

Abstract
Copper coexists with amyloid-β (Aβ) peptides at a high concentration in the senile plaques of Alzheimer’s disease (AD) patients and has been linked to oxidative damage associated with AD pathology. However, the origin of copper and the driving force behind its accumulation are unknown. We designed a sensitive fluorescent probe, Aβ(1–16)(Y10W), by substituting the tyrosine residue at position 10 in the hydrophilic domain of Aβ(1–42) with tryptophan. Upon mixing Cu(II), Aβ(1–16)(Y10W), and aliquots of Aβ(1–42) taken from samples incubated for different lengths of time, we found that the Cu(II) binding strength of aggregated Aβ(1–42) has been elevated by more than two orders of magnitude with respect to that of monomeric Aβ(1–42). Electron paramagnetic spectroscopic measurements revealed that the Aβ(1–42) aggregates, unlike their monomeric form, can seize copper from human serum albumin (HSA), an abundant copper-containing protein in brain and cerebrospinal fluid. The significantly elevated binding strength of the Aβ(1–42) aggregates can be rationalized by a Cu(II) coordination sphere constituted by three histidines from two adjacent Aβ(1–42) molecules. Our work demonstrates that the copper binding affinity by Aβ(1–42) is dependent on its aggregation state and provides new insight into how and why senile plaques accumulate copper in vivo.
INTRODUCTION
Senile plaques, comprising mainly amyloid β (Aβ) peptides1, 2 and constituting one of the pathological hallmarks of Alzheimer’s disease (AD), accumulate abnormally high concentrations of metals (e.g., Cu(II) at ~0.4 mM and Fe(III) at ~0.9 mM)3 in AD-afflicted brains. Efforts to understand the interaction between metals and Aβ have been made to correlate the presence and amounts of metals to AD pathogenesis.4, 5 In particular, copper has received a considerable amount of attention in the field, owing to its involvement in redox cycling to produce reactive oxygen species (ROS) and to exert possible oxidative damage in AD.4–13 A Raman spectral analysis of postmortem tissues from AD brains revealed that methionine residues of Aβ peptides are oxidized and the oxidation was attributed to ROS facilitated by copper redox cycling.14 Nevertheless, the origin and driving force behind copper accumulation in senile plaques have remained entirely unclear. At odds is that monomeric Aβ peptides bind copper with a moderate affinity constant (109–1010 M−1; or a dissociation constant Kd around nM).15–18 Such a binding strength is not sufficiently high for Aβ monomers to sequester copper from copper-transporting proteins.19, 20 Human serum albumin (HSA) is the major copper chelator and transporter protein in blood plasma,19, 20 with a Ka value around 1012 M−1 or a dissociation constant Kd value in the pM range.21, 22 It is also the major protein in the cerebrospinal fluid (CSF) that is known to be in contact with senile plaques.23 Although HSA is a viable source of copper, a recent study by Faller and coworkers showed that in vitro copper bound by monomeric Aβ can be readily taken out by HSA.24 Thus, the Aβ monomer is clearly not responsible for the copper accumulation in senile plaques. The Kd value of monomeric Aβ(1–42) was measured by Serell et al. to be 60 pM,18 which is substantially (1–5 orders of magnitude) lower than values determined by other studies (cf. Table S1 in the Supporting Information for a comprehensive list of the Kd values reported in the literature). Even such a binding strength was considered to be insufficient to seize copper from HSA.18
The high abundance of Aβ fibrils in senile plaques suggests the possibility that Aβ aggregates might possess a higher strength in binding copper and consequently be involved in the sequestration and enrichment of copper in senile plaques. The finding that senile plaques can be dissolved by strong copper chelators implies that copper helps stabilize Aβ aggregates.25 Thus far, there is a lack of direct evidence showing the aggregation-dependent enhancement of the copper binding strength of Aβ. It is worth noting that zinc, another metal accumulated in senile plaques, appears to bind to Aβ aggregates at a slightly higher (approximately two fold) affinity than to the Aβ monomers.26, 27
Here we report the use of a non-aggregating, copper-binding Aβ mutant, Aβ(1–16)(Y10W) (Table 1), as a fluorescent probe to monitor the elevated copper binding strength of aggregated Aβ. The intrinsic Tyr residue at position 10 of Aβ(1–42) has been widely used for fluorescence measurements of the copper binding constants of various Aβ peptides (cf. Table S1 and references therein). As shown in this work, substitution of the Tyr-10 of Aβ(1–16) with a Trp residue affords a much higher fluorescence quantum yield and a different wavelength from that of native Aβ(1–42). Furthermore, unlike Aβ(1–42), such a probe does not aggregate and become insoluble when complexed with Cu(II). Consequently, in competitive binding assay, the use of a soluble probe with a similar Cu(II) binding strength should be largely free of interferences such as loss of fluorescence accompanied by sedimentation of Aβ(1–42) aggregates or light scattering by suspended Aβ(1–42) aggregates. Owing to these advantages, competitive Cu(II) binding assays between this probe and Aβ(1–42) allowed us to observe a dramatic elevation of the apparent binding constant of Aβ(1–42) aggregates over its monomeric counterpart. This finding is further supported by electron paramagnetic resonance (EPR) measurements of copper sequestration from HSA by Aβ(1–42) aggregates. The elevation in the copper binding strength could be rationalized by a structure in which three histidine residues from two different Aβ molecules and the C-terminal carboxylic group in the Aβ aggregates could also provide a substantially stronger copper coordination sphere that is otherwise unavailable in the Aβ monomers.
Table 1.
Aβ peptides used in the study and their sequences+
| Peptide | Sequence |
|---|---|
| Aβ(1–16)(Y10W) | DAEFRHDSGWEVHHQK |
| Aβ(1–42) | DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAI IGLMVGGVVIA |
The boldface in the probe indicates the mutation of Tyr-10 with Trp and the hydrophobic segment of Aβ(1–42) is underlined.
METHODS
Chemicals and reagents
Aβ(1–42) was obtained from American Peptide Company Inc. (Sunnyvale, CA), and Aβ(1–16) and Aβ(1–16)(Y10W) were obtained from rPeptides Inc. (Bogart, GA). Aβ(1–16)(Y10W) synthesized in house with an automatic peptide synthesizer (Symphony Quartet, Tucson, AZ) was also used for some of the experiments. Acid- and globulin-free HSA (99% purity), sodium hydroxide, sulfuric acid, CuCl2, copper acetate, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and other chemicals were acquired from Sigma-Aldrich (St. Louis, MO). All of the aqueous solutions were prepared using water purified by a Simplicity® Water Purification System (Millipore, Bellerica, MA) to a resistivity of 18 MΩ·cm.
Sample preparation
Aβ(1–16) and Aβ(1–16)(Y10W) stock solutions (1–2 mM) were prepared by directly dissolving the lyophilized samples in 10 mM NaOH. They were then diluted with 10 mM HEPES buffer (pH 7.4) to desired concentrations. Prior to use, the Aβ(1–42) sample was treated with hexafluoroisopropanol (HFIP) to dissolve any pre-existing oligomers, and the solution was centrifuged for 30 min at 13000 rps to decant undissolved oligomers. The final solution was then lyophilized on a freeze-drier (VirTis Benchtop K, SP Scientific, Gardiner, NY). The Aβ(1–42) solutions were then prepared with our previously reported procedure.8, 9 Samples treated with the method by Teplow and coworkers28 were also used and did not show obvious difference than the HFIP-treated samples. The concentration of soluble peptides was determined with the UV-vis absorbance at 280 nm and the extinction coefficients of tryptophan (ε279 = 5400 cm M−1 for Aβ(1–16)(Y10W)) and tyrosine (ε276 = 1410 cm M−1 for Aβ(1–16) and Aβ(1–42)). To avoid denaturation, a high-purity HSA sample (99%) was accurately weighed to prepare fresh and concentrated stock solutions, and mixtures of HSA, Cu(II), and various Aβ(1–42) peptides were used immediately for the competitive EPR assays. The Cu(II) stock solution was prepared by dissolving 1 mM CuCl2 or copper acetate in 1 mM H2SO4 solution.
Fluorescence spectroscopy
Fluorescence measurements of Aβ(1–16) and Aβ(1–16)(Y10W) peptide solutions were carried out at room temperature using a Cary Eclipse spectrofluorimeter (Agilent Technologies, Santa Clara, CA). An excitation wavelength (λex) of 280 nm was used, and fluorescence intensities were recorded for Aβ at 307 nm and for Aβ(1–16)(Y10W) at 360 nm. The entrance and exit slits were 10 and 5 nm, respectively. Fluorescence measurements were performed at least three times, and the standard deviations were plotted as the error bars. For determining apparent Cu(II) binding constants of aggregated Aβ(1–42), samples were prepared following two procedures. In the first procedure, 1.5 mL of 12.5 μM Aβ(1–42) was prepared and incubated in a 37 °C water bath. An aliquot was removed from the incubation solution, mixed with equimolar Aβ(1–16)(Y10W) and/or Cu(II) before the fluorescence intensities of Aβ(1–16)(Y10W) were measured. The final Aβ(1–16)(Y10W) and Cu(II) concentrations were both 12.5 μM and the sampling times were 0, 20 min, 50 min, 3 h, 24 h and 48 h. In the second procedure, 12.5 μM Cu(II) and 12.5 μM Aβ(1–42) in 1.2 mL HEPES buffer was co-incubated. At different times of incubation aliquots of the mixture were withdrawn and mixed with 12.5 μM probe. The fluorescence intensity was monitored incrementally with a higher sampling frequency at the beginning of the aggregation. In both cases, to avoid light scattering by suspended aggregates during the fluorescence measurement, after the mixtures were allowed to stand for 10 min for equilibration, they were centrifuged and the supernatants were analyzed.
Atomic force microscopy
AFM images were obtained in air on an MFP-3D-SA microscope (Asylum Research, Santa Barbara, CA) equipped with the tapping mode. The AFM probes (MikroMasch, San Jose, CA) are single beam cantilevers. Aliquots were taken out of an Aβ(1–42) solution or an Aβ(1–42)/Cu(II) mixture at predetermined incubation times, cast onto Ni(II)-treated mica sheets, and kept in contact with the mica sheets for 15 min. Afterwards, the sheets were gently rinsed with water to remove salt, and then dried with nitrogen.
Size exclusion chromatography
Size exclusion chromatography was performed with a Waters 600 HPLC system equipped with a photodiode array detector (Model 2996). The mobile phase was 100 mM phosphate buffer (pH 7.4) and the flow rate was 0.2 mL/min. Two columns (GFC 2000 from Phenomenex Inc., Los Angeles, CA) were connected in series and retention times were calibrated with the following five standards: blue dextran (2000 kD), bovine serum albumin (66 kD), chymotrypsinogen A (25 kD), aprotinin (6.7 kD), and vitamin B12 (1.35 kD). From a 600 μL Aβ(1–42) solution incubated in a 37 °C water-bath, a 20-μL aliquot was taken at each specific incubation time and then injected into the HPLC columns. Elution of Aβ species was monitored with the detector set at 220 nm.
Electron paramagnetic resonance spectroscopy
EPR spectra were recorded using an X-band (~ 9.4 GHz) spectrometer (Elexsys E580) equipped with an HSQE resonator and a temperature controller (Oxford Instruments, Abingdon, England). EPR spectra were obtained at 123 K using N2 gas flow with a microwave power of 1 mW and a modulation amplitude of 5 G. The concentration of Cu(II) bound by HSA at pH 7.4 is proportional to the integrated peak area in the CW-EPR absorption spectrum. A control experiment performed in a 60 μM HSA solution showed rather small Cu(II) EPR peaks (<5% of HSA fully loaded with Cu(II)), suggesting that the HSA samples we prepared were largely free of copper contamination. To ensure that free Cu(II) was absent in the mixture of HSA and Cu(II), the Cu(II)/HSA ratio was maintained at 0.8/1. The frozen sample proportionality constant for Cu2+ was determined via titrations of CuCl2 with an excess of EDTA and 25% (V/V) glycerol. EPR spectra were baseline-corrected and peaks were integrated with LabVIEW 7 Express software (National Instruments, Austin, Texas). A sample of 76 or 306 μM Aβ(1–42) was loaded in an EPR quartz tube (4 mm O.D. and 3 mm I.D.) and incubated for 6 and 24 h at 37 °C. The solution was then mixed with the Cu(II)–HSA complex. Cu(II)–HSA was added to the pre-incubated Aβ(1–42) so that the final concentrations of HSA, Cu(II), and Aβ(1–42) were 61, 49, and 76 or 306 μM, respectively. All solutions were mixed thoroughly, sonicated for 2 min, and flash-frozen in liquid N2.
RESULTS
Aβ(1–42) aggregates have a substantially elevated copper binding strength in comparison with the monomer
The black curve in Figure 1A corresponds to the fluorescence spectrum of the Aβ(1–16)(Y10W) probe, which clearly exhibits an emission peak at 360 nm. With the addition of Cu(II), the Trp fluorescence of the probe is decreased (Figure S1A). This behavior is highly comparable to that of Aβ(1–16) and Aβ(1–42),15, 29 although the fluorescence signal is much stronger than the emissions of Aβ(1–16) and Aβ(1–42) at 307 nm. The Cu(II) binding constant of the probe can be measured by fitting the curve of fluorescence plotted against Cu(II) concentration (cf. Figure S1B and details of the fitting procedure in the Supporting Information).15, 29 When an equimolar amount of a freshly prepared Aβ(1–42) sample was mixed with the probe and Cu(II), about 50% of the fluorescence was quenched (red curve in Figure 1A). This suggests that the Cu(II) ions are equally distributed between Aβ(1–42) and the probe. The equal distribution is understandable since the Cu(II) binding strength of the probe is similar to that of Aβ(1–42) (cf. Figure S1 and description). Surprisingly, the fluorescence signal was partially recovered when Aβ(1–42) was pre-incubated for 20 min before being mixed with the probe (green curve in Figure 1A). More fluorescence recovery was observed after the Aβ(1–42) solution had been pre-incubated for a longer time (1440 min; blue curve in Figure 1A). In contrast, when the non-aggregating Aβ(1–16) sample was pre-incubated and then added into the solution, little recovery was observed (green and blue curves in Figure 1B). Since Aβ(1–42) is prone to rapid aggregation, the fluorescence recovery must have resulted from a stronger copper binding strength of Aβ(1–42) oligomers and other higher-ordered aggregates. To confirm the fidelity of using Aβ(1–16)(Y10W) to report on Cu(II) transfer, we added EDTA, a much stronger Cu(II) chelator, into a mixture of Aβ(1–16)(Y10W) and Cu(II) and observed a complete recovery of the probe fluorescence (Figure S2). We therefore conclude that from our experiments that, as the aggregation proceeds, Cu(II) initially bound by the probe is lost to the Aβ(1–42) aggregates.
Figure 1.
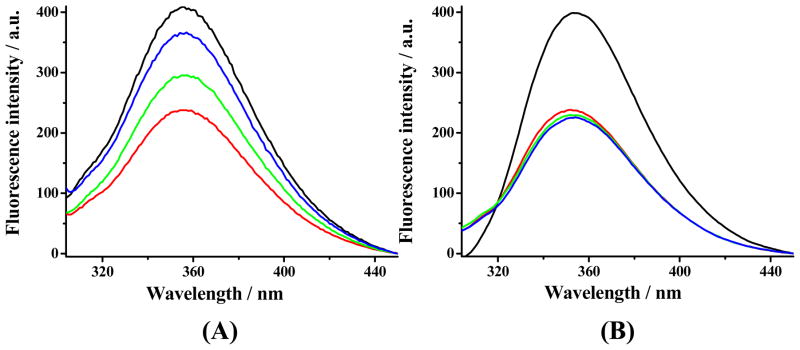
(A) Fluorescence spectra of the Aβ(1–16)(Y10W) probe without (black) and with additions of Cu(II) and aliquots of Aβ(1–42) taken from solutions incubated at 37 °C for 0 (red), 20 (green) and 1440 min (blue). (B) Control experiments using Aβ(1–16) instead of Aβ(1–42). The concentration of each species in the final solution was 12.5 μM.
We can define the collective binding strength of all Aβ(1–42) species (monomers and aggregates) to Cu(II) with the apparent (overall) binding constant, K:
| (1) |
| (2) |
where K′ is the Cu(II) binding constant of Aβ(1–16)(Y10W), which remains unchanged with time (cf. Figure S1). Since the binding constant of HEPES to copper (pK = 3.22)30 is six orders of magnitude smaller than that of the probe (pK = 9.49) and the Aβ peptides are in excess with respect to the Cu(II) concentration, the effect of HEPES buffer can be neglected.31 Let θw be the percentage of Aβ(1–16)(Y10W) bound to Cu(II) and the initial (formal) concentrations of Aβ(1–42), Aβ(1–16)(Y10W), and Cu(II) be A, L0 and C, respectively. K is related to K′ and θw as follows (cf. Supporting Information for the detailed derivation and the correction of the buffer effect in Figure S3):
| (3) |
At any given incubation time, θw can be readily obtained from the percentage of quenched fluorescence of the probe, (F0 is the fluorescence intensity of the probe-only solution and FL the fluorescence intensity of the probe in a mixture). Although the apparent binding constant varies with the Aβ(1–42) aggregation or incubation time, the formal concentration of the Aβ(1–42) monomer at the inception of incubation is known. Hence, using eq. 3, the apparent binding constant of a mixture of Aβ(1–42) aggregates and monomers can be accurately determined.
We took aliquots from an Aβ(1–42) solution incubated for different times and mixed them with the probe and Cu(II). Figure 2A reveals that the apparent binding constant of Aβ(1–42) aggregates has increased by ca. 20 times over a 24 h incubation and more than two orders of magnitude (ca. 120) over a 48 h incubation.
Figure 2.

Changes of apparent copper binding constant (A), morphology of Aβ(1–42) aggregates (inset), and distribution of copper (B) between the probe and Aβ(1–42) pre-incubated for different times. Cu(II) and the probe (12.5 μM each) were mixed with aliquots of Aβ(1–42) taken out from a continuously incubated solution. The formal (initial) Aβ(1–42) concentration in the incubated solution was 12.5 μM. In (B), the percentage of transfer is (θw0 − θwt)/θw0. The AFM images in the inset have areas of 5 μm × 2.5 μm.
Corresponding to the change in the apparent binding constant, atomic force microscopic (AFM) images (inset of Figure 2A) clearly display changes in the Aβ(1–42) aggregate morphology expected from the main Aβ(1–42) oligomerization/fibrillation pathway.32–34 In this pathway, the aggregation process begins with the formation of globular oligomers (cf. AFM images for 20 and 50 min of incubation), some of which subsequently transform into protofibrils (AFM image for 180 min incubation) and fibrils (images for 1440 and 2880 min). We also verified that the presence of the Aβ(1–16)(Y10W) does not alter the Aβ(1–42) aggregation/fibrillation pathway (cf. Figure S4). In addition, we followed the Aβ(1–42) aggregation/fibrillation process with the ThT assay (cf. Figure S5). Notice that the plateau in Figure S5 (ca. 24 h) is consistent with the time when fibrils were observed by AFM (cf. the inset of Figure 2 at 1440 min). The trend depicted in Figure S5 is in contrast to that in Figures 2, which does not show a lag phase. Therefore, it is evident that oligomers formed soon after the incubation begin to bind Cu(II). Furthermore, oligomers, protofibrils, and fibrils of Aβ(1–42), with a conformation different from that of the natively unstructured Aβ(1–42) monomer, possess a substantially stronger Cu(II) binding strength. We also plotted the percentage of Cu(II) transferred from the probe to Aβ(1–42) over the entire incubation period (Figure 2B) to illustrate the extent of copper transfer resulting from the elevated copper binding strength.
While the AFM image (inset of Figure 2) of the sample incubated for 20 min shows only a few globular oligomers, Figure 2B indicates that a considerable amount of Cu(II) (~20% of that originally bound by the probe) was lost. Thus, Cu(II) must be partially transferred to low-mass oligomers (i.e., dimers and pentamers), which are too small to be observed by AFM. The low-mass oligomers can be readily identified by size exclusion chromatography (Figure 3). Consistent with other reports,32, 35 the amount of soluble oligomers decreases with incubation time, while insoluble globular oligomers are formed as incubation prolongs (cf. the red and blue curves in Figure 3). Even after one day of incubation (yellow curve at 1440 min), a trace amount of monomers is still present in the solution, suggesting that Aβ fibrils remain in equilibrium with the monomeric Aβ. Overall, our results clearly indicate that the elevation of the Cu(II) binding strength in the incubated solution is proportional to the number of β-sheets inherent in all of the Aβ(1–42) oligomers (including the dimers) and aggregates.
Figure 3.

Size-exclusion chromatograms of 80 μM Aβ(1–42) solutions incubated for different times, with the monomer (~4.5 kD), dimer (~9 kD), pentamer (~20 kD), and a soluble oligomer (740 kD) identified. The small peak corresponding to 1.3–2 kD was verified by mass spectrometry as a cluster of minor impurities in the synthetic Aβ(1–42) sample. Notice that during the ca. 40-min separation of a fresh Aβ(1–42) solution (i.e., no incubation), a noticeable amount of dimers and pentamers had already formed.
Binding constants of Aβ(1–42) at different incubation times with Cu(II) present initially
We also co-incubated Cu(II) and Aβ(1–42) for different times. At predetermined incubation times, aliquots from this mixture were withdrawn and mixed with the probe for fluorescence measurements. Figure 4 shows the changes in the copper binding strength obtained by processing the fluorescence recovery data, together with the AFM images of the aggregates and distribution of copper between the probe and Aβ(1–42). Remarkably, both the trend and extent of changes in the K values (Figure 4A) are analogous to those in Figure 2A, even though amorphous aggregates are the predominate species at and beyond 3 h of incubation (inset of Figure 4A). Our observation is consistent with previous finding that copper binding to Aβ(1–42) alters the main aggregation pathway of Aβ(1–42) from fibrillation to the formation of amorphous aggregates.36 Nevertheless, the comparability of the K values deduced at intermediate and extended incubation times between Figures 2A and 4A suggest that the amorphous aggregates bind to Cu(II) almost as strongly as protofibrils and fibrils. Despite the current debate regarding whether fibrils evolve out of amorphous intermediates37 or are produced as a result of stacking of oligomers,37 it is believed that amorphous aggregates contain misfolded and β-sheet-rich Aβ(1–42) molecules.38 Again, the results in Figure 4 support our earlier contention that formation of β-sheets during the Aβ(1–42) aggregation is crucial to the elevation of the copper binding strength. We also conducted an experiment by continuously monitoring the fluorescence signals in a single solution of Cu(II), Aβ(1–16)(Y10W), and Aβ(1–42). The fluorescence recovery monitored as a function of incubation time is shown in Figure S6. The good comparability between Figure 4A and Figure S6 suggests that Aβ(1–16)(Y10W) does not alter the interaction of Cu(II) with Aβ(1–42) monomer and aggregates. In a separate experiment, we found that co-incubating Aβ(1–16)(Y10W) with Aβ(1–42) and Cu(II) does not alter the Aβ(1–42) aggregation pathway (i.e., amorphous aggregate formation caused by Cu(II); cf. Figure S7). However, monitoring the fluorescence recovery in a single solution prevents us from centrifuging the solution to remove large aggregates that can scatter light. As a consequence, light scattering (much like in the fluorescence scattering measurement of aggregation of amyloidogenic molecules34) is more serious in the solution beyond 24 h. Thus, we believe that the apparent binding constants deduced from Figure 4A are more reliable. Finally, we should add that, if all of the monomers in the incubated solution had been converted into fibrils or amorphous aggregates, the K values would be even greater than those shown in Figure 2 or Figure 4. Since in the yellow curve of Figure 3 monomers were still observed after 24 h of incubation, it is clear that the incubated solutions used to obtain Figures 2 and 4 were mixtures of Aβ(1–42) monomers, oligomers, and large aggregates.
Figure 4.

Changes of copper binding strength, morphology of the aggregates, and distribution of copper between the probe and Aβ(1–42) co-incubated for different times. (A) K values of Aβ(1–42) at various incubation times when Cu(II) was present from the beginning. Equimolar amounts (12.5 μM) of Aβ(1–42), Aβ(1–16)(Y10W), and Cu(II) were first mixed, followed by incubation at 37 °C for different times. (B) Percentages of Cu(II) transferred from the probe to Aβ(1–42) at different incubation times. Inset: aggregates imaged by AFM at the corresponding incubation times (image area = 5 μm × 2.5 μm).
Aβ(1–42) aggregates are capable of sequestering Cu(II) from HSA
With electron paramagnetic resonance (EPR), we investigated whether any Cu(II) transfer from HSA to Aβ aggregates is possible. Shown on the left of Figure 5 are EPR spectra of the Cu(II)–HSA complex (curve A), a freshly prepared Aβ(1–42) solution mixed with Cu(II)–HSA (curve B), and Cu(II)–HSA mixed with Aβ(1–16) (curve C). We have also obtained an EPR spectrum from a solution comprising Cu(II)–HSA and preformed Aβ(1–42) oligomers and protofibrils (curve D) as well as a spectrum of Cu(II) complexed with Aβ(1–42) oligomers and protofibrils (curve E). The right panel of Figure 5 is an overlay of EPR spectra A, D and E, which clearly exhibits the differences in g⊥ and g// peaks among the three solutions. The line shape of and parameters calculated from curve (A) (g// = 2.160, A// = 182 G for component 1) are consistent with those previously reported for the Cu(II)–HSA complex,24, 39, 40 with two negative peaks at 3329 and 3423 G and two distinctive hyperfine peaks. Notice that curves B and C are highly comparable to curve A, suggesting that neither Aβ(1–16) nor the Aβ(1–42) monomer can remove Cu(II) from HSA, a result expected from the aforementioned large difference in the binding strengths between monomeric Aβ and HSA. These data are also in line with the report by Faller’s group who showed that both monomeric Aβ(1–40) and Aβ(1–16) cannot seize Cu(II) from HSA.24 However, with the addition of a pre-incubated Aβ(1–42) solution into a Cu(II)–HSA solution, the negative peak at 3329 G increases at the expense of the negative peak at 3423 G (cf. right panel of Figure 5), concurrent with the appearance of three new hyperfine peaks (g// = 2.275 and A// = 168 G; cf. curve D for component 2 in the right panel of Figure 5). The parameters of these new peaks are consistent with those of the copper complex of aggregated Aβ(1–42) shown in curve E.41, 42 Therefore our data clearly demonstrate that pre-incubated Aβ(1–42) sample can sequester copper from the Cu(II)–HSA complex.
Figure 5.

Left: EPR spectra of (A) Cu(II)–HSA, (B) a mixture of Cu(II)–HSA and freshly prepared Aβ(1–42), (C) a mixture of Cu(II)–HSA and Aβ(1–16), (D) a mixture of Cu(II)–HSA and Aβ(1–42) aggregates (6 h incubation), and (E) the complex formed between Cu(II) and aggregated Aβ(1–42) (6 h incubation). Right panel: an overlay of spectra A, D, and E with the hyperfine peaks of the Cu(II)–Aβ(1–42) complex denoted as component 2 and Cu(II)–HSA as component 1. The arrow indicates the decrease of the negative peak at 3423. The concentrations of Cu(II), HSA, and Aβ were 49, 61, and 306 μM, respectively. Aggregation of Aβ(1–42) was performed by placing the sample inside the EPR tube and incubating it at 37 °C for 6 h.
We also estimated the copper binding constant (K) of aggregated Aβ(1–42) by deconvoluting the two components in curve D to yield a ratio of Cu(II) bound to HSA over that complexed by the Aβ(1–42) aggregates. An increase in the copper binding strength by about one order of magnitude was observed for Aβ(1–42) aggregates after a 6 h incubation (Table S2 in the Supporting Information), consistent with the results obtained by our fluorescence measurement. Both the EPR spectra shown in Figure 5 and the binding constants estimated from the competitive EPR assay (Table S2) have unequivocally validated the significantly elevated copper binding strength of aggregated Aβ(1–42).
Finally, we should add that, if all of the monomers in the incubated solution had been converted into fibrils or amorphous aggregates, the apparent affinity constants exhibited in Figure 2 or Figure 4 would be even greater. Since in Figure 3 Aβ(1–42) monomers were still observable even after 24 h of incubation, it is clear that the solutions shown in Figures 2 and 4 contained monomers at the beginning and mixtures of Aβ(1–42) monomers, oligomers, and large aggregates at later stages of the incubation. Thus, the affinity constants we measured only reflect portions of the true Cu(II) sequestration power of the Aβ(1–42) fibrils. Notice that in Figure 2A, the dissociation constant converted from the K value at 2880 min is about 3 pM, which is close to the Kd value of the Cu(II)–HSA complex.21 In Figure 5, the changes in the EPR spectra and parameters are likely to be caused solely by the Aβ(1–42) fibrils whose actual Kd value should be less than 3 pM.
DISCUSSION
Employing two independent methods (fluorescence and EPR competitive binding assays), we demonstrate that Aβ(1–42) oligomers and aggregates bind copper more strongly than their monomeric counterpart. Our finding contradicts the recent study by Sarell et al. who reported a dissociation constant (Kd) of ca. 60 pM for both monomeric and aggregated Aβ(1–42).18 Such a Kd value is at least more than an order of magnitude lower than values reported by many other studies for Aβ monomers even after buffer correction (see Table S1). As shown by our comparative studies detailed in the Supporting Information (cf. Figures S8, S9, and S10), the discrepancy stems from the uncertainties inherent in the Tyr-10 fluorescence measurement as well as in the sample preparative method. Specifically, changes in the Tyr-10 fluorescence of Aβ(1–42) at 307 nm can be caused by both Cu(II) binding and the precipitation and suspension of the Cu(II)-containing Aβ(1–42) aggregates in solution. The effects of the aggregate precipitation and suspension are particularly acute when large and insoluble aggregates are formed. The former process decreases the fluorescence intensity, while the latter process scatters light to cause signal fluctuation. When the relatively weak Tyr fluorescence is quenched to a low level, the interference of the adjacent Raman peak of water (313 nm) becomes more pronounced (cf. Figure S10A) and can also affect the accurate measurement of the fluorescence intensity. Our method allows the removal of the Cu(II)-containing Aβ(1–42) aggregates prior to the fluorescence measurements. Moreover, the fluorescence quantum yield of Trp is much higher than that of Tyr and Trp fluoresces at a longer wavelength. Consequently, the water Raman peak does not interfere (cf. Figure S10B) with the fluorescence of the Aβ(1–16)(Y10W) probe. We should also caution that sample pretreatment is crucial to the accurate measurement of the Cu(II)-binding affinity values of Aβ(1–42). We noticed that the pretreatment procedure employed in the work of Viles and coworkers is different from procedures used by other studies.18 We found that after such a pretreatment Aβ(1–42) samples generally contain a large number of oligomers (cf. Figure S8). These samples behave differently from those treated with the HFIP method.28, 43 As shown in Figure S9, the presence of oligomers at the beginning of Cu(II) addition results in the appearance of a higher Cu(II) binding strength. In other words, if in the initial solution oligomers are present at a high concentration, there will be little difference in the binding affinity values between the presumed “Aβ(1–42) monomers” and the actual oligomers. This factor, together with effects from other experimental conditions (buffer, pH, concentrations of competitive ligands, etc.; cf. the comprehensive review by Faller and co-workers31) must be taken into account in deducing the final affinity constants.
The metal binding strength increase in oligomers (including dimer) and other higher-ordered aggregates of Aβ(1–42) most likely originate from a tighter coordination structure that cannot be rendered by the monomer. His-6, His-13 or His-14, and the N-terminus are the three N-containing moieties that account for the 3N1O coordination sphere identified by EPR in the Cu(II) complex of the Aβ monomer.44, 45 Because His-13 and His-14 are adjacent to each other, they are on the opposite sides of the peptide backbone. Such an orientation prevents them from simultaneously participating in a tight Cu(II) binding when only one Aβ(1–42) monomer is involved.44, 46 Aβ(1–42) dimers are formed with a β-sheet comprising two pairs of β-strands withheld by hydrogen bonding.47, 48 Consequently, all of the metal binding sites are located in the N-termini that extend out of the β-sheet. Larger oligomers, protofibrils, and fibrils are produced from stacking of the β-sheet-containing oligomers (or “aggregation units”),49, 50 again positioning all of the hydrophilic N-termini on the same side of the stacked β-sheets.49 Although amorphous aggregates lack higher regularity for ordered β-sheet stacking, they have been shown to contain many β-sheets.38 Therefore, a plausible cause for the enhanced Cu(II) binding strength in both ordered and amorphous Aβ aggregates is that more histidine residues (one of the strongest metal-binding ligands among amino acid residues51) have become available for Cu(II) binding and are favorably spaced out to coordinate Cu(II). Binding by three histidine residues is a more stable configuration than those in other modes.52 As shown in our proposed model (Figure 6), upon aggregation His-13 in one Aβ molecule and His-14 from another can concurrently coordinate Cu(II), tightly locking the Cu(II) ion in position. In addition, the C-terminal carboxylic group is folded back and has been shown to provide additional ligation.52 Such a Cu(II) coordination sphere is also unlikely to exist in the monomeric form. Other possibilities can also contribute to the enhanced Cu(II) binding. For example, the ligand crowding brought about by the β-sheet formation could allow some amide groups or other dangling carboxylic groups to participate in additional binding. We also cannot rule out the possibility that the binding strength increase is caused by the change near the copper center from a more hydrophilic environment in the Aβ monomers to the more hydrophobic setting in the Aβ aggregates. The rather small increase (almost within experimental error) in the Zn(II) binding strength from Aβ monomers to aggregates26, 27 might also be resulted from one or more of the factors mentioned above.
Figure 6.

A model proposed for the Cu(II) coordination sphere in Aβ(1–42) oligomers and higher-ordered aggregates: Histidine residues from two different Aβ(1–42) molecules (denoted as Aβ1 and Aβ2, respectively) provide three of the four Cu(II)-binding ligands. One histidine is shown in purple while the other two in an adjacent Aβ(1–42) molecule in the same aggregate are depicted in grey.
The intermolecular coordination model posited above is supported by a growing body of evidence. For example, Pedersen et al. suggested that, unlike soluble Aβ(1–16), Aβ(1–40), which is also prone to β-sheet formation and aggregation, could rapidly form the Aβ–Cu(II)–Aβ linkage prior to oligomerization.53 We hypothesize that such a linkage might be preserved in the copper-complexing Aβ(1–42) oligomers and aggregates. Szalai and coworkers, using X-ray absorption spectroscopy, demonstrated that the Cu(II) coordination sphere in oligomers is different from that in monomers. Specifically, only two histidines participate in the Cu(II) binding, whereas three histidines are involved in the copper coordination by Aβ oligomers54 (even though it is not clear whether the three histidines are from the same or different Aβ molecules). From the present study, it is evident that the rapid formation of small, soluble oligomers (cf. chromatograms in Figure 3) synchronizes with the copper transfer from the probe to Aβ(1–42) aggregates.
The significantly elevated Cu(II) binding strength of Aβ aggregates has two major biological implications. First, it has been long proposed that overproduction of Aβ peptides (e.g., in the case of early onset AD55) and inefficient clearance are at least partially responsible for the Aβ accumulation/deposition and aggregation in vivo. Even when the regulation of cellular Cu(II) is normal, the higher Cu(II)-binding strength inherent in Aβ oligomers and fibrils due to Aβ overproduction and/or inefficient clearance would facilitate and aggravate Cu(II) sequestration from copper-transporting proteins such as HSA, which may explain why there exists a high level of copper in senile plaques.
The second implication is related to the high toxicity of diffusible oligomers of Aβ56, 57 and their enhanced copper binding strength. In normal brain Aβ monomer and its oligomeric forms are in an equilibrium and their relative abundance is dependent on how each species is metabolized (cleared). When copper homoeostasis is disrupted in AD,58 excess copper59 in cellular milieu is capable of binding Aβ oligomers and stabilizing the Aβ aggregates. Consequently, the equilibrium between the Aβ monomer and its aggregates is shifted to the direction of Aβ oligomerization/aggregation. Since the Aβ–Cu(II) complex catalyzes the ROS generation,5, 9, 10 the copper-containing oligomers could potentially behave as a mobile ROS generator, leading to an extensive oxidative stress that is commonly found in AD brain.14, 60 Given the significant role of albumin in regulating copper in normal brain and the possible link between albumin deficiency and AD development,40, 61, 62 in vivo Aβ aggregation may be triggered by albumin deficiency and/or its lower copper chelating capacity.59, 63 This is supported by an interesting fact that 60–90% of AD cases are associated with cerebral ischemia,64, 65 which has been shown to decrease the metal binding capacity of albumin.66 As a consequence, transfer of copper from albumin to Aβ aggregates is likely to be favored. Indeed, treatment of AD patients with plasma exchange, in which their albumin is replaced with that in donors’ plasma, is undergoing clinical trials and has shown promising results.67
CONCLUSION
In summary, our fluorescence and EPR measurements both clearly demonstrate that Aβ(1–42) oligomers and aggregates bind copper much more strongly than their monomeric counterpart. The novel application of a highly fluorescent, Aβ-derived probe enabled us to interrogate the increasing copper sequestration by aggregating Aβ(1–42) species. The Aβ(1–16)(Y10W) probe also mitigates uncertainties inherent in the use of the intrinsic Tyr-10 fluorescence of Aβ peptides for Cu(II) binding studies. The discovery of the aggregation-dependent copper binding of Aβ(1–42) suggests that Aβ(1–42) aggregates could be the species that accumulate copper and possibly other metals in senile plaques. The elevated Cu(II) binding strength of Aβ(1–42) aggregates strengthens the hypothesis of copper-induced oxidative stress in AD. Results from this study also manifest the importance of intact albumin and a possible linkage between disruption of copper homeostasis and AD pathology.
Supplementary Material
Table 2.
EPR parameters extracted from Figure 5
| Sample | Sample Description | A// | g// | g⊥ |
|---|---|---|---|---|
| A | Cu(II)–HSA | 182 G (1) | 2.160 (1) | 2.052 |
| B | Freshly prepared Aβ (1–42) + Cu(II)–HSA | 182 G (1) | 2.160 (1) | 2.052 |
| C | Aβ(1–16) + Cu(II)–HSA | 182 G (1) | 2.159 (1) | 2.052 |
| D | Cu(II)–HSA + Aggregated Aβ(1–42) (6 h incubation | 182 G (1) 168 G (2) |
2.161 (1) 2.275 (2) |
2.055 |
| E | Cu(II) + Aggregated Aβ(1–42) (incubated for 6 h) | 168 G (2) | 2.278 (2) | 2.059 |
The numbers in the parentheses denote components 1 and 2.
Acknowledgments
Funding: The work is partially supported by the National Institutes of Health (SC1NS070155-01 to FZ and GM065790 to GLM) and an NSF-RUI grant (No. 1112105 to FZ).
A portion of this research was performed with help from Dr. Eric Walter using EMSL, a national scientific user facility sponsored by the Department of Energy’s Office of Biological and Environmental Research and located in the Pacific Northwest National Laboratory (PNNL). We gratefully acknowledge Dr. Peter Z. Qin for the access to the EPR instrument at University of Southern California. We also thank Mr. Ding Li for drawing the model shown in Figure 6.
ABBREVIATIONS
- Aβ
Amyloid-β
- AD
Alzheimer’s disease
- ROS
Reactive oxygen species
- CSF
Cerebrospinal fluid
- HSA
Human serum albumin
- EPR
Electron paramagnetic resonance
- HEPES
4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid
- AFM
Atomic force microscopy
Footnotes
Notes:
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information: Detailed derivation of eq. 3, measurement of Ka of Aβ(1–16)(Y10W), effects of Aβ(1–42) sample treatments and the fluorescence measurement of Tyr-10 on accuracy of the Ka values and a Table listing Ka values of representative Aβ peptides. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298(5594):789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 2.Hardy JA, Higgins GA. Alzheimer’s disease: The amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 3.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 4.Smith DP, Smith DG, Curtain CC, Boas JF, Pilbrow JR, Ciccotosto GD, Lau TL, Tew DJ, Perez K, Wade JD, Bush AI, Drew SC, Separovic F, Masters CL, Cappai R, Barnham KJ. Copper-mediated amyloid-β toxicity is associated with an intermolecular histidine bridge. J Biol Chem. 2006;281:15145–15154. doi: 10.1074/jbc.M600417200. [DOI] [PubMed] [Google Scholar]
- 5.Huang XD, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JDA, Hanson GR, Stokes KC, Leopold M, Multhaup G, Goldstein LE, Scarpa RC, Saunders AJ, Lim J, Moir RD, Glabe C, Bowden EF, Masters CL, Fairlie DP, Tanzi RE, Bush AI. Cu(II) potentiation of Alzheimer Aβ neurotoxicity - Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem. 1999;274:37111–37116. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- 6.Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim YS, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- 7.Sparks DL, Schreurs BG. Trace amounts of copper in water induce β-amyloid plaques and learning deficits in a rabbit model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2003;100:11065–11069. doi: 10.1073/pnas.1832769100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang D, Li X, Liu L, Yagnik GB, Zhou F. Reaction rates and mechanism of the ascorbic acid oxidation by molecular oxygen facilitated by Cu(II)-containing amyloid-β complexes and aggregates. J Phys Chem B. 2010;114:4896–4903. doi: 10.1021/jp9095375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang D, Men L, Wang J, Zhang Y, Chickenyen S, Wang Y, Zhou F. Redox reactions of copper complexes formed with different β-amyloid peptides and their neuropathalogical relevance. Biochemistry. 2007;46:9270–9282. doi: 10.1021/bi700508n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guilloreau L, Combalbert S, Sournia-saquet A, Mazarguil H, Faller P. Redox chemistry of copper-amyloid-β: the generation of hydroxyl radical in the presence of ascorbate is linked to redox-potentials and aggregation state. ChemBioChem. 2007;8:1317–1325. doi: 10.1002/cbic.200700111. [DOI] [PubMed] [Google Scholar]
- 11.Hewitt N, Rauk A. Mechanism of hydrogen peroxide production by copper-bound amyloid beta peptide: a theoretical study. J Phys Chem B. 2009;113:1202–1209. doi: 10.1021/jp807327a. [DOI] [PubMed] [Google Scholar]
- 12.Hureau C, Coppel Y, Dorlet P, Solari PL, Sayen S, Guillon E, Sabater L, Faller P. Deprotonation of the Asp1-Ala2 peptide bond induces modification of the dynamic copper(II) environment in the amyloid-β peptide near physiological pH. Angew Chem Int Ed. 2009;48:9522–9525. doi: 10.1002/anie.200904512. [DOI] [PubMed] [Google Scholar]
- 13.Drew SC, Noble CJ, Masters CL, Hanson GR, Barnham KJ. Pleomorphic Copper Coordination by Alzheimer’s Disease Amyloid-β Peptide. J Am Chem Soc. 2009;131:1195–1207. doi: 10.1021/ja808073b. [DOI] [PubMed] [Google Scholar]
- 14.Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, Carey PR. Metal binding and oxidation of amyloid-β within isolated senile plaque cores: Raman microscopic evidence. Biochemistry. 2003;42:2768–2773. doi: 10.1021/bi0272151. [DOI] [PubMed] [Google Scholar]
- 15.Maiti N, Jiang D, Wain AJ, Patel S, Dinh KL, Zhou F. Mechanistic studies of Cu(II) binding to amyloid-beta peptides and the fluorescence and redox behaviors of the resulting complexes. J Phys Chem B. 2008;112:8406–8411. doi: 10.1021/jp802038p. [DOI] [PubMed] [Google Scholar]
- 16.Guilloreau L, Damian L, Coppel Y, Mazarguil H, Winterhalter M, Faller P. Structural and thermodynamical properties of Cu-II amyloid-beta 16/28 complexes associated with Alzheimer’s disease. J Biol Inorg Chem. 2006;11:1024–1038. doi: 10.1007/s00775-006-0154-1. [DOI] [PubMed] [Google Scholar]
- 17.Hong L, Bush WD, Hatcher LQ, Simon J. Determining thermodynamic parameters from isothermal calorimetric isotherms of the binding of macromolecules to metal cations originally chelated by a weak ligand. J Phys Chem B. 2008;112:604–611. doi: 10.1021/jp075747r. [DOI] [PubMed] [Google Scholar]
- 18.Sarell CJ, Syme CD, Rigby SEJ, Viles JH. Copper(II) binding to amyloid-beta fibrils of Alzheimer’s disease reveals a picomolar affinity: stoichiometry and coordination geometry are independent of Aβ oligomeric form. Biochemistry. 2009;48:4388–4402. doi: 10.1021/bi900254n. [DOI] [PubMed] [Google Scholar]
- 19.Linder MC. Biochemistry of copper. Plenum Press; New York: 1991. [Google Scholar]
- 20.Laussac JP, Sarkar B. Characterization of the copper (II) and Nickel(II) transport site of human serum albumin. Studies of copper (II) and nickel (II) binding to peptide 1–24 of human serum albumin by carbon-13 and proton NMR spectroscopy. Biochemistry. 1984;23:2832–2838. doi: 10.1021/bi00307a046. [DOI] [PubMed] [Google Scholar]
- 21.Rózga M, Sokołowska M, Protas AM, Bal W. Human serum albumin coordinates Cu(II) at its N-terminal binding site with 1 pM affinity. J Biol Inorg Chem. 2007;12:913–918. doi: 10.1007/s00775-007-0244-8. [DOI] [PubMed] [Google Scholar]
- 22.Masuoka J, Saltman P. Zinc(II) and copper(II) binding to serum albumin. A comparative study of dog, bovine, and human albumin. J Biol Chem. 1994;269:25557–25561. [PubMed] [Google Scholar]
- 23.Ida N, Hartmann T, Pantel J, Schröder J, Zerfass R, Förstl H, Sandbrink R, Masters CL, Beyreuther K. Analysis of heterogeneous βA4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J Biol Chem. 1996;271:22908–22914. doi: 10.1074/jbc.271.37.22908. [DOI] [PubMed] [Google Scholar]
- 24.Perrone L, Mothes E, Vignes M, Mockel A, Figueroa C, Miquel MC, Maddelein ML, Faller P. Copper transfer from Cu-Aβ to human serum albumin inhibits aggregation, radical production and reduces Aβ toxicity. Chembiochem. 2010;11:110–118. doi: 10.1002/cbic.200900474. [DOI] [PubMed] [Google Scholar]
- 25.Cherny RA, Legg JT, McLean CA, Fairlie DP, Huang X, Atwood CS, Beyreuther K, Tanzi RE, Masters CL, Bush AI. Aqueous dissolution of Alzheimer’s disease Aβ amyloid deposits by biometal depletion. J Biol Chem. 1999;274:23223–23228. doi: 10.1074/jbc.274.33.23223. [DOI] [PubMed] [Google Scholar]
- 26.Talmard C, Bouzan A, Faller P. Zinc binding to amyloid-β: isothermal titration calorimetry and Zn competition experiments with Zn sensors. Biochemistry. 2007;46:13658–13666. doi: 10.1021/bi701355j. [DOI] [PubMed] [Google Scholar]
- 27.Tõugu V, Karafin A, Palumaa P. Binding of zinc(II) and copper(II) to the full-length Alzheimer’s amyloid-beta peptide. J Neurochem. 2008;104:1249–1259. doi: 10.1111/j.1471-4159.2007.05061.x. [DOI] [PubMed] [Google Scholar]
- 28.Fezoui Y, Hartley DM, Harper JD, Khurana R, Walsh DM, Condron MM, Selkoe DJ, Lansbury PT, Jr, Fink AL, Teplow DB. An improved method of preparing the amyloid β-protein for fibrillogenesis and neurotoxicity experiments. Amyloid: Int J Exp Clin Invest. 2000;7:166–178. doi: 10.3109/13506120009146831. [DOI] [PubMed] [Google Scholar]
- 29.Syme CD, Nadal RC, Rigby SEJ, Viles JH. Copper binding to the amyloid-beta (Aβ) peptide associated with Alzheimer’s disease. J Biol Chem. 2004;279:18169–18177. doi: 10.1074/jbc.M313572200. [DOI] [PubMed] [Google Scholar]
- 30.Sokołowska M, Bal W. Cu(II) complexation by “non-coordinating” N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES buffer) J Inorg Biochem. 2005;99:1653–1660. doi: 10.1016/j.jinorgbio.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 31.Faller P, Hureau C. Bioinorganic chemistry of copper and zinc ions coordinated to amyloid-β peptide. Dalton Trans. 2009:1080–1094. doi: 10.1039/b813398k. [DOI] [PubMed] [Google Scholar]
- 32.Lee J, Culyba EK, Powers ET, Kelly JW. Amyloid-β forms fibrils by nucleated conformational conversion of oligomers. Nat Chem Biol. 2011;7(9):602–609. doi: 10.1038/nchembio.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie. Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 34.Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc Natl Acad Sci USA. 2003;100:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bush AI, Pettingell WH, Jr, Paradis MD, Tanzi RE. Modulation of Aβ adhesiveness and secretase site cleavage by zinc. J Biol Chem. 1994;269:12152–12158. [PubMed] [Google Scholar]
- 36.Ryu J, Girigoswami K, Ha C, Ku SH, Park CB. Influence of multiple metal ions on β-amyloid aggregation and dissociation on a solid surface. Biochemistry. 2008;47(19):5328–5335. doi: 10.1021/bi800012e. [DOI] [PubMed] [Google Scholar]
- 37.Liang Y, Lynn DG, Berland KM. Direct observation of nucleation and growth in amyloid self-assembly. J Am Chem Soc. 2010;132:6306–6308. doi: 10.1021/ja910964c. [DOI] [PubMed] [Google Scholar]
- 38.Maurer-Stroh S, Debulpaep M, Kuemmerer N, Lopez de la Paz M, Martins IC, Reumers J, Morris KL, Copland A, Serpell L, Serrano L, Schymkowitz JW, Rousseau F. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat Meth. 2010;7:237–242. doi: 10.1038/nmeth.1432. [DOI] [PubMed] [Google Scholar]
- 39.Valko M, Morris H, Mazúr M, Telser J, McInnes EJL, Mabbs FE. High-affinity binding site for copper(II) in human and dog serum albumins (an EPR study) J Phys Chem B. 1999;103:5591–5597. [Google Scholar]
- 40.Bal W, Christodoulou J, Sadler PJ, Tucker A. Multi-metal binding site of serum albumin. J Inorg Biochem. 1998;70:33–39. doi: 10.1016/s0162-0134(98)00010-5. [DOI] [PubMed] [Google Scholar]
- 41.Karr JW, Kaupp LJ, Szalai VA. Amyloid-β binds Cu2+ in a mononuclear metal ion binding site. J Am Chem Soc. 2004;126:13534–13538. doi: 10.1021/ja0488028. [DOI] [PubMed] [Google Scholar]
- 42.Karr JW, Szalai VA. Cu(II) binding to monomeric, oligomeric, and fibrillar forms of the Alzheimer’s disease amyloid-β peptide. Biochemistry. 2008;47(17):5006–5016. doi: 10.1021/bi702423h. [DOI] [PubMed] [Google Scholar]
- 43.Karr JW, Akintoye H, Kaupp LJ, Szalai VA. N-terminal deletions modify the Cu2+ binding site in amyloid-beta. Biochemistry. 2005;44:5478–5487. doi: 10.1021/bi047611e. [DOI] [PubMed] [Google Scholar]
- 44.El Khoury Y, Dorlet P, Faller P, Hellwig P. New insights into the coordination of Cu(II) by the amyloid-β 16 peptide from Fourier transform IR spectroscopy and isotopic labeling. J Phys Chem B. 2011;115:14812–14821. doi: 10.1021/jp207328y. [DOI] [PubMed] [Google Scholar]
- 45.Karr JW, Szalai VA. Roles of aspartate-1 in Cu(II) binding to the amyloid-β peptide of Alzheimer’s disease. J Am Chem Soc. 2007;129:3796–3797. doi: 10.1021/ja068952d. [DOI] [PubMed] [Google Scholar]
- 46.Shin BK, Saxena S. Substantial contribution of the two imidazole rings of the His13-His14 dyad to Cu(II) binding in amyloid-β(1–16) at physiological pH and its significance. J Phys Chem A. 2011;115:9590–9602. doi: 10.1021/jp200379m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamaguchi T, Yagi H, Goto Y, Matsuzaki K, Hoshino M. A disulfide-linked amyloid-β peptide dimer forms a protofibril-like oligomer through a distinct pathway from amyloid fibril formation. Biochemistry. 2010;49:7100–7107. doi: 10.1021/bi100583x. [DOI] [PubMed] [Google Scholar]
- 48.Hwang W, Zhang S, Kamm RD, Karplus M. Kinetic control of dimer structure formation in amyloid fibrillogenesis. Proc Natl Acad Sci USA. 2004;101:12916–12921. doi: 10.1073/pnas.0402634101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Döbeli H, Schubert D, Riek R. 3D structure of Alzheimer’s amyloid-β(1–42) fibrils. Proc Natl Acad Sci USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nelson R, Sawaya MR, Balbirnie M, Madsen AØ, Riekel C, Grothe R, Eisenberg D. Structure of the cross-β spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carrera F, Marcos ES, Merkling PJ, Chaboy JS, Muñoz-Páez A. Nature of metal binding sites in Cu(II) complexes with histidine and related N-coordinating ligands, as studied by EXAFS. Inorg Chem. 2004;43:6674–6683. doi: 10.1021/ic049699q. [DOI] [PubMed] [Google Scholar]
- 52.Parthasarathy S, Long F, Miller Y, Xiao Y, McElheny D, Thurber K, Ma B, Nussinov R, Ishii Y. Molecular-level examination of Cu2+ binding structure for amyloid fibrils of 40-residue Alzheimer’s β by solid-state NMR spectroscopy. J Am Chem Soc. 2011;133:3390–3400. doi: 10.1021/ja1072178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pedersen JT, Teilum K, Heegaard NH, Østergaard J, Adolph HW, Hemmingsen L. Rapid formation of a preoligomeric peptide–metal–peptide complex following copper(II) binding to amyloid-β peptides. Angew Chem Int Ed. 2011;50:2532–2535. doi: 10.1002/anie.201006335. [DOI] [PubMed] [Google Scholar]
- 54.Shearer J, Callan PE, Tran T, Szalai VA. Cu K-edge X-ray absorption spectroscopy reveals differential copper coordination within amyloid-β oligomers compared to amyloid-β monomer. Chem Comm. 2010;46:9137–9139. doi: 10.1039/c0cc02446e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marjanska M, Curran GL, Wengenack TM, Henry PG, Bliss RL, Poduslo JF, Jack CR, Jr, U3urbil K, Garwood M. Monitoring disease progression in transgenic mouse models of Alzheimer’s disease with proton magnetic resonance spectroscopy. Proc Natl Acad Sci USA. 2005;102:11906–11910. doi: 10.1073/pnas.0505513102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrel MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kayed B, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 58.Gaggelli E, Kozlowski H, Valensin D, Valensin G. Copper homeostasis and neurodegenerative disorders. Chem Rev. 2006;106:1995–2044. doi: 10.1021/cr040410w. [DOI] [PubMed] [Google Scholar]
- 59.Squitti R, Ventriglia M, Barbati G, Cassetta E, Ferreri F, Forno GD, Ramires S, Zappasodi F, Rossini PM. ‘Free’ copper in serum of Alzheimer’s disease patients correlates with markers of liver function. J Neural Transm. 2007;114:1589–1594. doi: 10.1007/s00702-007-0777-6. [DOI] [PubMed] [Google Scholar]
- 60.Barnham KJ, Masters CL, Bush AI. Neurodegenerative disease and oxidative stress. Nat Rev Drug Discov. 2004;3:205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- 61.Harris ED. Cellular copper transport and metabolism. Ann Rev Nutrit. 2000;20:291–310. doi: 10.1146/annurev.nutr.20.1.291. [DOI] [PubMed] [Google Scholar]
- 62.Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2006;4:176–185. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- 63.Clarke CS, Bannon FJ. Serum albumin in Down Syndrome with and without Alzheimer’s Disease. Ir J Med Sci. 2005;174:4–8. doi: 10.1007/BF03169121. [DOI] [PubMed] [Google Scholar]
- 64.Koistinaho M, Koistinaho J. Interactions between Alzheimer’s disease and cerebral ischemia-focus on inflammation. Brain Res Rev. 2005;48:240–250. doi: 10.1016/j.brainresrev.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 65.Little JR, Cook A, Cook SA, Maclntyre WJ. Microcirculatory obstruction in focal cerebral ischemia: Albumin and erythrocyte transit. Stroke. 1981;12:218–223. doi: 10.1161/01.str.12.2.218. [DOI] [PubMed] [Google Scholar]
- 66.Sbarouni E, Georgiadou P, Voudris V. Ischemia modification albumin changes-review and clinical implications. Clin Chem Lab Med. 2011;49:177–184. doi: 10.1515/CCLM.2011.037. [DOI] [PubMed] [Google Scholar]
- 67.Boada M, Ortiz P, Anaya F, Hernández I, Muñoz J, Núñez L, Olazarán J, Roca I, Cuberas G, Tárraga L, Buendia M, Pla RP, Ferre I, Páez A. Amyloi-targeted therapeutics in Alzheimer’s disease: use of human albumin in plasma exchange as a novel approach for Aβ mobilization. Drug News Perspect. 2009;22:325–339. doi: 10.1358/dnp.2009.22.6.1395256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.