

Abstract
Huntington Disease (HD) is an autosomal dominant neurological disorder characterized by motor, psychiatric and cognitive disturbances. Recent evidence indicates that the viability and function of cerebellar Purkinje cells (PCs) are compromised in an aggressive mouse model of HD. Here we investigate whether this is also the case in the HdhQ200 knock-in mouse model of HD. Using quantitative-real time-PCR and immunofluorescence, we observed a loss of the PC marker and calcium buffer calbindin in 50 week-old symptomatic mice. Reductions were also observed in parvalbumin and glutamic acid decarboxylase protein expression, most markedly in the molecular cell layer. Stereological analysis revealed an overall reduction in the PC population in HdhQ200/Q200 mice by nearly 40%, and loose patch electrophysiology of remaining PCs indicated a reduction in firing rate in HD mice compared to control littermates. Taken together, these data demonstrate that PC survival and function are compromised in a mouse model of adult-onset HD and suggest that further experiments should investigate the contribution of PC death and dysfunction to HD-associated motor impairment.
Keywords: cerebellum, gene expression, stereology, glutamic acid decarboxylase 67 (GAD67), calbindin (CALB), parvalbumin (PV), loose patch electrophysiology
Introduction
Huntington Disease (HD) is an autosomal-dominantly inherited neurodegenerative disorder characterized by a triad of symptoms including motor, cognitive, and psychiatric disturbances (Group 1993). Expression of the pathogenic protein, mutant huntingtin (htt), is ubiquitous throughout the brain and has been suggested to be involved in a number of aberrant processes including transcriptional dysregulation (Cha, Kosinski et al. 1998; Luthi-Carter, Hanson et al. 2002; Strand, Aragaki et al. 2005; Hodges, Strand et al. 2006; Imarisio, Carmichael et al. 2008; Bithell, Johnson et al. 2009), disruption of calcium signaling (Perry, Tallaksen-Greene et al. 2010; Giacomello, Hudec et al. 2011), and alterations of synaptic physiology (Klapstein, Fisher et al. 2001; Morton, Faull et al. 2001; Milnerwood and Raymond 2007; Cummings, Andre et al. 2009).
Studies have shown that the motor circuits of the basal ganglia are severely affected in this disease (Roos, Pruyt et al. 1985; Reiner, Albin et al. 1988; Andre, Cepeda et al. 2011) though there is evidence for morphological alterations in the cerebellum, including alterations in Purkinje cells (PCs), the main output neuron of the cerebellar cortex (Jeste, Barban et al. 1984; Rosas, Koroshetz et al. 2003; Fennema-Notestine, Archibald et al. 2004) in both patients (Rodda 1981; Jeste, Barban et al. 1984) and animal models of HD (Turmaine, Raza et al. 2000). Additionally, in an animal model of HD, deficits have been found in the levels of the neurotransmitter GABA in PCs (Reynolds, Dalton et al. 1999). Recent work using single cell RT-PCR has shown that there are transcriptional alterations specifically within PCs in an animal model of HD (Euler, Friedrich et al. 2012). However, there is little information regarding the extent to which PCs are lost in mouse models of HD and whether PC function is compromised.
We recently provided evidence for PC abnormalities in the R6/2 model of HD (Dougherty, Reeves et al. 2012); changes include a reduction in transcript and protein levels of GABAergic cell population components (glutamic acid decarboxylase 67 and parvalbumin) and a specific purkinje cell marker (calbindin). Furthermore, we found that PC loss was preceded by a significant reduction in PC spike rate. Taken together these data provide evidence that PC dysfunction is an important neuropathological phenotypic component of the R6/2 mouse model of HD.
Of note, the transgenic R6/2 line expresses an exon 1 fragment of mutant htt, resulting in a HD mouse model that displays an early disease onset and aggressive phenotype (Mangiarini, Sathasivam et al. 1996), so it is not clear whether the PC abnormalities we observed would be relevant for the understanding of adult-onset HD. Thus, we sought to determine whether PC loss and dysfunction are a part of the pathophysiology of HD in a mouse model of adult-onset HD. The knock-in HdhQ200 is particularly useful for this kind of study, as it expresses mutant htt in an appropriate genomic and protein context and recapitulates midlife onset and key progressive behavioral and pathological features of HD (Lin, Tallaksen-Greene et al. 2001). These include well-characterized progressive, measurable, and disease-relevant behavioral deficits and neurological abnormalities (Heng, Tallaksen-Greene et al. 2007; Heng, Duong et al. 2010) and, notably, robust striatal neuronal intranuclear inclusions by 20 weeks of age, a progressively deteriorating motor phenotype beginning at 50 weeks of age, and gross motor impairment present by 80 weeks of age (Heng, Duong et al. 2010). Previous studies have shown that a similar knock-in mouse (HdhQ150) develops a phenotype reminiscent of that seen in the commonly used R6/2 mouse model of HD albeit on a different time scale (Woodman, Butler et al. 2007). Additional reports have illustrated similarities in disease pathology in these mouse models including: aggregate formation (Sathasivam, Lane et al. 2010), motor abnormalities (Heng, Tallaksen-Greene et al. 2007), and physiological alterations in synaptic molecules (Heng, Detloff et al. 2009). It remains important to characterize similarities between these models as it is the knock-in model that seems to hold the most physiological relevance for the adult onset form of HD. Further, as the production of these knock-in mice involved a two-step gene targeting approach, only the size of the CAG repeat length differentiates the WT mouse from its knock-in littermates (Lin, Tallaksen-Greene et al. 2001).
In order to investigate whether PC survival and function are compromised in the HdhQ200 model, we performed molecular and electrophysiological characterizations of the alterations within the cerebellum. Here we show a reduction in transcript and protein levels of a specific PC marker (calbindin) and a PC-specific decrease in glutamic acid decarboxylase 67 and parvalbumin in symptomatic HdhQ200/Q200 mice. We also present data showing a reduction in PC number by over 50%, mirroring PC loss in the R6/2 mouse. Additionally, we see a reduction in spike rate in remaining PCs in symptomatic mice. These data indicate that PCs are vulnerable in the HdhQ200/Q200 knock-in model of HD and that this mouse model would be useful in determining the factors that contribute to PC loss and dysfunction in adult HD.
Methods
Animals
The Institutional Animal Care and Use Committee of the University of Alabama at Birmingham approved all experimental protocols. The HdhQ200 mice were maintained as described previously (Heng, Duong et al. 2010). Mice were bred using heterozygous HdhQ100 or HdhQ200 mating pairs in order to yield homozygous, heterozygous and wildtype (WT) mice. 50-week-old male and female mice were utilized for all experiments. These mice were housed in groups of up to 5 animals per cage with food and water ad libitum
Genotyping
Genotyping was performed as described in (Heng, Tallaksen-Greene et al. 2007). Briefly, DNA was isolated from tail samples using DNeasy Tissue Kit (Qiagen, Valencia, CA). PCR reactions utilized a Taq polymerase kit (Invitrogen, Carlsbad, CA) and primers (Invitrogen) specific to the CAG repeat in the Hdh locus. PCR products were visualized using agarose gel electrophoresis with ethidium bromide and read on a Gel Doc system (Bio-Rad, Hercules, CA). Periodically, tail samples were sent to Laragen (Laragen Inc., Los Angeles, CA, USA) for CAG sizing to confirm CAG stability and consistency of the CAG repeat number.
Gene Expression Analysis
Protocols were performed as described (Lucas, Markwardt et al. 2010). Briefly, mice were anesthetized with isoflourane and sacrificed by decapitation. Brains were removed and dissected by anatomical region. Tissues were flash frozen on dry ice and kept in the −80°C freezer until use. At least 12 hours prior to processing, tissues were placed in RNAlater-ICE (Ambion Austin, TX), cooled to −80°C, and kept at −20°C until homogenization. Homogenization was performed with a Tissue-Tearor homogenizer (Biospec Bartlesville, OK) in Trizol following the manufacturer’s instructions (Invitrogen Grand Island, NY). Taqman PCR was conducted with JumpStart Taq Readymix (Sigma St. Louis, MO) and Applied Biosystems (Carlsbad, California) primers for calbindin (Mm00486645_m1) and β-actin (Mm00607939_s1). Actin values were not different between groups (p>0.05).
Immunofluorescence
Mice were anesthetized with isofluorane and perfused intracardially with ice-cold 4% paraformaldehyde in phosphate buffered saline (PBS). Brains were removed and postfixed for an additional 24–72 hours. The samples were cryoprotected in graded sucrose over a 5 day period and then embedded and frozen in a mixture of 20% sucrose and Tissue-Tek O.C.T. Compound (Sakura Finetek USA, Inc. Torrance, CA) and stored at −80°C until use. Tissue blocks were sectioned at 30 μm, and sections were mounted onto charged slides (Fisher) and allowed to dry overnight before storage at −80°C. Slides were then thawed and washed with PBS followed by a one hour incubation in 10% serum (from the host of the secondary antibody) and PBS with 3% bovine serum albumin (BSA). The sections were then incubated overnight with a predetermined concentration of primary antibody and 5% serum in 0.3% TritonX PBS with 3% BSA. This was followed by washing and then a 2 hour incubation with the corresponding fluorescence-conjugated secondary antibody (Jackson ImmunoResearch West Grove, PA) and 5% serum in 0.3% TritonX PBS with 3% BSA. The slides were then immediately mounted using an antifade media containing DAPI, coverslipped and left to dry at room temperature overnight. When necessary, a Vector (Burlingame, CA) Mouse on Mouse Kit (Fluorescein Cat No. FMK-2201) was used to minimize background staining for experiments involving mouse-made antibodies. For comparison of immunofluorescence intensity between experimental groups, immunofluorescence was performed on approximately 16 sagittal sections/mouse throughout the extent of the cerebellum to initially evaluate staining patterns. Representative sections were chosen for imaging on a Leica confocal microscope (Leica Microsystems Inc; Buffalo Grove, IL); using wildtype cerebellum, intensity settings (gain and laser power) were set to allow clear visualization of purkinje cell bodies and processes. Then, PCs from HdhQ200 mice were visualized with the same settings. For evaluation of differences in intensity between the experimental groups in the PC layer and the molecular layer, a similar approach was utilized at a higher magnification.
Primary Antibody Information
Immunostaining was performed with the following antibodies: mouse anti-GAD67 (MAB5406, Millipore Billerica, MA) at a dilution of 1:500; mouse anti-Calb (C9848, Sigma) at a dilution of 1:2000; rabbit polyclonal anti-PV (PV25, Swant) at a dilution of 1:500; and goat polyclonal anti-mutant huntingtin (SC-8767, Santa Cruz) at a dilution of 1:100.
Stereology
Mice were anesthetized with isofluorane and perfused intracardially with ice-cold 4% paraformaldehyde in PBS. Brains and sections were prepared as described above for immunofluorescence. Sections were thawed and washed with distilled H2O for 5 minutes followed by a 10 minute incubation in hematoxylin (Sigma). Slides were washed in running H2O for 15 minutes followed by a 1 minute incubation in eosin (Sigma). Slides were then progressively dehydrated and cover-slipped using Permount (Fisher).
Cells were counted by an unbiased investigator blinded to animal genotype using StereoInvestigator (MicroBrightField, Inc Williston, VT). The optical fractionator method was used to generate an estimate of PCs counted in an unbiased selection of serial sections in a defined volume of the cerebellum. 30–35 μm thick sagittal tissue sections were serially cut with analysis performed on every twentieth section throughout the entire cerebellum of mice in each cohort (n=3/genotype). The number of PCs within the entire region was attained using the optical fractionator method with dissectors placed randomly according to a 263.68 × 212.13 μm grid. The dissector height was 26 μm and the counting frame was 60 × 60 μm. On average, 150 sites were counted per mouse. An estimation of cell population number was provided using overall raw counts and mean mounted section thickness (to account for tissue shrinkage).
Cell-Attached Loose Patch Recording
Mice were anesthetized with isoflourane and sacrificed by decapitation. Brains were placed in ice-cold artificial cerebrospinal fluid (ACSF) containing the following (in mM): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 25 NaHCO3 1.25 Na2HPO4 and 25 D-glucose with a pH of 7.4 and osmolality of 295 ± 5 mOsm. This external solution was bubbled with 95%O2/5%CO2. Sagittal cerebellar slices (300 μm thick) were cut using a Vibratome (7000 smz, Campden Instruments: Sarasota, FL). The resultant slices were allowed to rest for 60 min at room temperature (22–23°C); all experiments were then performed at room temperature. Slices were superfused continuously with oxygenated recording ACSF and viewed with an upright microscope (Zeiss Axio Examiner A1) using infrared-differential interference contrast optics. Loose patch recordings were acquired from visually-identified PCs using Axio Vision 4.8 software. Cellular activity was recorded using internal solution containing the following (in mM): 140 K-gluconate, 1 EGTA, 10 HEPES and 5 KCL, pH 7.3. Pipette tip resistance was 2–5 MΩ. The extracellular recording pipettes, containing the internal solution, were placed directly adjacent to the soma of the cell of interest. Positive pressure was applied throughout this process followed by a brief release of pressure to form a seal averaging approximately 45 MΩ. Recordings were obtained using an Axon CNS Molecular Devices amplifier (Multiclamp 700B), digitized at 20 kHz (Digidata 1440A), and filtered at 10 kHz. A 3 min gap-free protocol on Clampex 10.2 software was used for data collection. Event detection and analysis of event frequency and inter-event interval were performed semi-automatically using the program Clampfit 10.2. The detection threshold was set for analysis based on the event amplitude from a given cell.
Data Analyses
Data analyses for qRT-PCR, stereology, and loose patch electrophysiology were performed using Microsoft Excel and GraphPad Prism software. One-way ANOVAs were performed followed by post-hoc Tukey’s multiple comparison tests were utilized to assess statistical significance. Values were considered statistically significant when the p value was less than 0.05.
Results
Calbindin mRNA and protein is reduced in a knock-in model of HD
To determine whether PC loss was a possibility in the HdhQ(n) models of HD, we first determined whether there were alterations in transcript levels of the PC marker and calcium buffer calbindin (Calb) in mice homozygous for the mutant htt gene and with either 100 CAG (HdhQ100/Q100) or 200 CAG (HdhQ200/Q200) (n=10/group) at 50 weeks of age. There was a decrease in Calb transcript in the HdhQ200/Q200 mice as compared to WT littermate controls (Fig 1B; p<0.05). Additionally, there was a trend towards a reduction in the levels of Calb transcript in HdhQ100/Q100 compared to WT littermate controls (p<0.1). Assessment of haploid expression of the HdhQ200 mutant htt transgene revealed that heterozygotic expression resulted in a reduction in Calb (Fig 1B) from WT levels with no statistical difference between HdhQ200/+ and HdhQ200/Q200 mice. Considering that no alterations in Calb expression were observed in the HdhQ100/Q100, we chose to focus on the characterization of HdhQ200/Q200 and heterozygous (HdhQ200/+) mice as compared to age-matched WT mice.
Figure 1. HD mice exhibit reductions in calbindin mRNA expression.
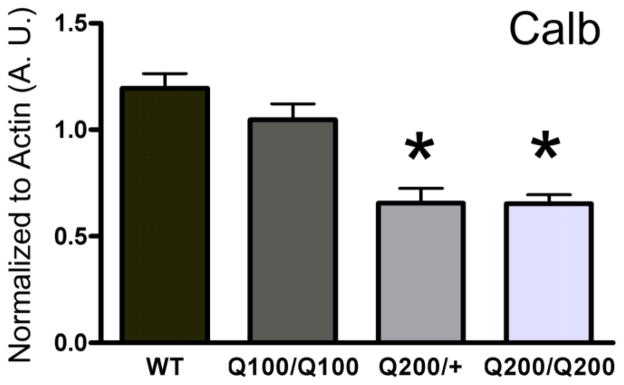
Using q-RT-PCR to measure transcript levels in cerebellum homogenates, expression of the purkinje marker calbindin (Calb) was significantly reduced in homozygous HdhQ200/Q200 mice at 50 weeks (n = 10/group, p<0.05). There was no alteration in the transcript level of Calb in homozygous HdhQ100/Q100 mice when compared to WT controls (n= 10/group; p<0.1). Assessment of haploid expression of the mutant htt transgene revealed that heterozygous expression resulted in a reduction in Calb from WT (as well as HdhQ100/Q100) levels with no statistical difference between HdhQ200/+ and HdhQ200/Q200 mice. One-way ANOVA was performed followed by post-hoc Tukey’s Multiple Comparison Tests with error bars representing SEM (*p<0.05).
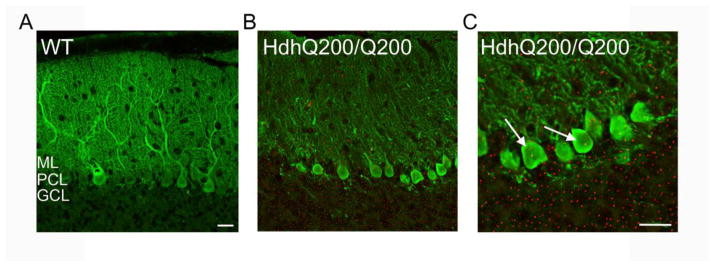
To determine whether Calb expression was reduced at the protein level, we performed immunofluorescence on WT and HdhQ200/Q200 mice with an antibody against Calb. Staining patterns revealed a reduction in staining intensity throughout the purkinje and molecular layers (Fig 2A–D). Staining intensity was most reduced in the molecular layer, potentially reflecting a decreased density of dendritic trees or a loss of calbindin in intact dendrites. To investigate this further, we evaluated the intensity of staining for two other proteins concentrated in PCs: glutamic acid decarboxylase (GAD67) (Fig 2 E–H) and parvalbumin (PV) (Fig 2I–L). There was a marked reduction in staining intensity for GAD67 and PV in the HdhQ200/Q200 mice as compared to WT mice especially in molecular layer (Fig 2G, H, K, L). However, while PC bodies were still visible with the Calb and GAD67 immunofluorescence, PV-positive cell bodies in the PC layer were almost non-existent, suggesting that PV is reduced within intact cell bodies (arrowheads, Fig 2J). Additionally, as evidenced by the PV staining, interneurons of the molecular layer were still present (arrows; Fig 2J). This staining pattern is strikingly similar to the one we previously observed in the R6/2 model (Dougherty, Reeves et al. 2012) as well as to the pattern reported in a model of spinocerebellar ataxia-1 (Vig, Subramony et al. 1998). Importantly, at this age, mutant htt inclusions were present within PCs of HdhQ200/Q200 mice (Fig 3).
Figure 2. Immunoreactivity for calbindin (Calb), GAD67, and parvalbumin (PV) is reduced in the Purkinje cell (PC) layer of the cerebellum of 50 week-old HdhQ200/Q200 mice.

In order to assess protein level changes in Calb, immunofluorescence was performed on HdhQ200/Q200. Staining revealed reductions of the protein levels of Calb (A–D) in HdhQ200/Q200 mice as compared to WT. Reductions in other GABAergic cell markers, GAD67 (E–H) and PV (I–J), were also evident as compared at WT. As exhibited by the intense staining pattern, the interneurons of the molecular layer remain PV+, arrows in J. PCs are still evident in (J) as highlighted by arrowheads though the PV protein levels appear to be reduced. Higher magnification images are shown highlighting reductions in the protein levels of the GABAergic cell markers tested in the neurophil (C, D, G, H, K, and L). ML: Molecular Layer. PCL: PC Layer. GCL: Granule Cell Layer. Representative images shown. (n=3 animals/group) Scale bars: 25 μm.
Figure 3. Huntingtin-positive inclusions are found in the PCs of HdhQ200 mice.
In order to localize mutant htt inclusions to specific cell types in these mice, immunofluorescence was performed with an antibody specific to mutant htt (in red) and calbindin (in green) for visualization of the PC layer. Very limited huntingtin staining was seen in the age matched WT (A). 50 week- old HdhQ200/Q200 mice exhibit staining throughout the cerebellum (B). Inclusions are seen in the interneurons of the molecular layer and throughout the PC layer (arrows in C). ML: Molecular Layer. PCL: PC Layer. GCL: Granule Cell Layer. Representative images shown. (n=3 animals/group) Scale bars: 25 μm
Homozygous HdhQ200 mice exhibit a reduction in PC number at 50 weeks of age
The loss of multiple PC markers in HdhQ200/Q200 mice suggested that the number of PCs may be reduced in this mouse model. In order to investigate whether alterations in PC-specific proteins were the result of reductions in cell numbers, we performed unbiased stereological counts on Hemotaxylin & Eosin-stained serial sections of the cerebellum (n=3/group). Counts revealed a significant reduction in the overall PC population in 50 week old HdhQ200/Q200 mice as compared to age-matched WT controls (Fig 4; p<0.05), while PC number was not significantly reduced in the heterozygous HdhQ200/+ mice. Assessment of the population number using mean section thickness gave an estimation of an average of 103018 cells in WT, 73559 cells in HdhQ200/+ and 64315 cells in HdhQ200/Q200 mice (38% reduction in PC number in HdhQ200/Q200 as compared to WT) (Fig. 4B).
Figure 4. HDhQ200 mice exhibit reductions in PC number.

A, Hematoxylin and Eosin staining was performed on 30 micron thick cerebellar sections from WT, HdhQ200/+, and HdhQ200/Q200 mice. Representative pictures are shown; arrowheads highlight positively stained PCs. Stereological analysis reveal a significant reduction in the number of PCs between WT and HdhQ200/Q200 mice at 50 weeks of age, quantification in B (n=3/group; *p<0.05). One-way ANOVA was performed followed by post-hoc Tukey’s Multiple Comparison Tests with error bars representing SEM. Scale bar: 100 μm.
Spike Frequency is reduced from the PCs of 50 week old homozygous HdhQ200 mice
As it is possible that the remaining PCs could compensate on a functional level for the loss of PCs, we next sought to determine whether the functional activities of the remaining PCs were altered in the HdhQ200 mice. Given that a change in firing frequency was an early robust measure of PC dysfunction in the R6/2 mice (Dougherty, Reeves et al. 2012), cell-attached loose patch electrophysiology was performed in 50 week old HdhQ200/Q200 and HdhQ200/+ mice (n=3–6 mice/group; 10–12 cells/group). Loose patch recordings revealed a significant reduction in spike frequency in the HdhQ200/Q200 mice as compared to age-matched WT controls (Fig 5; p<0.05), while there was no significant difference in firing rate in the HdhQ200/+ mice as compared to age-matched WT controls.
Figure 5. Decreased firing frequency from PCs is observed in HdhQ200 mice.
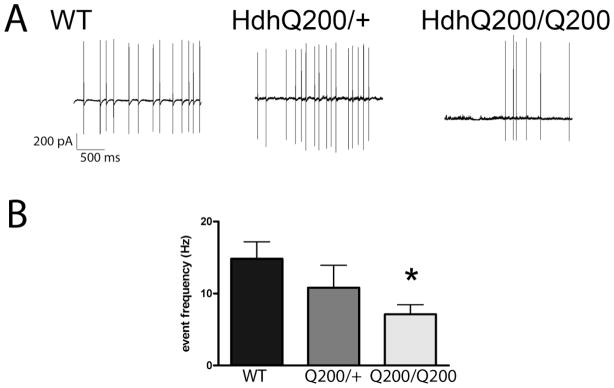
A. Using 50 week-old HdhQ200/+, HdhQ200/Q200 mice and age matched WT controls, loose patch recordings were performed on acute cerebellar slices (300 micron thick); sample traces are shown. Recordings lasted for 3 minutes and were then analyzed using clampex software, with quantification in B. There was a significant decrease in event number and interevent interval (data not shown) between the WT and HdhQ200/Q200 mice (*p<0.05). There was no significant difference between HdhQ200/+ and either group, though a trend was present between each (p<0.1; n= 3 animals and 10 cells/group). One-way ANOVA was performed followed by post-hoc Tukey’s Multiple Comparison Tests with error bars representing SEM.
Discussion
HD is a devastating neurological disorder characterized by cognitive, psychiatric and motor dysfunction. The motor component, termed chorea, along with the early observation of overt cell loss in the striatum, has previously led to a focus on the motor pathways of the basal ganglia as the source of this dysfunction (Lange, Thorner et al. 1976; Vonsattel, Myers et al. 1985). However, emerging evidence suggests that the main output neuron of the cerebellar cortex, the Purkinje Cell (PC), is dysfunctional in early onset HD. A recent study from our laboratory found that PCs are lost in the R6/2 model of HD (Dougherty, Reeves et al. 2012); in light of postmortem data indicating that signs of cell death are observed in PCs (Turmaine, Raza et al. 2000) and PC numbers are reduced in HD (Jeste, Barban et al. 1984), we hypothesized that PC number, and potentially function, would also be affected in mouse models of adult-onset HD.
We show that decreases in PC-specific transcript and protein levels occur as part of the disease etiology in the HdhQ200 mouse model of HD. Furthermore, we demonstrate that PCs are lost in symptomatic mice and that PCs are dysfunctional at a physiological level, with a dramatic reduction in firing rate. This demonstration of molecular and functional deficits in PCs directly mirrors what was observed in the severe R6/2 line that displays an early onset and aggressive phenotype. Importantly, these findings suggest that further experiments should investigate the contribution of PC dysfunction to HD-associated motor deficits.
Within the field, early studies suggested a role for the cerebellum in HD as shown by cerebellar atrophy in both patients and mouse models (Kageyama, Yamamoto et al. 2003; Fennema-Notestine, Archibald et al. 2004; Ruocco, Lopes-Cendes et al. 2006; Sakazume, Yoshinari et al. 2009; Nicolas, Devys et al. 2011). However, due to striatal atrophy and the marked loss of medium spiny neurons observed in HD patients and animal models, there has been a focus on the striatum in many studies of HD etiology. One major reason that the cerebellum is overlooked may be the lack of an overt cerebellum-associated motor phenotype. However, some work has shown that ataxia is present as part of the motor pathology (Koller and Trimble 1985). Further, it has been suggested that an ataxia-like dysfunction may be masked by the more obvious striatal motor pathologies. The vast majority of studies performed on the cerebellum explore alterations at the regional level. One caveat to this approach is that cerebellar neuronal populations are very diverse and, as a result, whole homogenate studies could dilute effects occurring in restricted cell populations. Recent PC single-cell transcription studies have shown mirroring reductions in mRNA levels of a transcription factor essential for PC development in HD and spinocerebellar ataxia type 1 (Serra, Duvick et al. 2006; Euler, Friedrich et al. 2012).
In addition to PC loss, reductions in the levels of the calcium buffer PV within intact cells would be expected to have major implications for the overall function of PCs. Studies have suggested that mature PCs lack functional NMDA receptors (Farrant and Cull-Candy 1991; Llano, Leresche et al. 1991) and therefore rely heavily on Ca2+ influx through voltage-gated Ca2+ channels and Ca2+ release from intracellular stores. As a potent calcium buffer, PV is an essential cytosolic moderator of excitotoxicity in the PCs. Loss of this buffer has been shown to result in an alteration in PC firing (Servais, Bearzatto et al. 2005) and a concomitant dysfunction in locomotor behavior (Farre-Castany, Schwaller et al. 2007). The reduction in molecules observed in the HdhQ200/+ suggests that haploid expression of the mutant htt allele is sufficient to cause the transcriptional alterations; though with the population reduction only trending towards a difference from WT, haploid mutant htt expression is not sufficient to cause the loss of cells or functional abnormalities.
The functional consequence of a reduction of PC firing, while not investigated here, could lead to alterations in firing from the deep cerebellar nuclei. Alterations in PC firing have been shown to result in propagation of misfired signals to the downstream cerebellar nuclei in other CAG repeat disorders involving the cerebellum (reviewed in (Shakkottai and Paulson 2009). Importantly, PCs have been shown to be particularly dependent on activity for survival (Morrison and Mason 1998); it is possible that a reduction in overall PC activity could contribute to the observed alteration in cell viability. Additionally, it would be interesting to investigate the morphology of the remaining PCs and whether the molecular layer of the cerebellum is thinner in HdhQ200/200 mice; while preliminary examination of spine density of remaining PCs indicates that there is no overt loss of spines in HdhQ200/200 PCs, a reduction in overall cell volume and/or spines could influence excitability.
Taken together, these data highlight the vulnerability of the PC population in a mouse model of adult-onset HD. Further investigation into the cell-autonomous versus non cell-autonomous effects of mutant htt on PC function and viability would increase our understanding of the role of PC dysfunction in the motor phenotype in HD with the ultimate goal of improving motor function and quality of life in people with HD.
Table 1.
Quantitative RT-PCR specifications
| Sample/Template | details |
|---|---|
|
| |
| Source | mouse cerebellum |
| Method of Preservation | dry ice/RNAlater-ICE |
| Storage time | <6 mo prior to isolation |
| Handling | Fresh |
| Extraction Method | TriZol |
| RNA: DNA-free | Intron-spanning primers, DNAse step |
| Concentration | Nanodrop |
|
| |
| Assay optimisation/validation | |
|
| |
| Accession number | Calb: Mm00486645_ml |
| Actin: Mm00607939_sl | |
| Amplicon Results | Calbindin: Chr.4: 15808411 – 15833856 (length 124) |
| Actin: Chr.5: 143664795 – 143668403 (length 115) | |
| Priming conditions | Oligo-dT |
| PCR efficiency | Calb: 93.6; Actin: 87.6 |
|
| |
| RT/PCR | |
|
| |
| Protocols | in text |
| Reagents | in text |
| NTC | undetectable |
| NAC | undetectable |
| Positive controls | none |
|
| |
| Data Analysis | |
|
| |
| Specialist software | calibrator method; BioRad CFX Manager Version 1.6 |
| Statistical justification | biological replicates |
| Transparent, validated normalisation | normalization to actin |
Acknowledgments
This work was funded by National Institutes of Health (NIH) Grant 1R01NS070009-01 (R.M.C.). Imaging studies and stereology were supported by NIH Neuroscience Blueprint Core Grant NS57098 to the University of Alabama at Birmingham. We would also like to acknowledge Dr. David Standaert and his laboratory for microscopy training and access to the Leica confocal microscope, and Dr. John Hablitz for guidance with electrophysiology techniques and access to the electrophysiology rig.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andre VM, Cepeda C, et al. Differential electrophysiological changes in striatal output neurons in Huntington’s disease. J Neurosci. 2011;31(4):1170–1182. doi: 10.1523/JNEUROSCI.3539-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bithell A, Johnson R, et al. Transcriptional dysregulation of coding and non-coding genes in cellular models of Huntington’s disease. Biochem Soc Trans. 2009;37(Pt 6):1270–1275. doi: 10.1042/BST0371270. [DOI] [PubMed] [Google Scholar]
- Cha JH, Kosinski CM, et al. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc Natl Acad Sci U S A. 1998;95(11):6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings DM, V, Andre M, et al. Alterations in cortical excitation and inhibition in genetic mouse models of Huntington’s disease. J Neurosci. 2009;29(33):10371–10386. doi: 10.1523/JNEUROSCI.1592-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty SE, Reeves JL, et al. Disruption of Purkinje cell function prior to huntingtin accumulation and cell loss in an animal model of Huntington disease. Exp Neurol. 2012;236(1):171–178. doi: 10.1016/j.expneurol.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty SE, Reeves JL, et al. Disruption of Purkinje cell function prior to huntingtin accumulation and cell loss in an animal model of Huntington Disease. Exp Neurol. 2012 doi: 10.1016/j.expneurol.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euler P, Friedrich B, et al. Gene expression analysis on a single cell level in Purkinje cells of Huntington’s disease transgenic mice. Neurosci Lett. 2012;517(1):7–12. doi: 10.1016/j.neulet.2012.03.080. [DOI] [PubMed] [Google Scholar]
- Farrant M, Cull-Candy SG. Excitatory amino acid receptor-channels in Purkinje cells in thin cerebellar slices. Proc Biol Sci. 1991;244(1311):179–184. doi: 10.1098/rspb.1991.0067. [DOI] [PubMed] [Google Scholar]
- Farre-Castany MA, Schwaller B, et al. Differences in locomotor behavior revealed in mice deficient for the calcium-binding proteins parvalbumin, calbindin D-28k or both. Behav Brain Res. 2007;178(2):250–261. doi: 10.1016/j.bbr.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Fennema-Notestine C, Archibald SL, et al. In vivo evidence of cerebellar atrophy and cerebral white matter loss in Huntington disease. Neurology. 2004;63(6):989–995. doi: 10.1212/01.wnl.0000138434.68093.67. [DOI] [PubMed] [Google Scholar]
- Giacomello M, Hudec R, et al. Huntington’s disease, calcium, and mitochondria. Biofactors. 2011;37(3):206–218. doi: 10.1002/biof.162. [DOI] [PubMed] [Google Scholar]
- Group, H. s. D. C. R. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72(6):971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Heng MY, Detloff PJ, et al. In vivo evidence for NMDA receptor-mediated excitotoxicity in a murine genetic model of Huntington disease. J Neurosci. 2009;29(10):3200–3205. doi: 10.1523/JNEUROSCI.5599-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng MY, Duong DK, et al. Early autophagic response in a novel knock-in model of Huntington disease. Hum Mol Genet. 2010;19(19):3702–3720. doi: 10.1093/hmg/ddq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng MY, Tallaksen-Greene SJ, et al. Longitudinal evaluation of the Hdh(CAG)150 knock-in murine model of Huntington’s disease. J Neurosci. 2007;27(34):8989–8998. doi: 10.1523/JNEUROSCI.1830-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges A, Strand AD, et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet. 2006;15(6):965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- Imarisio S, Carmichael J, et al. Huntington’s disease: from pathology and genetics to potential therapies. Biochem J. 2008;412(2):191–209. doi: 10.1042/BJ20071619. [DOI] [PubMed] [Google Scholar]
- Jeste DV, Barban L, et al. Reduced Purkinje cell density in Huntington’s disease. Exp Neurol. 1984;85(1):78–86. doi: 10.1016/0014-4886(84)90162-6. [DOI] [PubMed] [Google Scholar]
- Kageyama Y, Yamamoto S, et al. A case of adult-onset Huntington disease presenting with spasticity and cerebellar ataxia, mimicking spinocerebellar degeneration. Rinsho Shinkeigaku. 2003;43(1–2):16–19. [PubMed] [Google Scholar]
- Klapstein GJ, Fisher RS, et al. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington’s disease transgenic mice. J Neurophysiol. 2001;86(6):2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- Koller WC, Trimble J. The gait abnormality of Huntington’s disease. Neurology. 1985;35(10):1450–1454. doi: 10.1212/wnl.35.10.1450. [DOI] [PubMed] [Google Scholar]
- Lange H, Thorner G, et al. Morphometric studies of the neuropathological changes in choreatic diseases. J Neurol Sci. 1976;28(4):401–425. doi: 10.1016/0022-510x(76)90114-3. [DOI] [PubMed] [Google Scholar]
- Lin CH, Tallaksen-Greene S, et al. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum Mol Genet. 2001;10(2):137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- Llano I, Leresche N, et al. Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron. 1991;6(4):565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- Lucas EK, Markwardt SJ, et al. Parvalbumin deficiency and GABAergic dysfunction in mice lacking PGC-1alpha. J Neurosci. 2010;30(21):7227–7235. doi: 10.1523/JNEUROSCI.0698-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, et al. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet. 2002;11(17):1911–1926. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87(3):493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Milnerwood AJ, Raymond LA. Corticostriatal synaptic function in mouse models of Huntington’s disease: early effects of huntingtin repeat length and protein load. J Physiol. 2007;585(Pt 3):817–831. doi: 10.1113/jphysiol.2007.142448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison ME, Mason CA. Granule neuron regulation of Purkinje cell development: striking a balance between neurotrophin and glutamate signaling. J Neurosci. 1998;18(10):3563–3573. doi: 10.1523/JNEUROSCI.18-10-03563.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AJ, Faull RL, et al. Abnormalities in the synaptic vesicle fusion machinery in Huntington’s disease. Brain Res Bull. 2001;56(2):111–117. doi: 10.1016/s0361-9230(01)00611-6. [DOI] [PubMed] [Google Scholar]
- Nicolas G, Devys D, et al. Juvenile Huntington disease in an 18-month-old boy revealed by global developmental delay and reduced cerebellar volume. Am J Med Genet A. 2011;155A(4):815–818. doi: 10.1002/ajmg.a.33911. [DOI] [PubMed] [Google Scholar]
- Perry GM, Tallaksen-Greene S, et al. Mitochondrial calcium uptake capacity as a therapeutic target in the R6/2 mouse model of Huntington’s disease. Hum Mol Genet. 2010;19(17):3354–3371. doi: 10.1093/hmg/ddq247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Albin RL, et al. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988;85(15):5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds GP, Dalton CF, et al. Brain neurotransmitter deficits in mice transgenic for the Huntington’s disease mutation. J Neurochem. 1999;72(4):1773–1776. doi: 10.1046/j.1471-4159.1999.721773.x. [DOI] [PubMed] [Google Scholar]
- Rodda RA. Cerebellar atrophy in Huntington’s disease. J Neurol Sci. 1981;50(1):147–157. doi: 10.1016/0022-510x(81)90049-6. [DOI] [PubMed] [Google Scholar]
- Roos RA, Pruyt JF, et al. Neuronal distribution in the putamen in Huntington’s disease. J Neurol Neurosurg Psychiatry. 1985;48(5):422–425. doi: 10.1136/jnnp.48.5.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas HD, Koroshetz WJ, et al. Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology. 2003;60(10):1615–1620. doi: 10.1212/01.wnl.0000065888.88988.6e. [DOI] [PubMed] [Google Scholar]
- Ruocco HH, Lopes-Cendes I, et al. Clinical presentation of juvenile Huntington disease. Arq Neuropsiquiatr. 2006;64(1):5–9. doi: 10.1590/s0004-282x2006000100002. [DOI] [PubMed] [Google Scholar]
- Sakazume S, Yoshinari S, et al. A patient with early onset Huntington disease and severe cerebellar atrophy. Am J Med Genet A. 2009;149A(4):598–601. doi: 10.1002/ajmg.a.32707. [DOI] [PubMed] [Google Scholar]
- Sathasivam K, Lane A, et al. Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington’s disease. Hum Mol Genet. 2010;19(1):65–78. doi: 10.1093/hmg/ddp467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra HG, Duvick L, et al. RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell. 2006;127(4):697–708. doi: 10.1016/j.cell.2006.09.036. [DOI] [PubMed] [Google Scholar]
- Servais L, Bearzatto B, et al. Mono- and dual-frequency fast cerebellar oscillation in mice lacking parvalbumin and/or calbindin D-28k. Eur J Neurosci. 2005;22(4):861–870. doi: 10.1111/j.1460-9568.2005.04275.x. [DOI] [PubMed] [Google Scholar]
- Shakkottai VG, Paulson HL. Physiologic alterations in ataxia: channeling changes into novel therapies. Arch Neurol. 2009;66(10):1196–1201. doi: 10.1001/archneurol.2009.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand AD, Aragaki AK, et al. Gene expression in Huntington’s disease skeletal muscle: a potential biomarker. Hum Mol Genet. 2005;14(13):1863–1876. doi: 10.1093/hmg/ddi192. [DOI] [PubMed] [Google Scholar]
- Turmaine M, Raza A, et al. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington’s disease. Proc Natl Acad Sci U S A. 2000;97(14):8093–8097. doi: 10.1073/pnas.110078997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig PJ, Subramony SH, et al. Reduced immunoreactivity to calcium-binding proteins in Purkinje cells precedes onset of ataxia in spinocerebellar ataxia-1 transgenic mice. Neurology. 1998;50(1):106–113. doi: 10.1212/wnl.50.1.106. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, et al. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44(6):559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Woodman B, Butler R, et al. The Hdh(Q150/Q150) knock-in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain Res Bull. 2007;72(2–3):83–97. doi: 10.1016/j.brainresbull.2006.11.004. [DOI] [PubMed] [Google Scholar]