

Abstract
The liver lymphocyte population is enriched with natural killer (NK) cells, which play a key role in host defense against viral infection and tumor transformation. Recent evidence from animal models suggests that NK cells also play an important role in inhibiting liver fibrosis by selectively killing early or senescence activated hepatic stellate cells (HSCs) and by producing the anti-fibrotic cytokine IFN-γ. Furthermore, clinical studies have revealed that human NK cells can kill primary human HSCs and that the ability of NK cells from HCV patients to kill HSCs is enhanced and correlates inversely with the stages of liver fibrosis. IFN-α treatment enhances, while other factors (e.g., alcohol, TGF-β) attenuate, the cytotoxicity of NK cells against HSCs, thereby differentially regulating liver fibrogenesis. In addition, the mouse liver lymphocyte population is also enriched for natural killer T (NKT) cells, whereas human liver lymphocytes have a much lower percentage of NKT cells. Many studies suggest that NKT cells promote liver fibrogenesis by producing pro-fibrotic cytokines such as IL-4, IL-13, hedgehog ligands, and osteopontin; however, NKT cells may also attenuate liver fibrosis under certain conditions by killing HSCs and by producing IFN-γ. Finally, the potential for NK and NKT cells to be used as therapeutic targets for anti-fibrotic therapy is discussed.
Keywords: viral hepatits, alcoholic liver disease, NKG2D, NAFLD, IFN-gamma
1. Introduction
Liver fibrosis and its end-stage consequence, cirrhosis, represent the final common pathway of virtually all chronic liver diseases and affect hundreds of millions of people worldwide. Despite impressive advancements in the field, no treatment options currently exist to cure fibrosis, with the exception of liver transplantation. The complex mechanisms driving the progression of chronic liver injury to fibrosis are not fully understood because of the multifaceted and often paradoxical intercellular relationships. Accumulating evidence suggests that hepatic stellate cell (HSC) activation during liver injury is a key step in the development of liver fibrosis [1–5]. In a healthy liver, HSCs are quiescent and play a central role in the storage of retinol (vitamin A compound). After injury, HSCs become activated and transdifferentiate into matrix-producing cells termed myofibroblasts. The activation of HSCs is controlled by many types of cytokines, growth factors, immune cells, and other factors [1–5]. Among the immune cells involved, macrophages have been shown not only to contribute to the pathogenesis of liver fibrosis but also to promote liver fibrosis resolution [6]. Dendritic cells (DCs) were reported to exacerbate liver fibrosis via the production of TNF-α by an early paper [7], but a recent study using more specific DC markers suggests that DCs ameliorate liver fibrosis by promoting fibrosis regression via the secretion of matrix metalloproteinase-9 [8]. Additionally, the functions of natural killer (NK) and NKT cells in liver fibrogenesis have recently received great attention because these cells are enriched among liver lymphocytes and are also markedly altered in various liver diseases [9]. In this review, we highlight recent advances in the understanding of the functions of NK and NKT cells that are important for the pathogenesis of liver fibrogenesis and will briefly discuss NK and NKT cells as potential therapeutic targets for anti-fibrotic therapy.
2. NK and NKT cell biology
NK cells are lymphocytes of the innate immune system that recognize and kill infected and tumorigenic cells. These cells represent a third lineage of lymphoid cells that is distinct from T and B cells. Unlike B and T cells, NK cells do not express an antigen receptor. Instead, they rely upon an array of cell surface receptors to detect changes in the expression of host cell surface molecules that typically appear on a variety of ‘stressed cells’, including microbe- or virus-infected cells, transformed cells, and injured cells [10]. The decision to kill a cell is made based on the net balance of signals delivered by inhibitory and activating receptor molecules that are expressed on NK cells. The inhibitory receptors include killer Ig-like receptors (KIRs) and Ly-49A and CD94/NKG2 receptors that recognize MHC class I molecules (inhibitory ligands) expressed on nearly all normal cells, which subsequently inactivate NK cell function. Thus, NK cells do not kill normal host cells. The stimulatory receptors present on NK cells include NKp46, NKp30 and NKp44, which are collectively referred to as natural cytotoxicity receptors, NKG2D, and DNAX accessory molecule-1 (CD226) [10–12]. Among these, NKG2D is the most well-defined receptor, which binds its ligands (e.g., MICA/B and ULBP in humans, RAE-1 and MULT1 in mice) that are expressed on target cells and subsequently promotes NK cell activation. After activation, NK cells can directly kill target cells via the exocytosis of perforin- and granzyme-containing cytoplasmic granules. NK cells can also kill target cells in a perforin-independent manner by utilizing FAS ligand, TNF-α, and TNF-related apoptosis-inducing ligands (TRAIL). Additionally, production of many cytokines (particularly IFN-γ, TNF-α, IL-10, IL-22) and chemokines (including MIP-1α and -β and RANTES) is another important mechanism by which NK cells regulate target cells and immune responses [13, 14].
NKT cells are a heterogeneous group of T lymphocytes that recognize lipid antigens presented by the nonclassical MHC class I-like molecule CD1 [15]. Human tissues express five distinct isoforms of CD1, including CD1a, -b, -c, -d, and -e, whereas mice only express CD1d. The CD1d-dependent NKT cells can be grouped into two types of cells: type I and type II NKT cells. Type I NKT cells, which are also called classical or invariant NKT (iNKT) cells because they express an invariant T cell receptor α (TCR-α) chain, comprise 95% of liver NKT cells. Type II NKT cells express diverse TCRs and make up less than 5% of liver NKT cells. Upon activation by lipid antigens such as α-galactosylceramide (α-GalCer), iNKT cells are able to produce large quantities of IFN-γ, IL-4, IL-13, TNF-α, IL-17, and many other cytokines. Activated iNKT cells also produce cytotoxic mediators such as perforin, Fas ligand, and TRAIL to kill target cells. Although activation of iNKT cells by the exogenous lipid antigen α-GalCer has been extensively investigated, the endogenous ligands that activate NKT cells remain largely unknown.
3. Hepatic NK and NKT cells
NK cells in the liver, which were originally called “Pit” cells in rats, are located in the hepatic sinusoids in close vicinity to liver non-parenchymal cells and represent a unique organ-associated NK cell population [16]. Under normal physiological conditions, hepatic NK cells have a rapid turnover, with an estimated residence time of 1 to 2 weeks in the liver. Because their proliferative activity is very low, the hepatic NK cell population is continuously replenished from an extra-hepatic source of stem cells, which is most likely located in the bone marrow [16]. The frequency of NK cells amongst liver lymphocytes is much higher than that of peripheral blood lymphocytes; in mice, 10% of hepatic lymphocytes are NK cells, while 30–50% of liver lymphocytes from rat and human livers are NK cells [9]. Various pathological conditions (such as viral infection, acute and chronic inflammation) as well as treatments with biological response modifiers that activate the immune system (such as polyinosinic-polycytidylic acid [poly I:C] or IFNs) significantly increase the number of NK cells in the liver (see review [9] and reference therein). At present, the mechanisms underlying the enrichment of NK cells in the liver are not fully understood, although it is believed that adhesion to sinusoidal endothelial cells is an important step in the recruitment of NK cells from the vascular compartment into the liver [17]. Such adhesion is regulated by cell adhesion molecules, which mediate cell-to-cell and cell-to-matrix interactions. In humans, several cell adhesion molecules, such as CD11a/CD18, CD2, CD54, CD56, and CD58, have been detected on the surface of NK cells [18], and blocking these molecules with neutralizing antibodies markedly decreases the number of NK cells in the liver [17].
After migrating into the liver, peripheral NK cells develop into liver-specific NK cells with unique features, such as higher levels of cytotoxicity against different tumor target cells, compared to NK cells from other organs [19, 20]. Such liver-specific NK cell development is believed to be controlled by the liver sinusoidal microenvironment. For example, human NK cells extracted from normal donor liver perfusates from a living donor liver transplantation were able to kill human hepatocellular carcinoma cell line HepG2 cells, an effect which was further enhanced by IL-2 treatment; however, NK cells from recipient livers with cirrhosis showed impaired anti-tumor activity even in the presence of IL-2 stimulation [19]. At present, how liver microenvironment affects liver-specific NK cell development remains unclear.
In addition to NK cells, NKT cells are also enriched amongst liver lymphocytes, as approximately 30–35% of mouse liver lymphocytes are NKT cells, and 5–10% of rat and human liver lymphocytes are NKT cells, which are significantly larger frequencies than that observed for peripheral blood lymphocytes (<5% NKT cells) [9]. NKT cells not only directly kill target cells but also produce a wide variety of cytokines, thereby playing diverse roles in controlling liver injury, fibrosis, regeneration, and hepatocarcinogenesis [9].
4. Anti-fibrotic effect of NK cells
NK cell killing of activated HSCs was first demonstrated by two different groups using mouse models in 2006 [21, 22], and this observation was later confirmed by many additional studies both in animal models [23–32] and patients [28, 31, 33–36] (Table 1). Collectively, these findings suggest that NK cells selectively kill early activated (transitional) or senescence-activated HSCs but do not kill quiescent or fully activated (myofibroblasts) HSCs (Fig. 1). In addition, NK cells also produce IFN-γ, which then induces HSC apoptosis and cell cycle arrest and subsequently inhibits liver fibrosis (Fig. 1).
Table 1.
The major findings related to the anti-fibrotic effects of NK cells
| The references | Animal model or human studies | Major findings |
|---|---|---|
| Radaeva et al. [21] 2006 | DDC-feeding or CCl4 treatment in mice; mouse NK-HSC co-culture |
In vivo depletion of NK cells exacerbated, while activation of NK cells ameliorated, liver fibrosis in mouse models. In vitro co-culture with NK cells killed activated HSCs through NKG2D- and TRAIL-dependent mechanisms. |
| Melhem et al. [22] 2006 | CCl4 treatment in mice; human NK-LX2 co-culture |
In vivo liver fibrosis was exacerbated in mice with NK cell deficiency. In vitro co-culture with NK cells killed activated HSCs in a granzyme B-dependent manner. |
| Jeong et al. [23] 2006 | CCl4 treatment in mice; mouse NK-HSC co-culture | Activation of STAT1 by IFN-γ augmented NK cell killing of HSCs, thereby attenuating liver fibrosis. |
| Radaeva et al. [24] 2007 | Mouse NK-HSC co-culture | Co-culture with NK cells selectively killed early activated HSCs that expressed elevated levels of the NK cell-activating ligand retinoic acid inducible early gene 1 (RAE1). |
| Krizhanovsky et al. [25] 2008 | CCl4 treatment in mice; co- culture of NK-IMR-90 fibroblasts or primary human myofibroblasts | The number of senescence-activated HSCs in CCl4-treated mice was increased after depletion of NK cells but was decreased following administration of the NK cell activator poly I:C. In vitro co-culture with NK cells killed senescence-activated HSCs. |
| Jeong et al. [26] 2008 | Chronic ethanol feeding plus CCl4 treatment in mice | Feeding mice with an ethanol diet abrogated the anti-fibrotic effect of NK cells and IFN-γ. |
| Hintermann et al. [27] 2010 | CCl4 treatment in mice | CXCL10-deficient mice had more NK cells in the liver and were resistant to CCl4-induced liver fibrosis. |
| Yoshida et al. [33] 2011 | Peripheral lymphocytes from HCV patients | Blood NK cell percentages were decreased in HCV patients and inversely correlated with liver fibrosis progression, which was monitored by counting platelets. |
| Muhanna e al [28] 2011 | Human NK-LX2 co-culture; CCl4 treatment in mice | Knockdown of the NK cell inhibitory receptor iKIR stimulated NK cells and promoted their antifibrogenic activity in mice and human HSC co-cultures. |
| Jeong et al. [29] 2011 | CCl4 treatment in mice; mouse NK-HSC co-culture | Fully activated HSCs were resistant to NK cell killing via the production of TGF-β that inhibited NK cells. |
| Hou et al. [30] 2012 | Schistosoma japonicum egg-induced liver fibrosis in mice; mouse NK-HSC co- culture |
In vivo depletion of NK cells enhanced S japonicum-induced liver fibrosis, while activation of NK cells inhibited it. In vitro co-culture with NK cells killed activated HSCs. |
| Sagiv A et al. [32] 2012 | Human NK-HSC co-culture; CCl4 treatment in mice | Co-culture with NK cells killed senescence-activated HSCs through a perforin-dependent mechanism. Perforin KO mice had increased liver fibrosis compared to WT mice after chronic CCl4 administration. |
| Gur et al. [31] 2012 | CCl4 treatment in mice; mouse NK-HSC co-culture; human NK-HSC co-culture | High levels of NCR1 (murine orthologue of the NKp46) were detected on murine HSCs. NCR1 KO mice were more susceptible to CCl4-induced liver fibrosis. In vitro co-culture with murine NK cells killed mouse HSCs in an NCR1-dependent manner. Co-culture with human NK cells killed human HSCs in an NKp46-dependent manner. |
| Glassner et al. [34] 2012 | Human NK-HSC co-culture; liver biopsies from HCV patients | NK cells from HCV patients were highly efficient at inducing apoptosis of activated primary human HSCs in vitro in TRAIL-, FasL- and NKG2D-dependent manners. An inverse correlation of liver fibrosis stages and the ability of NK cells to induce HSC apoptosis in HCV patients was observed. |
| Kramer et al. [35] 2012 | Analyses of intra-and extra-hepatic NK cells from HCV patients; fibrosis was determined on biopsies | Accumulation of NKp46high NK cells was found in the HCV- infected livers. NKp46high NK cells had higher levels of cytolytic activity and IFN-γ secretion than NKp46dim NK cells, and the frequency of intrahepatic NKp46high NK cells was inversely correlated with fibrosis stages. |
| Eisenhardt et al. 2012 | Analyses of peripheral NK cells from HCV patients; co-culture primary human HSCs-HCV patient NK cells | In healthy controls, CXCR3+CD56bright NK cells had stronger activity against HSCs than CXCR3−CD56dim NK cells. Chronic HCV infection was associated with an increased frequency of CXCR3+CD56bright NK cells, but these cells showed impaired degranulation and impaired IFN-γ secretion in response to HSCs. |
Fig. 1.
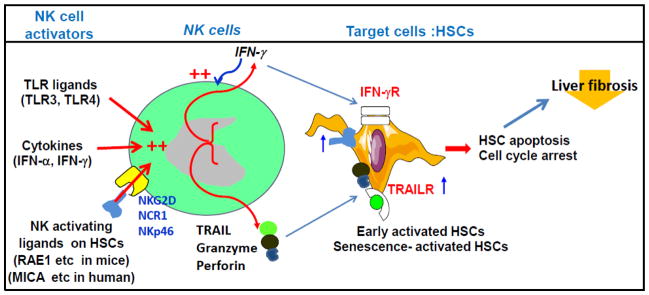
Mechanisms underlying the anti-fibrotic effect of NK cells. NK cells can be activated by several TLR ligands and cytokines. Early and senescence-activated HSCs express elevated levels of NK cell-activating ligands that bind several NK activating receptors on NK cells, including NKG2D (mice and human), NCR1 (mice), and NKp46 (human), and subsequently induce NK cell activation. Activated NK cells release cytotoxic mediators that selectively kill early or senescence-activated HSCs but not quiescent or fully activated HSCs. Activated NK cells also produce IFN-γ, which not only directly induces HSC death but also further enhances NK cell cytotoxicity against HSCs. Quiescent and fully activated HSCs do not express elevated NK cell-activating ligands and are resistant to NK cell killing.
4.1. NK cells selectively kill early activated but not quiescent or fully activated HSCs: In vivo and in vitro evidence
In a healthy liver, HSCs are quiescent and store retinol, but in response to liver injury, they are activated and converted into highly proliferative, contractile myofibroblast-like cells [3]. Activation of HSCs in an injured liver gives rise to an array of cells at different stages of activation and trans-differentiation that display transitional phenotypes, gene expression, and functions, which can be identified by several markers. Among these, Desmin, an intermediate filament typical of contractile cells, has been widely used as the “gold standard” for identifying all stages of HSCs in rodent livers, although its expression in humans is unreliable [3]. As illustrated in Fig. 2A, immunostaining with Desimin show numerous processes in the quiescent HSCs, while the early activated HSCs in the fibrotic mouse livers induced by 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) are identified as small round cells with a scant cytoplasm that lack the processes. The myofiboblast-like cells are revealed as elongated shape and localized in the portal fibrotic areas.
Fig. 2.
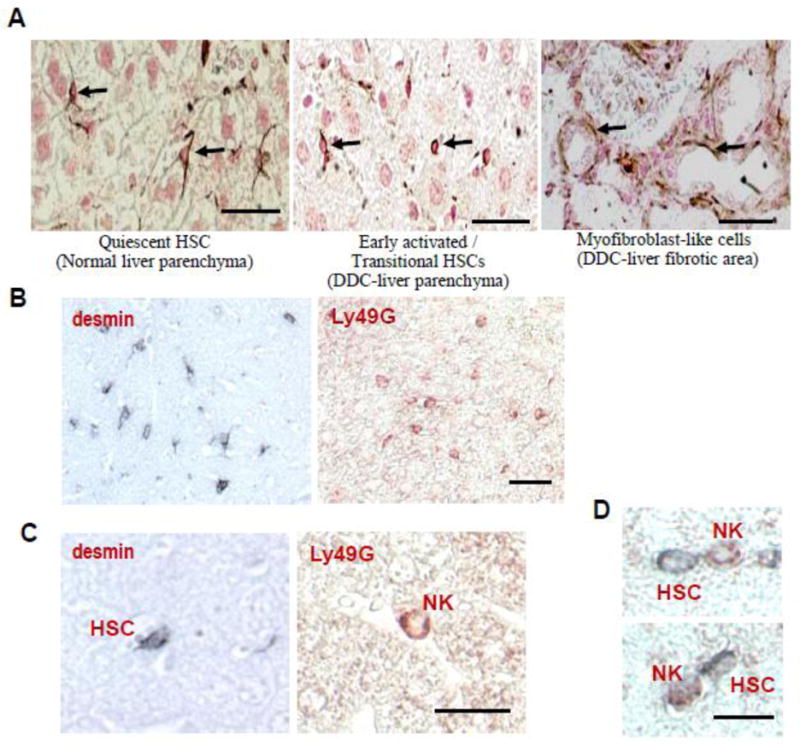
In vivo evidence for the contact between NK cells and early activated HSCs. (A): Identification of early activated/transitional HSCs and NK cells in the fibrotic livers from DDC-fed mice. Under the light microscope, early activated desmin-positive cells (arrows in the middle panel) appear as small cells with scant cytoplasm and lack numerous processes typical for quiescent HSCs (arrows in the left panel). Bars, 50 μm. (B): Mice were fed a DDC diet for 2 weeks. Liver sections were stained with desmin and Ly49G antibodies to identify HSCs and NK cells, respectively. Positive staining was developed using either 3,3′-diaminobenzidine containing nickel chloride (black staining for HSCs) or 3-amino-9-ethylcarbazole (red staining for NK cells). Note that positive cells occupied a similar area of the liver parenchyma, including zones 2 and 3. (C): Higher magnification of HSCs and NK cells located in the perisinusoidal space of Disse and in the hepatic sinusoid, respectively. (D): Double immunostaining of NK cells and HSCs demonstrating that Ly49G-positive NK cells (red) are located in close proximity to desmin-positive (black) early activated HSCs. Bars, 50 μm.
Immunohistochemistry analyses have shown in vivo that NK cells kill early activated, but not quiescent or fully activated, HSCs. First, the number of early activated desmin positive HSCs with an oval shape was significantly decreased in DDC-fed mice after administration of the NK cell activator poly I:C (Radaeva and Gao, unpublished data). Second, immunohistochemistry analyses show that early activated HSCs and NK cells have similar distributions throughout zones II and III of the liver parenchyma but do not reside in the periportal fibrotic area (Figs. 2B–C). Third, the direct contact between NK cells and early activated HSCs are often observed in the injured liver (Fig. 2D).
In vitro cell co-culture and cytotoxicity assays clearly demonstrate that NK cells kill early activated, but not quiescent or fully activated, HSCs (Fig. 1) [24]. Quiescent HSCs are spontaneously activated when cultured on plastic dishes, and the activation of HSCs can be divided into early and chronic stages of activation based on cell morphology and gene expression. HSCs cultured for 4–7 days become characteristically early activated HSCs and gradually lose their stores of retinol, whereas cells cultured for long periods of time (21 days) become fully activated HSCs with myofibroblast-like functionality. In vitro cytotoxicity assays show that NK cells only kill day 5–7 cultured HSCs but not freshly isolated quiescent HSCs or day 21-cultured HSCs, suggesting that NK cells selectively kill early activated HSCs [24]. Furthermore, we have provided evidence suggesting that during activation, early activated HSCs produce retinoic acid, which upregulates the NK cell activating ligand retinoic acid inducible gene 1 (RAE1) expression on HSCs. RAE1 binds NKG2D on NK cells and subsequently activate NK cells to kill the early activated HSCs through TRAIL- and NKG2D-dependent mechanisms [21, 24]. In contrast, chronically activated HSCs or myofibroblasts lose their cytoplasmic stores of retinol and do not produce RA and RAE1, thereby gaining resistance to NK cell killing. Similar to mouse models, NK cells from HCV-infected patients effectively induce the apoptosis of activated HSCs through TRAIL-, Fas L-, and NKG2D-dependent mechanisms [34]. In addition, TRAIL receptor expression is elevated in HSCs after activation [37], which likely also contributes to the increased sensitivity of these activated HSCs to NK cell killing.
Apart from NKG2D, the NK cell activating receptor NKp46 and its mouse ortholog NCR1 are also involved in controlling liver fibrosis through the killing of primary human and mouse HSCs, respectively [31]. NKp46, a unifying marker for NK cells across mammalian species, recognizes viral hemagglutinins and unknown cellular ligands [38]. Recently, Gur et al. [31] demonstrated that, in the absence of NKp46, non-activated NK cell killing of HSCs was completely abolished, although activated NK cells remained able to kill HSCs, suggesting that NKp46 plays a critical role in mediating the non-activated NK cell killing of HSCs and that other receptors (such as NKG2D) contribute to the activated NK cell killing of HSCs.
Furthermore, the increased sensitivity of activated HSCs to NK cell killing may also be due to changes in NK cell inhibitory ligand expression [22]. Following CCl4-induced fibrosis, activated HSCs lose expression of the MHC-1 antigen, which is an NK cell inhibitory ligand that suppresses NK cell function by binding the inhibitory killer-cell immunoglobulin-like receptors (iKIRs) on NK cells. Consequently, these activated HSCs become sensitive to NK cell killing [22]. The important role of iKIRs in attenuating the NK cell-mediated anti-fibrotic effect is supported by the finding that silencing iKIR via the transfection of iKIR siRNA enhances NK cell killing of HSCs and restrains liver fibrosis [28].
4.2. NK cells kill senescence-activated HSCs
Activated HSCs can become senescent and demonstrate stable cell cycle arrest, reduced levels of extracellular matrix components, and the upregulation of extracellular matrix-degrading enzymes, which is an important step in limiting the fibrogenic response to tissue damage [25, 39]. Senescent HSCs accumulate in the fibrotic scar area after chronic liver injury. Depletion of NK cells increased the number of senescent HSCs, while activation of NK cells by injection of poly I:C reduced those senescent HSCs in the fibrotic livers from chronic CCl4-treated mice [25]. This is because senescence-activated HSCs express elevated levels of NK cell-activating ligands and become sensitive to NK cell killing [25]. Such NK cell killing is mediated via the granule exocytosis pathway [32], which is different from the TRAIL pathway involved in the NK cell killing of early activated HSCs [21].
4.3. NK cells restrict liver fibrosis by producing cytokines (e.g., IFN-γ)
Production of a large amount of IFN-γ is a hallmark of activated NK cells and mediates many of the functions of NK cells, such as anti-viral and anti-tumor defense. Recent evidence suggests that production of IFN-γ also contributes significantly to the anti-fibrotic effect of NK cells by directly inducing HSC apoptosis and cell cycle arrest [23, 40] and enhancing NK cell killing of activated HSCs [21] (Fig. 1).
In addition to IFN-γ, NK cells also produce many other cytokines and chemokines; however, the mechanisms by which these mediators affect the anti-fibrotic effects of NK cells remain largely unknown. For example, blood-derived NK cells and mucosa-associated lymphoid tissue-residing NK cells (also called NK-22 cells) produce IL-22 [41], a cytokine that has been shown to protect against liver injury and inhibit liver fibrosis [42, 43]. Thus, it would be interesting to examine whether activated NK cells from diseased livers produce IL-22 and whether the production of IL-22 contributes to the anti-fibrotic effect of NK cells.
5. The Diverse and Complex Roles of NKT cells in liver fibrogenesis
In contrast to the consistent results of NK cell-mediated anti-fibrotic effects, the functions of NKT cells in the pathogenesis of liver fibrosis seem more complex and likely play diverse roles due to following reasons. First, there are several types of NKT cells, including type I and type II NKT cells, which play different roles and sometimes opposing functions in the liver [9, 44, 45]. Second, the detection of NKT cells after activation is more difficult than that of NK cells because of the rapid downregulation of NKT cell markers and/or apoptosis after activation [46, 47]. Third, the mechanisms by which NKT cells are activated by endogenous ligands and cytokines in vivo remain largely unknown [48]. Fourth, after activation, NKT cells become tolerant and nonresponsive to subsequent stimulation [49–51]. Finally, activated NKT cells can produce large amounts of both anti-fibrotic (e.g., IFN-γ) and pro-fibrotic (e.g., IL-4, IL-13, hedgehog ligands, and osteopontin) cytokines, as well as many other cytokines, chemokines, and mediators that can differentially regulate liver fibrogenesis (Fig. 3).
Fig. 3.
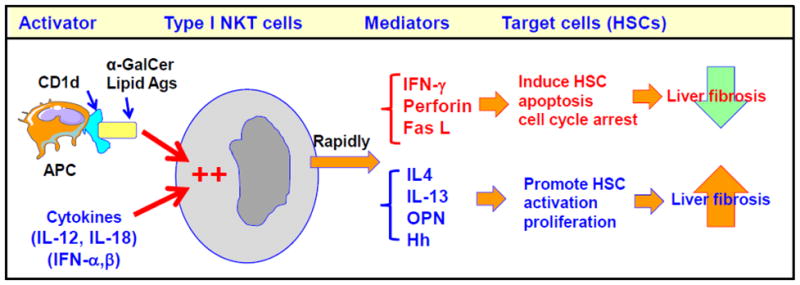
Mechanisms underlying the complex roles of NKT cells in liver fibrogenesis. NKT cells can be activated by lipid antigens (such as α-GalCer) presented by CD1d on antigen presenting cells (APCs). In addition, several cytokines can also activate NKT cells. Activated NKT cells rapidly produce a wide variety of anti-fibrotic and pro-fibrotic mediators that attenuate and enhance liver fibrosis, respectively. The net effect of NKT cells is reflected by the balance between the anti- and pro-fibrotic effects of NKT cells. Thus, NKT cells may play different roles in the control of liver fibrogenesis in different stages and different types of liver diseases. OPN: osteopontin; Hh: hedgehog ligands.
Table 2 lists the major recent studies that have evaluated the roles of NKT cells in liver fibrogenesis. The first study discussing NKT cells and liver fibrosis was conducted in HCV patients in 2004 [52], and the authors reported that the number of intrahepatic iNKT cells, the hepatic expression of CD1d, and the production of pro-fibrotic IL-4 and IL-13 cytokines by iNKT cells were significantly increased in HCV patients with cirrhosis. Based on these findings, the authors concluded that iNKT cells responded to the progressive liver damage caused by chronic HCV infection and subsequently promoted liver fibrosis via the production of pro-fibrotic cytokines. Investigation into the roles of NKT cells in liver fibrogenesis using animal models has begun only recently (Table 2). Most of these studies relied on two strains of NKT-deficient mice: J 18 KO mice (with a deficiency in type I iNKT cells) and CD1d KO mice (with a deficiency in both type I iNKT and type II NKT cells).
Table 2.
The major findings related to NKT cells and liver fibrosis
| The references | Models | Major findings | Suggested functions |
|---|---|---|---|
| de Lalla et al. [52] 2004 | Analyses of circulating and liver iNKT in HCV patients | iNKT cells were increased in chronically HBV- or HCV-infected livers with preferential production of profibrotic IL-4 and IL-13 cytokines. In vitro CD1d-dependent activation of iNKT cells from healthy donors elicited IL-4 and IL-13. | Pro-fibrotic |
| Park et al. [53] 2009 | CCl4 treatment in mice | Jα18 KO mice had a higher grade of fibrosis than WT mice at 2 weeks, but not 4 weeks, after CCl4 injection. Administration of the iNKT activator α-GalCer accelerated CCl4-induced acute liver injury and fibrosis but had no effects in the chronic model. Incubation with NKT cells killed HSCs in vitro. | Pro-fibrotic Anti-fibrotic |
| Miyagi et al. [66] 2010 | Mouse model (HFD feeding) | Jα18 KO mice were more susceptible to HFD-induced inflammation and fibrosis (on a BALB/c background). | Anti-fibrotic |
| Syn et al. [63] 2010 | Mouse model (MCD feeding); Human NASH cirrhotic livers | NASH cirrhotic livers had higher numbers of NKT cells compared to healthy livers. Patched-deficient mice with an accumulation of liver NKT cells had increased liver fibrosis, while CD1d KO mice had decreased liver fibrosis in a model of MCD feeding. | Pro-fibrotic |
| Jin et al. [68] 2011 | Mouse model (HBV TG mice) | NKT cells were over-activated in chronic CCl4-treated HBV TG mice. Depletion of NKT cells reduced, while adaptive transfer of NKT cells enhanced, CCl4-induced liver injury and fibrosis. | Pro-fibrotic |
| Ishikawa et al. [54] 2011 | Mouse model (chronic TAA injection) | CD1d KO mice had decreased liver inflammation, injury, and fibrosis compared to WT mice after TAA administration. | Pro-fibrotic |
| Syn et al. [67] 2012 | Mouse model (MCD diet feeding) | Jα18 KO and CD1d KO mice had significantly reduced hedgehog and osteopontin expression and less fibrosis compared to WT mice in a model of MCD diet feeding. | Pro-fibrotic |
5.1. CCl4-induced liver fibrosis model
The functions of iNKT cells in liver fibrogenesis have been extensively investigated in a mouse model of CCl4-induced liver fibrosis in Jα18 KO mice [53]. This study revealed that Jα18 KO mice were more susceptible to CCl4-induced acute liver injury and inflammation, suggesting that iNKT cells protect against CCl4-induced acute liver injury. Such protection is likely mediated via the iNKT suppression of HSC activation and cytokine production. In contrast, treatment with α-GalCer induced strong iNKT cell activation and enhanced CCl4-induced acute liver injury and fibrosis. In a model of chronic liver injury induced by chronic CCl4 administration, Jα18 KO and wild-type mice had comparable degree of liver injury, with only a slightly higher grade of liver fibrosis in Jα18 KO mice than wild-type mice at 2 weeks, but not 4 weeks, after CCl4 injection. Chronicα-GalCer treatment induced iNKT tolerance and had little effect on liver injury and fibrosis in the model of chronic CCl4 challenge. In summary, iNKT cells seem to play diverse roles in controlling liver fibrogenesis. Different degrees of iNKT activation, including acute and chronic iNKT activation, may differentially regulate the development and progression of liver fibrogenesis.
5.2. Thioacetamide (TAA)-induced liver fibrosis
Chronic TAA treatment is another model of chemical-induced liver fibrosis. Ishikawa et al. [54] reported that CD1dKO mice were resistant to TAA-induced liver injury, inflammation, and fibrosis. These authors proposed that CD1d-restricted NKT cells, including type I (iNKT) and type II NKT cells, not only produce IL-4 and IL-13, which directly induce HSC activation and liver fibrosis, but also produce IFN-γ, which stimulates macrophage activation and liver inflammation, thereby promoting liver injury and liver fibrosis.
5.3. Nonalcoholic Fatty Liver Disease (NAFLD)-associated liver fibrosis
The role of NKT cells in high fat diet (HFD)-induced obesity, insulin resistance, and fatty liver disease has been extensively investigated in C56BL/6 mice, but the results have been controversial [55–59]. In addition, in murine models of obesity-associated fatty liver, hepatic NKT cells are decreased [60–62], whereas human NAFLD is associated with increased NKT cell accumulation in the liver [63–65]. The reasons underlying such opposite changes in liver NKT cells are not clear but may be related to differences in the pathogenesis and severity of liver disease in mouse fatty liver and human NAFLD. It is believed that the accumulation of NKT cells in human NAFLD is due to hepatic hedgehog pathway activation, and such NKT cell accumulation exacerbates liver fibrosis [63].
C57BL/6 mice are the best strain to study HFD-induced obesity and insulin resistance, although these animals do not develop HFD-induced fibrosis. In contrast, BALB/c mice are resistant to HFD-induced obesity and insulin resistance but are sensitive to HFD-induced fibrosis. Feeding BALB/c mice with a HFD induced mild liver inflammation and fibrosis, which was exacerbated in Jα18 KO mice on a BALB/c background [66]. This result suggests that iNKT cells play a hepatoprotective and anti-fibrotic role in HFD-induced NAFLD in mice on a BALB/c background.
Feeding a methionine choline-deficient (MCD) diet induces fatty liver, inflammation, and fibrosis in rodents, which mimics the severe form of nonalcoholic steatohepatitis (NASH) that occurs in some humans. Using this model, Dr. Diehl’s group demonstrated that NKT cells play an important role in promoting liver fibrogenesis via the production of hedgehog ligands and osteopontin that promote HSC activation and fibrogenesis [63, 67].
5.4. HBV-transgenic mice
HBV-transgenic mice, C57BL/6J-TgN (AlbHBV)44Bri, which contain HBV genome S, pre-S, and X domains, develop spontaneous liver fibrosis with enhanced expression of collagen I, matrix metalloproteinase, and tissue inhibitor of metalloproteinase [68]. HBV-transgenic mice were also shown to be more susceptible to CCl4-induced liver injury and fibrosis, with an accumulation and overactivation of NKT cells in the liver [68]. Moreover, depletion of NKT cells reduced CCl4-induced liver injury and fibrosis in the HBV-transgenic mice [68], which suggests that NKT cells promote liver fibrosis in this model via the production of IL-4 and IL-13 [68].
6. NK cells and liver fibrosis in human liver diseases
Current studies from animal models suggest that NK cells inhibit liver fibrosis. Interestingly, several recent studies have implied that NK cells may play an even greater role in controlling liver fibrosis in patients with chronic liver diseases compared to that observed using mouse models. First, human liver lymphocytes are approximately 30–50% NK cells, which is a much higher frequency than that of the mouse liver lymphocyte population (approximately 10% NK cells). Second, liver NK cells are not significantly altered in most models of liver fibrosis in rodents, whereas the functions of NK cells are significantly altered under many conditions in patients with liver disease, such as viral hepatitis, IFN-α therapy, chronic alcohol consumption, and cirrhosis. Herein, we discuss the potential implications of NK cells in the pathogenesis of liver fibrosis in patients.
6.1. Viral hepatitis
The activation and function of NK cells in the pathogenesis of HCV infection have been extensively investigated in the past five years. Most of these studies have suggested that during HCV infection, virally-infected hepatocytes produce IFN-α/β and other cytokines (e.g., IL-12, IL-15, IL-18), which induce peripheral blood NK cell activation and enhance NK cell cytotoxicity [35, 69–74]. In addition, IFN-α therapy has been shown to rapidly induce the activation of peripheral NK cells in HCV patients [74–77]. However, the regulation of intrahepatic NK cells during chronic HCV infection remains obscure due to the availability of liver biopsy and the lack of appropriate control liver samples. A number of studies [35, 70, 76] have shown that intrahepatic NK cells possess greater cytotoxic activity than peripheral blood NK cells in HCV patients, which is consistent with previous findings showing that healthy livers contain NK cells with higher basal levels of cytotoxicity compared to peripheral NK cells [19, 20]. Interestingly, a recent study reported that in comparison to intrahepatic NK cells from patients undergoing surgery for uncomplicated gallstone, intrahepatic NK cells from patients with chronic HCV infection demonstrated lower levels of cytotoxicity [78]. This result suggests that intrahepatic NK cell activity during HCV infection is suppressed compared to that in healthy livers, despite the fact that these cells exhibited higher activity than peripheral NK cells. Thus, further studies are needed to confirm these findings.
The anti-viral function of NK cells has been well documented for many years. Thus, activated NK cells likely play an important role in controlling/preventing HCV infection by killing HCV-infected hepatocytes, inhibiting HCV replication, or by priming the adaptive immune response [73, 79, 80]. Recently, Dr. Nattermann’s group published three interesting papers, which suggest that human NK cells also play a critical role in restricting liver fibrosis in HCV patients by killing HSCs [34–36].
In their first study [34], peripheral blood CD56+CD3− NK cells from untreated HCV RNA(+) patients, IFN-α-treated patients, and healthy controls were purified and subsequently incubated with activated primary human HSCs. This incubation induced NK cell degranulation/activation, with only negligible secretion of IFN-γ and TNF-α, and subsequently promoted HSC apoptosis through TRAIL-, FasL-, and NKG2D-dependent mechanisms. Peripheral blood CD56+CD3− NK cells from IFN-α-treated patients demonstrated the highest level of cytotoxicity against human HSCs, followed by those from untreated HCV RNA(+) patients and healthy controls. In addition, the ability of NK cells to induce HSC apoptosis correlated inversely with the stages of liver fibrosis [34].
In the second paper published by this group [35], peripheral blood and intrahepatic CD56+CD3− NK cells from patients with chronic HCV, NASH, or autoimmune hepatitis, were purified and analyzed for the expression of NKp46, a major human NK cell activating receptor. Peripheral blood and intrahepatic NKp46high NK cells demonstrated higher cytolytic activity against HSCs and stronger IFN-γ secretion in vitro than NKp46Dim NK cells. Liver lymphocytes were also associated with a higher frequency of NKp46high cells but lower frequency of NKp46dim cells compared to those present in the peripheral blood. Moreover, the frequency of intra-hepatic NKp46High NK cells was inversely correlated with HCV RNA levels and fibrosis stages.
In the latest paper from Dr. Nattermann’s group [36], the expression of CXCR3 was analyzed on peripheral blood NK cells from HCV patients and healthy controls. Based on CXCR3 expression, NK cells were divided into CXCR3+ and CXCR3− NK cell groups. In healthy controls, CXCR3+CD56bright NK cells displayed stronger cytotoxicity activity against HSCs than CXCR3+CD56dim NK cells. However, in HCV patients, both CXCR3+CD56bright and CXCR3+CD56dim NK cells had similar activities, which were lower than that of CXCR3+CD56bright NK cells from healthy controls. This seems not in agreement with the conclusion from their first study that CD56+CD3− NK cells from HCV patients were more effective in induction of HSC apoptosis than NK cells from healthy controls [34].
Taken together, these clinical studies suggest that activated human HSCs can stimulate human NK cell activation, the latter of which conversely kills activated human HSCs, and this process plays an important role in inhibiting liver fibrosis. IFN-α treatment increases the ability of human NK cells from HCV patients to kill activated HSCs. However, the differences in cytotoxicity against HSCs between NK cells from HCV-infected patients and normal healthy controls were somewhat inconsistent between studies [34–36], although this may be explained by the different subsets of NK cells that were examined, the different treatments of NK cells, and the patients with different stages of fibrosis who were investigated in these studies.
6.2. Alcoholic liver disease
Chronic alcohol consumption is a leading cause of chronic liver disease, which can lead to liver fibrosis and cirrhosis [81]. It has also been well documented that alcohol consumption accelerates the progression of liver fibrosis in patients with chronic hepatitis C. Multiple mechanisms have been implicated in the pathogenesis of alcoholic liver fibrosis and the alcohol acceleration of liver fibrosis in HCV patients [81–83]. Because chronic alcohol consumption suppresses NK cell function, it is plausible that such inhibition contributes to the progression of alcoholic liver fibrosis. Indeed, feeding mice an ethanol diet markedly inhibited NK cell cytotoxicity against HSCs and exacerbated liver fibrosis [26], and this result was due to ethanol inhibition of TRAIL, NKG2D, and IFN-γ expression by NK cells [26]. In addition, it would be interesting to determine whether the ability of NK cells to kill HSCs is also suppressed in patients with alcoholic liver disease or HCV patients with alcohol consumption and whether IFN-α treatment can restore the cytotoxicity of NK cells against HSCs.
6.3. Cirrhosis
Despite NK cell-mediated protection against fibrosis, chronic liver injury still leads to fibrosis and cirrhosis, which suggests that HSCs may employ specific mechanisms to escape from NK cell-mediated lysis under many conditions. One such mechanism appears to involve fully activated HSC and myofibroblast production of high levels of TGF-β, which is one of the most potent inhibitors for NK cells and prevents NK cell killing of HSCs [29].
7. NKT cells and liver fibrosis in human liver diseases
Mouse liver lymphocytes are approximately 30–40% NKT cells, whereas human liver lymphocyte populations may contain much lower percentages of NKT cells. The percentage of NKT (CD56+CD3+) cells amongst human liver lymphocytes has been shown to be highly variable, ranging from 3% to 15% [63, 84], and CD1d-dependent NKT cells were found to be make up <1% of human liver lymphocytes [84, 85]. Although treatment of mice with α-GalCer induces significant liver inflammation and injury [50], phase I/II clinical trials revealed that injection of α-GalCer did not produce any signs of liver injury in humans [85–87]. This result suggests that human liver lymphocytes may contain a low percentage of NKT cells and that these cells may play a less significant role in the pathogenesis of liver fibrosis in patients.
NKT cells from HCV patients without cirrhosis produce both anti-fibrotic (IFN-γ) and pro-fibrotic (IL-4 and IL-13) cytokines, whereas NKT cells from cirrhotic HCV patients preferentially produce IL-4 and IL-13. Thus, it is thought that NKT cells promote liver fibrosis in HCV patients [52]. The number of NKT cells is significantly elevated in NASH-related cirrhosis, and such elevation is believed to promote liver fibrogenesis in patients with NASH [63]. Because these cells produce a wide variety of cytokines and cytotoxic mediators, NKT cells likely play complex and even opposing roles in controlling liver fibrogenesis in patients with chronic liver diseases with different stages and different etiologies. Further clinical studies are needed to clarify such complex functions of NKT cells.
8. NK/NKT cells as potential therapeutic targets for anti-fibrotic therapy
Studies in the last 6 years from animal models and patients clearly indicate that NK cells have strong anti-fibrotic activity, which suggests that the induction of NK cell activity could represent a novel strategy to treat liver fibrosis [88]. Several cytokines are known to stimulate NK cell activation, including IFN-α/β, IL-2, IL-12, IL-15, IL-18, and IFN-γ. Among these, IFN-α/β has the greatest ability to induce NK cell activation, and IFN-α therapy has been shown to potentiate NK cell killing of activated HSCs in HCV patients [34]. Thus, IFN-α, as an NK activator and a direct inhibitor of HSCs, is a promising option for anti-fibrotic therapy in patients with viral hepatitis or other etiologies. Indeed, early studies revealed that IFN-α therapy improved liver fibrosis in HCV patients who responded to anti-viral therapy, as well as in non-responders who did not clear HCV infection [89, 90]. However, a recent large, multicenter 5-year trial of low-dose IFN-α showed no benefit of this therapy on liver fibrosis in patients with chronic HCV infection and advanced fibrosis with or without cirrhosis [91]. This lack of beneficial effect of low-dose of IFN-α therapy may be due to the observation that advanced fibrosis is associated with high levels of TGF-β, which inhibits a low-dose of IFN-α-mediated activation of NK cells [29]. Moreover, many current ongoing anti-fibrotic clinical trials are examining the effects of IFN-α alone or IFN-α in combination with other drugs [92]. It would be interesting to include studies during these trials to determine whether peripheral and intrahepatic NK cells are activated after IFN-α therapy and if such activation is correlated with an improvement in liver fibrosis in patients. IL-12 and IL-18 are also strong NK cell activators, but treatment with IL-12 and IL-18 induces significant liver inflammation and injury [93] and is therefore not a good strategy to treat liver fibrosis.
In addition to NK cell activators, many factors have been shown to attenuate NK cell cytotoxicity against HSCs. For example, TGF-β, alcohol consumption, and NK cell inhibitory receptors were reported to suppress NK cell killing of HSCs [26, 28, 29]. Removal of these inhibitory factors may present additional promising strategies for the treatment liver fibrosis. For example, siRNA knockdown of iKIR enhanced NK cell function and promoted NK cell anti-fibrogenic activity in mice and in co-culture with human NK-HSCs, and this technique has been proposed as a therapeutic option for anti-fibrotic therapy [28].
The NKT activator α-GalCer was also used in phase I/II clinical trials to treat viral hepatitis but did not show any anti-viral effects [86, 87]. However, liver fibrosis was not investigated in these studies. Because these cells produce a large variety of cytokines and cytotoxic mediators that play complex and possibly opposing roles in modulating liver fibrosis, NKT cells will be difficult to use as a therapeutic target for anti-fibrotic treatments.
Highlights.
The liver lymphocyte population is enriched with NK and NKT cells
NK cells inhibit liver fibrosis by killing activated HSCs and by producing IFN-γ
IFN-α therapy enhances, while alcohol and TGF-β inhibit, NK cell killing of HSCs
NKT cells produce pro-fibrotic cytokines to promote liver fibrosis
NKT cells also produce anti-fibrotic cytokines to inhibit liver fibrosis
Abbreviations
- α-GalCer
α-galactosylceramide
- DC
Dendritic cell
- DDC
3,5-diethoxycarbonyl-1,4-dihydrocollidine
- HSC
hepatic stellate cell
- iNKT
invariant NKT
- KIR
killer Ig-like receptor
- MCD diet
methionine choline-deficient diet
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NK cell
natural killer cell
- NKT cell
natural killer T cell
- RAE-1
retinoic acid inducible gene 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pinzani M, Macias-Barragan J. Update on the pathophysiology of liver fibrosis. Expert Rev Gastroenterol Hepatol. 2010;4:459–472. doi: 10.1586/egh.10.47. [DOI] [PubMed] [Google Scholar]
- 5.Marra F, Aleffi S, Galastri S, Provenzano A. Mononuclear cells in liver fibrosis. Semin Immunopathol. 2009;31:345–358. doi: 10.1007/s00281-009-0169-0. [DOI] [PubMed] [Google Scholar]
- 6.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Connolly MK, Bedrosian AS, Mallen-St Clair J, Mitchell AP, Ibrahim J, Stroud A, Pachter HL, Bar-Sagi D, Frey AB, Miller G. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-alpha. J Clin Invest. 2009;119:3213–3225. doi: 10.1172/JCI37581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiao J, Sastre D, Fiel MI, Lee UE, Ghiassi-Nejad Z, Ginhoux F, Vivier E, Friedman SL, Merad M, Aloman C. Dendritic cell regulation of carbon tetrachloride-induced murine liver fibrosis regression. Hepatology. 2012;55:244–255. doi: 10.1002/hep.24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao B, Radaeva S, Park O. Liver natural killer and natural killer T cells: immunobiology and emerging roles in liver diseases. J Leukoc Biol. 2009;86:513–528. doi: 10.1189/jlb.0309135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 11.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3:781–790. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 12.Ortaldo JR, Young HA. Mouse Ly49 NK receptors: balancing activation and inhibition. Mol Immunol. 2005;42:445–450. doi: 10.1016/j.molimm.2004.07.024. [DOI] [PubMed] [Google Scholar]
- 13.Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- 14.Robertson MJ. Role of chemokines in the biology of natural killer cells. J Leukoc Biol. 2002;71:173–183. [PubMed] [Google Scholar]
- 15.Kronenberg M. Toward an understanding of NKT cell biology: progress and paradoxes. Annu Rev Immunol. 2005;23:877–900. doi: 10.1146/annurev.immunol.23.021704.115742. [DOI] [PubMed] [Google Scholar]
- 16.Vanderkerken K, Bouwens L, De Neve W, Van den Berg K, Baekeland M, Delens N, Wisse E. Origin and differentiation of hepatic natural killer cells (pit cells) Hepatology. 1993;18:919–925. doi: 10.1002/hep.1840180425. [DOI] [PubMed] [Google Scholar]
- 17.Luo D, Vanderkerken K, Bouwens L, Kuppen PJ, Baekeland M, Seynaeve C, Wisse E. The role of adhesion molecules in the recruitment of hepatic natural killer cells (pit cells) in rat liver. Hepatology. 1996;24:1475–1480. doi: 10.1002/hep.510240629. [DOI] [PubMed] [Google Scholar]
- 18.Lotzova E. Definition and functions of natural killer cells. Nat Immun. 1993;12:169–176. [PubMed] [Google Scholar]
- 19.Ishiyama K, Ohdan H, Ohira M, Mitsuta H, Arihiro K, Asahara T. Difference in cytotoxicity against hepatocellular carcinoma between liver and periphery natural killer cells in humans. Hepatology. 2006;43:362–372. doi: 10.1002/hep.21035. [DOI] [PubMed] [Google Scholar]
- 20.Vermijlen D, Luo D, Froelich CJ, Medema JP, Kummer JA, Willems E, Braet F, Wisse E. Hepatic natural killer cells exclusively kill splenic/blood natural killer-resistant tumor cells by the perforin/granzyme pathway. Journal of leukocyte biology. 2002;72:668–676. [PubMed] [Google Scholar]
- 21.Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130:435–452. doi: 10.1053/j.gastro.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 22.Melhem A, Muhanna N, Bishara A, Alvarez CE, Ilan Y, Bishara T, Horani A, Nassar M, Friedman SL, Safadi R. Anti-fibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J Hepatol. 2006;45:60–71. doi: 10.1016/j.jhep.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 23.Jeong WI, Park O, Radaeva S, Gao B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology. 2006;44:1441–1451. doi: 10.1002/hep.21419. [DOI] [PubMed] [Google Scholar]
- 24.Radaeva S, Wang L, Radaev S, Jeong WI, Park O, Gao B. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am J Physiol Gastrointest Liver Physiol. 2007;293:G809–816. doi: 10.1152/ajpgi.00212.2007. [DOI] [PubMed] [Google Scholar]
- 25.Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeong WI, Park O, Gao B. Abrogation of the antifibrotic effects of natural killer cells/interferon-gamma contributes to alcohol acceleration of liver fibrosis. Gastroenterology. 2008;134:248–258. doi: 10.1053/j.gastro.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hintermann E, Bayer M, Pfeilschifter JM, Luster AD, Christen U. CXCL10 promotes liver fibrosis by prevention of NK cell mediated hepatic stellate cell inactivation. J Autoimmun. 2010;35:424–435. doi: 10.1016/j.jaut.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muhanna N, Abu Tair L, Doron S, Amer J, Azzeh M, Mahamid M, Friedman S, Safadi R. Amelioration of hepatic fibrosis by NK cell activation. Gut. 2011;60:90–98. doi: 10.1136/gut.2010.211136. [DOI] [PubMed] [Google Scholar]
- 29.Jeong WI, Park O, Suh YG, Byun JS, Park SY, Choi E, Kim JK, Ko H, Wang H, Miller AM, Gao B. Suppression of innate immunity (natural killer cell/interferon-gamma) in the advanced stages of liver fibrosis in mice. Hepatology. 2011;53:1342–1351. doi: 10.1002/hep.24190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hou X, Yu F, Man S, Huang D, Zhang Y, Liu M, Ren C, Shen J. Negative regulation of Schistosoma japonicum egg-induced liver fibrosis by natural killer cells. PLoS Negl Trop Dis. 2012;6:e1456. doi: 10.1371/journal.pntd.0001456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gur C, Doron S, Kfir-Erenfeld S, Horwitz E, Abu-Tair L, Safadi R, Mandelboim O. NKp46-mediated killing of human and mouse hepatic stellate cells attenuates liver fibrosis. Gut. 2012;61:885–893. doi: 10.1136/gutjnl-2011-301400. [DOI] [PubMed] [Google Scholar]
- 32.Sagiv ABA, Yon M, Simon J, Lowe SW, Krizhanovsky V. Granule exocytosis mediates immune surveillance of senescent cells. Oncogene. 2012 doi: 10.1038/onc.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshida K, Ohishi W, Nakashima E, Fujiwara S, Akahoshi M, Kasagi F, Chayama K, Hakoda M, Kyoizumi S, Nakachi K, Hayashi T, Kusunoki Y. Lymphocyte subset characterization associated with persistent hepatitis C virus infection and subsequent progression of liver fibrosis. Hum Immunol. 2011;72:821–826. doi: 10.1016/j.humimm.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 34.Glassner A, Eisenhardt M, Kramer B, Korner C, Coenen M, Sauerbruch T, Spengler U, Nattermann J. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab Invest. 2012;92:967–977. doi: 10.1038/labinvest.2012.54. [DOI] [PubMed] [Google Scholar]
- 35.Krämer BKC, Kebschull M, Glässner A, Eisenhardt M, Nischalke HD, Alexander M, Sauerbruch T, Spengler U, Nattermann J. NKp46(High) expression defines a NK cell subset that is potentially involved in control of HCV replication and modulation of liver fibrosis. Hepatology. 2012 doi: 10.1002/hep.25804. in press. [DOI] [PubMed] [Google Scholar]
- 36.Eisenhardt M, Glassner A, Kramer B, Korner C, Sibbing B, Kokordelis P, Nischalke HD, Sauerbruch T, Spengler U, Nattermann J. The CXCR3(+)CD56Bright Phenotype Characterizes a Distinct NK Cell Subset with Anti-Fibrotic Potential That Shows Dys-Regulated Activity in Hepatitis C. PLoS One. 2012;7:e38846. doi: 10.1371/journal.pone.0038846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taimr P, Higuchi H, Kocova E, Rippe RA, Friedman S, Gores GJ. Activated stellate cells express the TRAIL receptor-2/death receptor-5 and undergo TRAIL-mediated apoptosis. Hepatology. 2003;37:87–95. doi: 10.1053/jhep.2003.50002. [DOI] [PubMed] [Google Scholar]
- 38.Walzer T, Blery M, Chaix J, Fuseri N, Chasson L, Robbins SH, Jaeger S, Andre P, Gauthier L, Daniel L, Chemin K, Morel Y, Dalod M, Imbert J, Pierres M, Moretta A, Romagne F, Vivier E. Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46. Proc Natl Acad Sci U S A. 2007;104:3384–3389. doi: 10.1073/pnas.0609692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schnabl B, Purbeck CA, Choi YH, Hagedorn CH, Brenner D. Replicative senescence of activated human hepatic stellate cells is accompanied by a pronounced inflammatory but less fibrogenic phenotype. Hepatology. 2003;37:653–664. doi: 10.1053/jhep.2003.50097. [DOI] [PubMed] [Google Scholar]
- 40.Rockey DC, Maher JJ, Jarnagin WR, Gabbiani G, Friedman SL. Inhibition of rat hepatic lipocyte activation in culture by interferon-gamma. Hepatology. 1992;16:776–784. doi: 10.1002/hep.1840160325. [DOI] [PubMed] [Google Scholar]
- 41.Wolk K, Sabat R. Interleukin-22: A novel T- and NK-cell derived cytokine that regulates the biology of tissue cells. Cytokine Growth Factor Rev. 2006;17:367–380. doi: 10.1016/j.cytogfr.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 42.Kong XFD, Wang H, Hong F, Bertola A, Wang FS, Gao B. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis. Hepatology. 2012 doi: 10.1002/hep.25744. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meng F, Wang K, Aoyama T, Grivennikov S, Paik Y, Scholten D, Cong M, Iwaisako K, Liu X, Zhang M, Österreicher C, Stickel F, Ley K, Brenner D, Kisseleva T. IL-17 signaling in inflammatory cells, Kupffer cells and Hepatic Stellate cells exacerbates liver fibrosis. Gastroenterology. 2012 doi: 10.1053/j.gastro.2012.05.049. On line. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santodomingo-Garzon T, Swain MG. Role of NKT cells in autoimmune liver disease. Autoimmun Rev. 2011;10:793–800. doi: 10.1016/j.autrev.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 45.Notas G, Kisseleva T, Brenner D. NK and NKT cells in liver injury and fibrosis. Clin Immunol. 2009;130:16–26. doi: 10.1016/j.clim.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 46.Harada M, Seino KI, Wakao H, Sakata S, Ishizuka Y, Ito T, Kojo S, Nakayama T, Taniguchi M. Down-regulation of the invariant Valpha14 antigen receptor in NKT cells upon activation. Int Immunol. 2004;16:241–247. doi: 10.1093/intimm/dxh023. [DOI] [PubMed] [Google Scholar]
- 47.Eberl G, MacDonald HR. Rapid death and regeneration of NKT cells in anti-CD3epsilon- or IL-12-treated mice: a major role for bone marrow in NKT cell homeostasis. Immunity. 1998;9:345–353. doi: 10.1016/s1074-7613(00)80617-2. [DOI] [PubMed] [Google Scholar]
- 48.Venkataswamy MM, Porcelli SA. Lipid and glycolipid antigens of CD1d-restricted natural killer T cells. Semin Immunol. 2010;22:68–78. doi: 10.1016/j.smim.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jung KKM, Park C, Choi YH, Jeon Y, Park SH, Seo SK, Jin D, Choi I. The protective role of VSIG4 expressed on kupffer cells during immune-mediated liver injury by inducing tolerance of liver T- and NKT-cells. Hepatology. 2012 doi: 10.1002/hep.25906. [DOI] [PubMed] [Google Scholar]
- 50.Biburger M, Tiegs G. Alpha-galactosylceramide-induced liver injury in mice is mediated by TNF-alpha but independent of Kupffer cells. J Immunol. 2005;175:1540–1550. doi: 10.4049/jimmunol.175.3.1540. [DOI] [PubMed] [Google Scholar]
- 51.Parekh VV, Wilson MT, Olivares-Villagomez D, Singh AK, Wu L, Wang CR, Joyce S, Van Kaer L. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J Clin Invest. 2005;115:2572–2583. doi: 10.1172/JCI24762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Lalla C, Galli G, Aldrighetti L, Romeo R, Mariani M, Monno A, Nuti S, Colombo M, Callea F, Porcelli SA, Panina-Bordignon P, Abrignani S, Casorati G, Dellabona P. Production of profibrotic cytokines by invariant NKT cells characterizes cirrhosis progression in chronic viral hepatitis. J Immunol. 2004;173:1417–1425. doi: 10.4049/jimmunol.173.2.1417. [DOI] [PubMed] [Google Scholar]
- 53.Park O, Jeong WI, Wang L, Wang H, Lian ZX, Gershwin ME, Gao B. Diverse roles of invariant natural killer T cells in liver injury and fibrosis induced by carbon tetrachloride. Hepatology. 2009;49:1683–1694. doi: 10.1002/hep.22813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ishikawa S, Ikejima K, Yamagata H, Aoyama T, Kon K, Arai K, Takeda K, Watanabe S. CD1d-restricted natural killer T cells contribute to hepatic inflammation and fibrogenesis in mice. J Hepatol. 2011;54:1195–1204. doi: 10.1016/j.jhep.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 55.Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KS, Gao B, Lee CH, Kersten S, Qi L. Activation of natural killer T cells promotes M2 Macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4 (IL-4)/STAT6 protein signaling axis in obesity. J Biol Chem. 2012;287:13561–13571. doi: 10.1074/jbc.M112.350066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mantell BS, Stefanovic-Racic M, Yang X, Dedousis N, Sipula IJ, O’Doherty RM. Mice lacking NKT cells but with a complete complement of CD8+ T-cells are not protected against the metabolic abnormalities of diet-induced obesity. PLoS One. 2011;6:e19831. doi: 10.1371/journal.pone.0019831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Satoh M, Andoh Y, Clingan CS, Ogura H, Fujii S, Eshima K, Nakayama T, Taniguchi M, Hirata N, Ishimori N, Tsutsui H, Onoe K, Iwabuchi K. Type II NKT cells stimulate diet-induced obesity by mediating adipose tissue inflammation, steatohepatitis and insulin resistance. PLoS One. 2012;7:e30568. doi: 10.1371/journal.pone.0030568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tajiri K, Shimizu Y. Role of NKT Cells in the Pathogenesis of NAFLD. Int J Hepatol. 2012;2012:850836. doi: 10.1155/2012/850836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kotas ME, Lee HY, Gillum MP, Annicelli C, Guigni BA, Shulman GI, Medzhitov R. Impact of CD1d deficiency on metabolism. PLoS One. 2011;6:e25478. doi: 10.1371/journal.pone.0025478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Z, Soloski MJ, Diehl AM. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology. 2005;42:880–885. doi: 10.1002/hep.20826. [DOI] [PubMed] [Google Scholar]
- 61.Li Z, Oben JA, Yang S, Lin H, Stafford EA, Soloski MJ, Thomas SA, Diehl AM. Norepinephrine regulates hepatic innate immune system in leptin-deficient mice with nonalcoholic steatohepatitis. Hepatology. 2004;40:434–441. doi: 10.1002/hep.20320. [DOI] [PubMed] [Google Scholar]
- 62.Kremer M, Thomas E, Milton RJ, Perry AW, van Rooijen N, Wheeler MD, Zacks S, Fried M, Rippe RA, Hines IN. Kupffer cell and interleukin-12-dependent loss of natural killer T cells in hepatosteatosis. Hepatology. 2010;51:130–141. doi: 10.1002/hep.23292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, Witek RP, Choi SS, Guy CD, Fearing CM, Teaberry V, Pereira FE, Adams DH, Diehl AM. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology. 2010;51:1998–2007. doi: 10.1002/hep.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tajiri KSY, Tsuneyama K, Sugiyama T. Role of liver-infiltrating CD3+CD56+ natural killer T cells in the pathogenesis of nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. 2009 doi: 10.1097/MEG.0b013e32831bc3d6. In press. [DOI] [PubMed] [Google Scholar]
- 65.Adler M, Taylor S, Okebugwu K, Yee H, Fielding C, Fielding G, Poles M. Intrahepatic natural killer T cell populations are increased in human hepatic steatosis. World J Gastroenterol. 2011;17:1725–1731. doi: 10.3748/wjg.v17.i13.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miyagi T, Takehara T, Uemura A, Nishio K, Shimizu S, Kodama T, Hikita H, Li W, Sasakawa A, Tatsumi T, Ohkawa K, Kanto T, Hiramatsu N, Hayashi N. Absence of invariant natural killer T cells deteriorates liver inflammation and fibrosis in mice fed high-fat diet. J Gastroenterol. 2010;45:1247–1254. doi: 10.1007/s00535-010-0272-y. [DOI] [PubMed] [Google Scholar]
- 67.Syn WKAK, Swiderska M, Michelotti GA, Liaskou E, Pang H, Xie G, Philips G, Chan IS, Karaca GF, Pereira TD, Chen Y, Mi Z, Kuo PC, Choi SS, Guy CD, Abdelmalek MF, Diehl AM. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut. 2012 doi: 10.1136/gutjnl-2011-301857. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jin Z, Sun R, Wei H, Gao X, Chen Y, Tian Z. Accelerated liver fibrosis in hepatitis B virus transgenic mice: involvement of natural killer T cells. Hepatology. 2011;53:219–229. doi: 10.1002/hep.23983. [DOI] [PubMed] [Google Scholar]
- 69.Amadei B, Urbani S, Cazaly A, Fisicaro P, Zerbini A, Ahmed P, Missale G, Ferrari C, Khakoo SI. Activation of natural killer cells during acute infection with hepatitis C virus. Gastroenterology. 2010;138:1536–1545. doi: 10.1053/j.gastro.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ahlenstiel G, Titerence RH, Koh C, Edlich B, Feld JJ, Rotman Y, Ghany MG, Hoofnagle JH, Liang TJ, Heller T, Rehermann B. Natural killer cells are polarized toward cytotoxicity in chronic hepatitis C in an interferon-alfa-dependent manner. Gastroenterology. 2010;138:325–335. e321–322. doi: 10.1053/j.gastro.2009.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oliviero B, Varchetta S, Paudice E, Michelone G, Zaramella M, Mavilio D, De Filippi F, Bruno S, Mondelli MU. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology. 2009;137:1151–1160. doi: 10.1053/j.gastro.2009.05.047. [DOI] [PubMed] [Google Scholar]
- 72.Golden-Mason LSA, Bambha KM, Cheng L, Rosen HR. Race- and gender-related variation in NKp46 expression associated with differential anti-HCV immunity. Hepatlogy. 2012 doi: 10.1002/hep.25771.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Golden-Mason L, Cox AL, Randall JA, Cheng L, Rosen HR. Increased natural killer cell cytotoxicity and NKp30 expression protects against hepatitis C virus infection in high-risk individuals and inhibits replication in vitro. Hepatology. 2010;52:1581–1589. doi: 10.1002/hep.23896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Edlich B, Ahlenstiel G, Azpiroz AZ, Stoltzfus J, Noureddin M, Serti E, Feld JJ, Liang TJ, Rotman Y, Rehermann B. Early changes in interferon signaling define natural killer cell response and refractoriness to interferon-based therapy of hepatitis C patients. Hepatology. 2012;55:39–48. doi: 10.1002/hep.24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stegmann KA, Bjorkstrom NK, Veber H, Ciesek S, Riese P, Wiegand J, Hadem J, Suneetha PV, Jaroszewicz J, Wang C, Schlaphoff V, Fytili P, Cornberg M, Manns MP, Geffers R, Pietschmann T, Guzman CA, Ljunggren HG, Wedemeyer H. Interferon-alpha-induced TRAIL on natural killer cells is associated with control of hepatitis C virus infection. Gastroenterology. 2010;138:1885–1897. doi: 10.1053/j.gastro.2010.01.051. [DOI] [PubMed] [Google Scholar]
- 76.Ahlenstiel G, Edlich B, Hogdal LJ, Rotman Y, Noureddin M, Feld JJ, Holz LE, Titerence RH, Liang TJ, Rehermann B. Early changes in natural killer cell function indicate virologic response to interferon therapy for hepatitis C. Gastroenterology. 2011;141:1231–1239. 1239 e1231–1232. doi: 10.1053/j.gastro.2011.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stegmann KA, Bjorkstrom NK, Ciesek S, Lunemann S, Jaroszewicz J, Wiegand J, Malinski P, Dustin LB, Rice CM, Manns MP, Pietschmann T, Cornberg M, Ljunggren HG, Wedemeyer H. Interferon alpha-stimulated natural killer cells from patients with acute hepatitis C virus (HCV) infection recognize HCV-infected and uninfected hepatoma cells via DNAX accessory molecule-1. J Infect Dis. 2012;205:1351–1362. doi: 10.1093/infdis/jis210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Varchetta SMD, Mantovani S, Oliviero B, Cremonesi E, Ludovisi S, Michelone G, Alessiani M, Rosati R, Montorsi M, Mondelli MU. Impaired intrahepatic natural killer cell cytotoxic function in chronic hepatitis C virus infection. Hepatology. 2012 doi: 10.1002/hep.25723.. [DOI] [PubMed] [Google Scholar]
- 79.Pelletier S, Drouin C, Bedard N, Khakoo SI, Bruneau J, Shoukry NH. Increased degranulation of natural killer cells during acute HCV correlates with the magnitude of virus-specific T cell responses. J Hepatol. 2010;53:805–816. doi: 10.1016/j.jhep.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang SH, Huang CX, Ye L, Wang X, Song L, Wang YJ, Liang H, Huang XY, Ho WZ. Natural killer cells suppress full cycle HCV infection of human hepatocytes. J Viral Hepat. 2008;15:855–864. doi: 10.1111/j.1365-2893.2008.01014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572–1585. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gao B. Alcohol and hepatitis virus interactions in liver pathology. Comprehensive Handbook of Alcohol related Pathology. 2005;2:819–832. [Google Scholar]
- 83.Szabo G, Wands JR, Eken A, Osna NA, Weinman SA, Machida K, Joe Wang H. Alcohol and hepatitis C virus--interactions in immune dysfunctions and liver damage. Alcohol Clin Exp Res. 2010;34:1675–1686. doi: 10.1111/j.1530-0277.2010.01255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kenna T, Mason LG, Porcelli SA, Koezuka Y, Hegarty JE, O’Farrelly C, Doherty DG. NKT cells from normal and tumor-bearing human livers are phenotypically and functionally distinct from murine NKT cells. J Immunol. 2003;171:1775–1779. doi: 10.4049/jimmunol.171.4.1775. [DOI] [PubMed] [Google Scholar]
- 85.Motohashi S, Ishikawa A, Ishikawa E, Otsuji M, Iizasa T, Hanaoka H, Shimizu N, Horiguchi S, Okamoto Y, Fujii S, Taniguchi M, Fujisawa T, Nakayama T. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res. 2006;12:6079–6086. doi: 10.1158/1078-0432.CCR-06-0114. [DOI] [PubMed] [Google Scholar]
- 86.Veldt BJ, van der Vliet HJ, von Blomberg BM, van Vlierberghe H, Gerken G, Nishi N, Hayashi K, Scheper RJ, de Knegt RJ, van den Eertwegh AJ, Janssen HL, van Nieuwkerk CM. Randomized placebo controlled phase I/II trial of alpha-galactosylceramide for the treatment of chronic hepatitis C. J Hepatol. 2007;47:356–365. doi: 10.1016/j.jhep.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 87.Woltman AM, Ter Borg MJ, Binda RS, Sprengers D, von Blomberg BM, Scheper RJ, Hayashi K, Nishi N, Boonstra A, van der Molen R, Janssen HL. Alpha-galactosylceramide in chronic hepatitis B infection: results from a randomized placebo-controlled Phase I/II trial. Antivir Ther. 2009;14:809–818. doi: 10.3851/IMP1295. [DOI] [PubMed] [Google Scholar]
- 88.Gao B, Radaeva S, Jeong WI. Activation of natural killer cells inhibits liver fibrosis: a novel strategy to treat liver fibrosis. Expert Rev Gastroenterol Hepatol. 2007;1:173–180. doi: 10.1586/17474124.1.1.173. [DOI] [PubMed] [Google Scholar]
- 89.Shiratori Y, Imazeki F, Moriyama M, Yano M, Arakawa Y, Yokosuka O, Kuroki T, Nishiguchi S, Sata M, Yamada G, Fujiyama S, Yoshida H, Omata M. Histologic improvement of fibrosis in patients with hepatitis C who have sustained response to interferon therapy. Ann Intern Med. 2000;132:517–524. doi: 10.7326/0003-4819-132-7-200004040-00002. [DOI] [PubMed] [Google Scholar]
- 90.Guerret S, Desmouliere A, Chossegros P, Costa AM, Badid C, Trepo C, Grimaud JA, Chevallier M. Long-term administration of interferon-alpha in non-responder patients with chronic hepatitis C: follow-up of liver fibrosis over 5 years. J Viral Hepat. 1999;6:125–133. doi: 10.1046/j.1365-2893.1999.00148.x. [DOI] [PubMed] [Google Scholar]
- 91.Di Bisceglie AM, Shiffman ML, Everson GT, Lindsay KL, Everhart JE, Wright EC, Lee WM, Lok AS, Bonkovsky HL, Morgan TR, Ghany MG, Morishima C, Snow KK, Dienstag JL. Prolonged therapy of advanced chronic hepatitis C with low-dose peginterferon. N Engl J Med. 2008;359:2429–2441. doi: 10.1056/NEJMoa0707615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cohen-Naftaly M, Friedman SL. Current status of novel antifibrotic therapies in patients with chronic liver disease. Therap Adv Gastroenterol. 2011;4:391–417. doi: 10.1177/1756283X11413002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ono S, Ueno C, Seki S, Matsumoto A, Mochizuki H. Interleukin-12 and -18 induce severe liver injury in mice recovered from peritonitis after sublethal endotoxin challenge. Surgery. 2003;134:92–100. doi: 10.1067/msy.2003.189. [DOI] [PubMed] [Google Scholar]