were performed at room temperature in the same buffer used for cleavage assays and were initiated by adding 1/10 volume of 20 mM TAMRA-FP (ActivX Biosciences, Inc.) dissolved in anhydrous dimethylsulfoxide. Reactions were quenched by addition of SDS-loading buffer, samples were subjected to SDS-PAGE, and protein was detected by staining with Coomassie Blue and fluorescence was detected using a Typhoon fluorescent imager.
Supplementary Material
Acknowledgments
Supported by NIH grant AI-16892 (R.T.S.). R.V.M. was supported by an NIH postdoctoral fellowship (GM097972). We thank B. Stinson, A. Olivares, J. Sohn, A. Nager, A. de Regt, and S. Kim for helpful discussions.
Footnotes
Author contributions. R.V.M. performed all experiments. R.V.M. and R.T.S. contributed to experimental design, interpretation, and writing the manuscript.
The authors declare no competing financial interests.
References
- 1.Singh N, Kuppili RR, Bose K. The structural basis of mode of activation and functional diversity: a case study with HtrA family of serine proteases. Arch Biochem Biophys. 2011;516:85–96. doi: 10.1016/j.abb.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Grau S, et al. Implications of the serine protease HtrA1 in amyloid precursor protein processing. Proc Natl Acad Sci USA. 2005;102:6021–6026. doi: 10.1073/pnas.0501823102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coleman HR, Chan CC, Ferris F, Chew EY. Age-related macular degeneration. Lancet. 2008;372:1835–1845. doi: 10.1016/S0140-6736(08)61759-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milner JM, Patel A, Rowan AD. Emerging roles of serine proteinases in tissue turnover in arthritis. Arthritis Rheum. 2008;58:3644–3656. doi: 10.1002/art.24046. [DOI] [PubMed] [Google Scholar]
- 5.Vande Walle L, Lamkanfi M, Vandenabeele P. The mitochondrial serine protease HtrA2/Omi: an overview. Cell Death Differ. 2008;15:453–460. doi: 10.1038/sj.cdd.4402291. [DOI] [PubMed] [Google Scholar]
- 6.Chien J, Campioni M, Shridhar V, Baldi A. HtrA serine proteases as potential therapeutic targets in cancer. Curr Cancer Drug Targets. 2009;9:451–468. doi: 10.2174/156800909788486704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hara K, et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med. 2009;360:1729–1739. doi: 10.1056/NEJMoa0801560. [DOI] [PubMed] [Google Scholar]
- 8.Bowden MA, et al. High-temperature requirement factor A3 (Htra3): a novel serine protease and its potential role in ovarian function and ovarian cancers. Mol Cell Endocrinol. 2010;327:13–18. doi: 10.1016/j.mce.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Walsh NP, Alba BM, Bose B, Gross CA, Sauer RT. OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell. 2003;113:61–71. doi: 10.1016/s0092-8674(03)00203-4. [DOI] [PubMed] [Google Scholar]
- 10.Alba BM, Gross CA. Regulation of the Escherichia coli sigma-dependent envelope stress response. Mol Microbiol. 2004;52:613–619. doi: 10.1111/j.1365-2958.2003.03982.x. [DOI] [PubMed] [Google Scholar]
- 11.Waller PR, Sauer RT. Characterization of degQ and degS, Escherichia coli genes encoding homologs of the DegP protease. J Bacteriol. 1996;178:1146–1153. doi: 10.1128/jb.178.4.1146-1153.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Las Peñas A, Connolly L, Gross CA. The sigmaE-mediated response to extracytoplasmic stress in Escherichia coli is transduced by RseA and RseB, two negative regulators of sigmaE. Mol Microbiol. 1997;24:373–385. doi: 10.1046/j.1365-2958.1997.3611718.x. [DOI] [PubMed] [Google Scholar]
- 13.Missiakas D, Mayer MP, Lemaire M, Georgopoulos C, Raina S. Modulation of the Escherichia coli sigmaE (RpoE) heat-shock transcription-factor activity by the RseA, RseB and RseC proteins. Mol Microbiol. 1997;24:355–371. doi: 10.1046/j.1365-2958.1997.3601713.x. [DOI] [PubMed] [Google Scholar]
- 14.Campbell EA, et al. Crystal structure of Escherichia coli σE with the cytoplasmic domain of its anti-σ RseA. Mol Cell. 2003;11:1067–1078. doi: 10.1016/s1097-2765(03)00148-5. [DOI] [PubMed] [Google Scholar]
- 15.Ades SE, Grigorova IL, Gross CA. Regulation of the alternative sigma factor sigma(E) during initiation, adaptation, and shutoff of the extracytoplasmic heat shock response in Escherichia coli. J Bacteriol. 1999;185:2512–2519. doi: 10.1128/JB.185.8.2512-2519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanehara K, Ito K, Akiyama Y. YaeL (EcfE) activates the sigma(E) pathway of stress response through a site-2 cleavage of antisigma(E), RseA. Genes Dev. 2002;16:2147–2155. doi: 10.1101/gad.1002302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akiyama Y, Kanehara K, Ito K. RseP (YaeL), an Escherichia coli RIP protease, cleaves transmembrane sequences. EMBO J. 2004;23:4434–4442. doi: 10.1038/sj.emboj.7600449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, et al. Cleavage of RseA by RseP requires a carboxyl-terminal hydrophobic amino acid following DegS cleavage. Proc Natl Acad Sci USA. 2009;106:14837–14842. doi: 10.1073/pnas.0903289106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flynn JM, Levchenko I, Sauer RT, Baker TA. Modulating substrate choice: the SspB adaptor delivers a regulator of the extracytoplasmic-stress response to the AAA+ protease ClpXP for degradation. Genes Dev. 2004;18:2292–2301. doi: 10.1101/gad.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaba R, Grigorova IL, Flynn JM, Baker TA, Gross CA. Design principles of the proteolytic cascade governing the σE-mediated envelope stress response in Escherichia coli: keys to graded buffered and rapid signal transduction. Genes Dev. 2007;21:124–136. doi: 10.1101/gad.1496707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilken C, Kitzing K, Kurzbauer R, Ehrmann M, Clausen T. Crystal structure of the DegS stress sensor: How a PDZ domain recognizes misfolded protein and activates a protease. Cell. 2004;117:483–494. doi: 10.1016/s0092-8674(04)00454-4. [DOI] [PubMed] [Google Scholar]
- 22.Zeth K. Structural analysis of DegS, a stress sensor of the bacterial periplasm. FEBS Lett. 2004;569:351–358. doi: 10.1016/j.febslet.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 23.Sohn J, Grant RA, Sauer RT. Allosteric activation of DegS, a stress sensor PDZ protease. Cell. 2007;131:572–583. doi: 10.1016/j.cell.2007.08.044. [DOI] [PubMed] [Google Scholar]
- 24.Sohn J, Grant RA, Sauer RT. Allostery is an intrinsic property of the protease domain of DegS: implications for enzyme function and evolution. J Biol Chem. 2010;285:34039–34047. doi: 10.1074/jbc.M110.135541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 26.Sohn J, Sauer RT. OMP peptides modulate the activity of DegS protease by differential binding to active and inactive conformations. Mol Cell. 2009;33:64–74. doi: 10.1016/j.molcel.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sohn J, Grant RA, Sauer RT. OMP peptides activate the DegS stress-sensor protease by a relief of inhibition mechanism. Structure. 2009;17:1411–1421. doi: 10.1016/j.str.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hasselblatt H, et al. Regulation of the sigmaE stress response by DegS: how the PDZ domain keeps the protease inactive in the resting state and allows integration of different OMP-derived stress signals upon folding stress. Genes Dev. 2007;21:2659–2670. doi: 10.1101/gad.445307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui Q, Karplus M. Allostery and cooperativity revisited. Protein Sci. 2008;17:1295–1307. doi: 10.1110/ps.03259908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.del Sol A, Tsai CJ, Ma B, Nussinov R. The origin of allosteric functional modulation: multiple pre-existing pathways. Structure. 2009;17:1042–1050. doi: 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci USA. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kimmel JL, Reinhart GD. Isolation of an individual allosteric interaction in tetrameric phosphofructokinase from Bacillus stearothermophilus. Biochemistry. 2001;40:11623–11629. doi: 10.1021/bi010844a. [DOI] [PubMed] [Google Scholar]
- 33.Schneider F, Hammarström P, Kelly JW. Transthyretin slowly exchanges subunits under physiological conditions: A convenient chromatographic method to study subunit exchange in oligomeric proteins. Protein Sci. 2001;10:1606–1613. doi: 10.1110/ps.8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clausen T, Kaiser M, Huber R, Ehrmann M. HTRA proteases: regulated proteolysis in protein quality control. Nat Rev Mol Cell Biol. 2011;12:152–162. doi: 10.1038/nrm3065. [DOI] [PubMed] [Google Scholar]
- 35.Creighton TE. Proteins, Structures and Molecular Properties. 2. W.H. Freeman & Co; New York: 1993. [Google Scholar]
- 36.Eigen M. Kinetics of reaction control and information transfer in enzymes and nucleic acids. Nobel Symp. 1967;5:333–369. [Google Scholar]
- 37.Hammes GG, Wu CW. Regulation of enzyme activity. The activity of enzymes can be controlled by a multiplicity of conformational equilibria. Science. 1971;172:1205–1211. doi: 10.1126/science.172.3989.1205. [DOI] [PubMed] [Google Scholar]
- 38.Hagan CL, Silhavy TJ, Kahne D. β-Barrel membrane protein assembly by the Bam complex. Annu Rev Biochem. 2011;80:189–210. doi: 10.1146/annurev-biochem-061408-144611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.