

Abstract
Mice overexpressing TLR7 (TLR7.1 mice) are a model of SLE pathogenesis and exhibit peripheral myeloid expansion. We show that TLR7.1 mice have a dramatic expansion of splenic cells that derive from granulocyte/macrophage progenitors (GMP) compared to WT mice. In the bone marrow, TLR7.1 mice exhibited hallmarks of emergency myelopoiesis and contained a discrete population of Sca-1+ GMP, termed emergency GMP (eGMP), that are more proliferative and superior myeloid precursors than classical Sca-1− GMP. The emergency myelopoiesis and peripheral myeloid expansion in TLR7.1 mice was dependent on type I IFN signaling. TLR7 agonist administration to non-transgenic mice also drove type I IFN-dependent emergency myelopoiesis. TLR7.1 plasmacytoid DC were cell-intrinsically activated by TLR7 overexpression and constitutively produced type I IFN directly ex vivo. This work shows that type I IFN can act upon myeloid progenitors to promote the development of eGMP, which leads to an expansion of their progeny in the periphery.
Introduction
The hematopoietic system is capable of rapidly increasing myeloid cell output to combat pathogens. Accordingly, viral, bacterial and parasitic infection results in significant reprogramming of steady state hematopoiesis (1). This demand-driven hematopoietic state is a component of “emergency myelopoiesis,” a process in which myeloid effector cells are rapidly generated in the bone marrow (BM) and shunted to the periphery (2).
The mechanisms governing the shift from steady state hematopoiesis to emergency myelopoiesis are not completely understood. In vitro studies indicate that TLR signaling in hematopoietic progenitor cells can intrinsically alter their development into different hematopoietic lineages (3). TLR signaling may also indirectly influence hematopoiesis by inducing production of cytokines, including the type I IFN family. Type I IFN can act directly on quiescent Long-Term Hematopoietic Stem Cells (LT-HSC) and cause them to enter the cell cycle (4), but the role of type I IFN in influencing myeloid development is unknown. Here, we use mice with myeloid expansion due to overexpression of TLR7 through a bacterial artificial chromosome transgene (TLR7.1 mice), TLR7 agonist administration and in vitro culture of myeloid progenitor cells (5) to show that type I IFN driven by chronic TLR7 signaling can drive emergency myelopoiesis.
Materials and Methods
Mice
C57BL/6 mice were purchased from Charles River or bred in our vivarium. TLR7.1 mice (5) and IFNARKO mice were provided by S. Bolland (NIH) and D.B. Stetson (University of Washington), respectively. Mixed BM chimeric mice were generated by lethally irradiating C57BL/6 x B6.SJL F1 mice and reconstituting them with a 1:1 ratio of C57BL/6 (CD45.2) and B6.SJL (CD45.1) or TLR7.1 (CD45.2) and B6.SJL (CD45.1) BM cells. Mice were housed at the University of Washington or Benaroya Research Institute. Experiments were performed under IACUC-approved protocols.
Cell isolation
Lineage-negative progenitor cells from 2 femurs and tibias were enriched from RBC-lysed BM using a Lineage Cell Depletion Kit (Miltenyi Biotec). Biotin-conjugated mAbs to CD11b, Gr1, F4/80, CD11c, CD3 and NK1.1 were added to the lineage-negative cell suspensions for negative gating during flow cytometry. Splenocytes were isolated by collagenase (type IV, Worthington)/DNase (Sigma Aldrich) digestion followed by calcium chelation and RBC lysis.
Flow cytometry, cell sorting, BrdU and TLR7 agonist administration
For surface staining, 2–4 × 106 cells were blocked with fluorescently labeled (lineage negative progenitors) or unlabeled (spleen) anti-CD16/32 mAb followed by the addition of mAbs as indicated. Samples were incubated at 4C for 30 minutes, except for anti-CD34 stains for 60–90 min. All mAbs were from eBioscience, Biolegend or Invitrogen. Data were acquired using a LSRII (BD Biosciences) and analyzed using FlowJo (TreeStar). Doublets were excluded before gating on live cells using FSC and SSC. For BrdU incorporation, mice were injected i.p. with 1 mg of BrdU (Sigma Aldrich) 1 hour before sacrifice. BrdU incorporation was assayed by using the BD BrdU Flow Kit procedure (BD Biosciences). Where indicated, 10 μg of the TLR7 agonist gardiquimod (Invivogen) was administered i.v. every other day over the course of 6 days.
Quantitative real-time PCR (qPCR)
Samples were prepared as previously described (6). Primer sequences are listed in Supplemental Fig. 2J.
Bio-plex assay
Serum samples were analyzed using Bio-Plex Pro Mouse Cytokine 23-plex Assay with a Bio-Plex 200 system (Bio-rad).
Colony forming unit assays
250 GMP or eGMP from WT or TLR7.1 mice were sorted and plated in duplicate for 6 days in Methocult GF M3534 (Stemcell Technologies). Colonies were scored on day 5. On day 6, cells were extracted and quantified by flow cytometry using polystyrene counting beads (Polysciences, Inc.).
Data presentation and statistical analysis
Graphs and histograms were generated using Prism (GraphPad) using a two-tailed Student’s t-test or one-way ANOVA with a tukey post-test, as noted. Error bars correspond to SEM. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Results and Discussion
TLR7.1 mice have differential expansion of splenic myeloid populations
TLR7.1 mice were previously shown to develop severe splenomegaly characterized by the expansion of cells expressing the surface markers CD11c and CD11b (5), which mark a variety of myeloid populations. We more precisely defined the myeloid expansion in TLR7.1 mice and observed that TLR7.1 mice exhibited an increased frequency and absolute number of inflammatory monocytes and neutrophils in their spleens compared to WT mice (Supplemental Fig. 1A). Unexpectedly, we found that TLR7.1 mice exhibited a diminished percentage of splenic plasmacytoid dendritic cells (pDC), CD8α+ DC and CD8α−33D1+ DC, but an increased absolute number compared to WT mice (Supplemental Fig. 1B). We also identified a CD11c+MHCII+ DC population lacking expression of CD8α and 33D1 that was increased by percentage and absolute number in TLR7.1 mice compared to WT mice (Supplemental Fig. 1B). CD8α−33D1− DC expressed high levels of CD11b, Sirpα, and CX3CR1, but low levels of the endothelial marker ESAM (Supplemental Fig. 1C), indicating they belong to the granulocyte/macrophage progenitor (GMP)-derived ESAMlo subset of CD11b+ DC (7).
The paradoxical decrease in percentage and increase in absolute number of pDC, CD8α+ DC and CD8α−33D1+ DC was due to the significant increase in spleen size and cellularity in TLR7.1 mice compared to WT mice (Supplemental Fig. 1D). To resolve a clearer picture of splenic myeloid homeostasis in TLR7.1 mice, we calculated the fold change in cell number of each myeloid population between TLR7.1 and WT mice. This showed that GMP-derived inflammatory monocytes, neutrophils and CD8α−33D1− DC were dramatically expanded (20–25x increase), whereas common dendritic cell progenitor (CDP)-derived CD8α−33D1+ DC, CD8α+ DC and pDC were modestly increased (2–3x increase) in TLR7.1 mice compared to WT mice (Supplemental Fig. 1E).
TLR7.1 mice exhibit emergency myelopoiesis
In the steady state, myeloid cells develop in a step-wise fashion from LT-HSC to short term-HSC (ST-HSC) to multipotent progenitor cells (MPP), which together compose the “LSK” (Lineage−Sca1+c-Kit+) fraction of the BM. MPP mature into common myeloid progenitors (CMP) or lymphoid-primed MPP, that further develop through a series of increasingly committed progenitor cells, such as the GMP and CDP, to give rise to mature myeloid cells (8). During inflammation, hematopoiesis shifts toward emergency myelopoiesis to rapidly generate more effector cells. Due to the preferential expansion of GMP progeny in TLR7.1 mice, we hypothesized that TLR7 overexpression caused a permanent state of emergency myelopoiesis leading to peripheral myeloid expansion.
TLR7.1 mice had an increased absolute number of LSK cells compared to WT mice, with significant increases in each of the three progenitors that comprise this compartment, LT-HSC, ST-HSC and MPP, identified using either Flk2/CD135 (Fig. 1A, 1B) or the SLAM family member CD150 (data not shown). TLR7.1 mice had fewer CMP and more GMP compared to WT mice using steady state gating strategies (Fig. 1A, 1C). Strikingly, TLR7.1 mice exhibited a significant increase in Sca-1+ GMP, identified as CD34+CD16/32+ cells within the LSK gate, compared to WT mice (Fig. 1A, 1D). We termed these Sca-1+ GMP “emergency GMP” (eGMP) as they appear during emergency myelopoiesis in this system, as well as during Ehrlichia muris and Plasmodium chabaudi infection (9, 10). Therefore, TLR7.1 mice are undergoing emergency myelopoiesis in the BM, characterized by an increase in LSK cells, a decrease in CMP, an expansion of GMP and the appearance of eGMP. The number of CDP and common lymphoid precursor cells (CLP), which have been shown to have DC potential (3), were similar in WT and TLR7.1 mice (Supplemental Fig. 1F).
Fig. 1. TLR7.1 mice exhibit emergency myelopoiesis in the bone marrow.

(A) Gating strategy and representative flow cytometry plots for BM LSK, CMP, GMP and eGMP. (B) Number of LT-HSC, ST-HSC and MPP from 3–5 experiments. (C, D) Number of CMP, GMP (C) and eGMP (D) from 5 independent experiments. Significance determined by two-tailed, unpaired student’s t-test.
Cell-extrinsic changes in myeloid development in TLR7.1 mice
The alterations in myelopoiesis and peripheral myeloid homeostasis in TLR7.1 mice suggested that TLR7 overexpression might drive a cell-intrinsic expansion of hematopoietic progenitor cells. We first examined TLR7 expression in hematopoietic progenitors by qPCR. Tlr7 mRNA was expressed in LSK, CMP and GMP in WT mice and overexpressed 4–8 fold in TLR7.1 mice (Supplemental Fig. 2A). Additionally, Tlr7 mRNA was expressed in mature WT myeloid cells in the spleen and BM and overexpressed 3–12 fold in TLR7.1 mice.
To assess whether TLR7 overexpression drives cell-intrinsic myeloid expansion in TLR7.1 mice, we generated mixed BM chimeric mice in which lethally irradiated hosts were reconstituted with congenically marked WT and TLR7.1 BM at a 1:1 ratio. Both TLR7.1:WT mixed BM chimeric mice and WT mice reconstituted with 100% TLR7.1 BM exhibited splenomegaly, showing that splenic expansion occurs when TLR7 is overexpressed only in hematopoietic cells (Supplemental Fig. 2B). TLR7.1:WT mixed BM chimeric mice exhibited a phenotype similar to TLR7.1 mice including increases in GMP and eGMP in the BM and expansion of inflammatory monocytes and neutrophils in the spleen compared to control chimeras (data not shown). However, we saw no advantage of TLR7.1 over WT BM hematopoietic progenitors (Supplemental Fig. 2C). This was surprising as LSK, CMP and GMP have been shown to express TLR, and in vitro TLR ligation can cause the myeloid differentiation of these progenitors (3). We also saw no advantage of TLR7.1 cells in producing neutrophils or monocytes (data not shown). Our observation that TLR7 signals in BM progenitor cells do not drive myeloid differentiation in vivo leaves open the question of why LSK, CMP and GMP express TLR and if there particular situations where TLR signaling can intrinsically drive myeloid differentiation in vivo.
TLR7.1 mice exhibit a type I IFN signature in BM myeloid progenitors
Because emergency myelopoiesis in TLR7.1 mice was cell-extrinsic, we looked for alterations in serum cytokines in TLR7.1 mice. We found no differences in cytokines with known roles in myelopoiesis (Supplemental Fig. 2D and data not shown). Next, we asked whether type I IFN, a family of cytokines implicated in early hematopoiesis, inflammation and SLE, but not included in our cytokine panel, might be increased in the BM of TLR7.1 mice. TLR7.1 mice have an IFN signature in the spleen (5), though we were unable to detect IFNα or IFNβ protein in the serum of TLR7.1 mice by ELISA or bioassay (data not shown). Therefore, we focused on whether type I IFN was acting directly on hematopoietic progenitor cells in TLR7.1 mice. We sorted total LSK, CMP and GMP from WT and TLR7.1 BM and assayed these cells by qPCR for expression of interferon-stimulated genes (ISG). TLR7.1 progenitors exhibited higher expression of ISG than WT cells (Fig. 2A), showing that LSK and myeloid progenitor cells in TLR7.1 mice have been exposed to and responded to type I IFN in vivo.
Fig. 2. TLR7.1 myeloid progenitors exhibit a type I IFN signature in the bone marrow where pDC are constitutively activated and generating type I IFN mRNA.

(A) ISG mRNA from BM progenitors, sorted as in Fig. 1A and assayed by qPCR. Data are from 2 independent experiments. (B) BM pDC from WT and TLR7.1 mice were gated on as CD11b−Siglec-H+BST-2+ and a histogram of MHCII expression is shown, representative of 5 independent experiments. (C) BM pDC from mixed BM chimeras generated with a 1:1 ratio of TLR7.1 and B6.SJL BM were gated as CD45.2+ and CD45.1+, respectively. MHCII expression on CD45.2+ and CD45.1+ BM pDC from the same mouse are shown, representative of 3 independent experiments. (D) BM cells were sorted (pDC as in Fig. 2B; inflammatory monocytes CD11b+Ly6C+Ly6G-; neutrophils CD11b+Ly6C+Ly6G+) and used for qPCR. Data are representative of two experiments. n.d. not detected.
Bone marrow pDC are cell-intrinsically activated by TLR7 overexpression and constitutively produce type I IFN
The ISG expression in TLR7.1 myeloid progenitors suggested that increases in type I IFN in the BM environment may cause the myeloid expansion seen in TLR7.1 mice. We hypothesized that pDC in the BM, which express the highest amounts of Tlr7 mRNA in both WT and TLR7.1 mice (Supplemental Fig. 2A), were generating type I IFN constitutively due to TLR7 overexpression. TLR7.1 BM pDC had increased expression of MHCII compared to WT BM pDC (Fig. 2B, Supplemental Fig. 2E), supporting the hypothesis that TLR7 overexpression leads to pDC activation. Analysis of mixed BM chimeras showed that within the same mouse, TLR7.1 BM pDC had higher surface MHCII than WT BM pDC (Fig. 2C, Supplemental Fig. 2F). Therefore, BM pDC were cell-intrinsically activated due to TLR7 overexpression.
Next, we examined whether TLR7.1 BM pDC were generating type I IFN. pDC, inflammatory monocytes and neutrophils were sorted from the BM of WT and TLR7.1 mice and prepared for qPCR directly ex vivo. TLR7.1 BM pDC were generating higher amounts of Ifna1 and Ifnb mRNA in vivo compared to WT BM pDC (Fig. 2D). Neither BM inflammatory monocytes nor neutrophils were constitutively producing type I IFN mRNA (Fig. 2D). These data indicate that TLR7 overexpression drives cell-intrinsic activation of BM pDC and the generation of type I IFN that may then act upon BM hematopoietic progenitor cells. It is interesting to speculate that these professional type I IFN-producing cells are found in the BM to mediate type I IFN-dependent changes in hematopoiesis.
Lack of type I IFN signaling limits emergency myelopoiesis in TLR7.1 mice
To directly test whether type I IFN is responsible for the peripheral myeloid expansion and emergency myelopoiesis observed in TLR7.1 mice, we bred TLR7.1 mice to mice lacking the type I IFN receptor (IFNARKO). The weight and overall cellularity of the spleens of TLR7.1xIFNARKO mice were significantly reduced compared to TLR7.1 mice, though they were still increased compared to WT mice (Fig. 3A and data not shown). The increase in absolute number of TLR7.1 inflammatory monocytes and neutrophils compared to WT was largely dependent on type I IFN signaling, whereas the number of CD8α−33D1− DC, though significantly lower in TLR7.1xIFNARKO mice than TLR7.1 mice, was more modestly reduced (Fig. 3B).
Fig. 3. TLR7.1 emergency myelopoiesis depends on type I IFN signaling.

(A) Spleen weight. (B) Number of splenic myeloid cells were gated on as in Fig. S1A and B. (C–D) BM progenitor cells were gated on as in Fig. 1A. All data represent the mean of 12–15 mice assessed in at least 3 independent experiments. Significance determined by one-way ANOVA with a tukey post-test.
As we observed mitigation of the TLR7.1 peripheral myeloid expansion in TLR7.1xIFNARKO mice, we postulated that the emergency myelopoiesis observed in TLR7.1 mice would be diminished. Consistent with this hypothesis, the increased number of LSK cells in TLR7.1 mice was almost completely reduced to WT numbers in TLR7xIFNARKO mice (Supplemental Fig. 2G). The reduction in CMP numbers in TLR7.1 mice was reversed in the TLR7.1xIFNARKO mice, but the increase in GMP numbers was not affected by the lack of type I IFN signaling (Fig. 3C). Strikingly, eGMP were nearly absent in TLR7.1xIFNARKO mice (Fig. 3D). Therefore, as emergency myelopoiesis and peripheral myeloid expansion seen in the TLR7.1 mice largely depends upon type I IFN signaling, we believe that TLR7.1 mice represent a model of chronic type I IFN-driven myeloid expansion.
TLR7 agonist administration drives emergency myelopoiesis in WT, but not IFNARKO mice
To assess whether chronic TLR7 signaling can drive type I IFN-dependent emergency myelopoiesis in a non-transgenic system, the TLR7 agonist gardiquimod was serially administered to WT and IFNARKO mice for 6 days. A significant increase in eGMP was observed in WT, but not IFNARKO, mice treated with TLR7 agonist (Fig. 4A, 4B). Further, other hematopoietic alterations consistent with the process of emergency myelopoiesis, such as increases in LSK, decreases in CMP and increases in GMP were observed in WT mice treated with TLR7 agonist (Supplemental Fig. 2H). This approach validated our observations made in the TLR7.1 system that TLR7-driven type I IFN can act upon bone marrow progenitor cells to drive emergency myelopoiesis.
Fig. 4. Serial TLR7 agonist administration drives type I IFN-dependent emergency myelopoiesis.

(A) Identification of eGMP in WT and IFNARKO mice treated with or without 10 μg gardiquimod i.v. every other day for 6 days. (B) Number of eGMP. Data show 6 mice per group from 2 independent experiments with each dot representing an individual mouse. Significance determined by one-way ANOVA with a tukey post-test.
eGMP produce more myeloid cells in vitro and are more proliferative than GMP in vivo
We hypothesized that type I IFN-dependent eGMP are better myeloid progenitors than GMP. To test this prediction, equivalent numbers of WT GMP, TLR7.1 GMP and TLR7.1 eGMP were sorted and plated in methylcellulose media containing IL-3, IL-6 and SCF, which support differentiation of granulocyte and macrophage colonies. eGMP generated more total colonies, and a greater proportion of these colonies were mixed-lineage granulocyte/macrophage CFU (CFU-GM), than GMP (Fig. 5A and Supplemental Fig. 2I). eGMP-derived colonies were also much larger than those generated by GMP (data not shown). Strikingly, TLR7.1 eGMP produced approximately 4-fold more cells than WT or TLR7.1 GMP (Fig. 5B, 5C). GMP yielded similar colony numbers, proportions of CFU-GM and cell numbers whether they derived from WT or TLR7.1 mice, suggesting that GMP exposed to type 1 IFN, but that do not express Sca-1, do not increase their differentiative capacity. These data demonstrate that eGMP are superior progenitors of myeloid cells compared to GMP.
Fig. 5. eGMP are superior myeloid precursors and more proliferative than GMP.
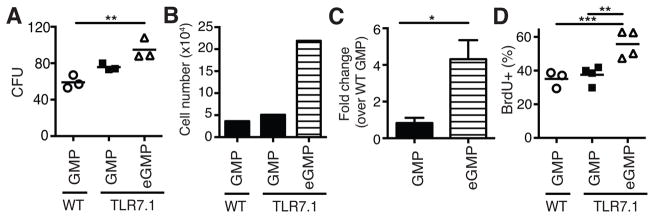
(A–C) 250 WT GMP, TLR7.1 GMP or TLR7.1 eGMP were plated in methylcellulose media with SCF, IL-3 and IL-6. (A) Number of colony forming units (CFU) after 5 days. (B) Absolute cell number generated after 6 days in culture from 1 experiment. (C) Fold change in cell yield over WT GMP. (A, C) Data are representative of cells sorted from 3 WT or TLR7.1 mice in 2 independent experiments. (D) Percent BrdU+ GMP and eGMP in WT and TLR7.1 mice 1 h after injection of BrdU. Data are representative of 2 experiments. Each dot represents an individual mouse with bar representing the mean. (A–D) Significance determined by one-way ANOVA with a tukey post-test, two-tailed, paired or unpaired student’s t-test, where appropriate.
Our in vivo observations and our in vitro CFU data suggest that eGMP possess different functional properties than GMP that allow them to more efficiently generate mature myeloid cells. GMP were identified by flow cytometry as being Sca-1−, whereas eGMP were Sca-1+. Interestingly, Sca-1 expression has been shown to correlate with cell cycle activity in hematopoietic progenitor cells during some settings of emergency hematopoiesis (4, 11). Significantly more TLR7.1 eGMP had progressed through S phase of the cell cycle during a one hour BrdU pulse than GMP from either WT or TLR7.1 mice (Fig. 5D). This increased proliferation defines a functional difference between eGMP and GMP that may allow eGMP to more efficiently generate myeloid progeny. Sca-1, a gpi-anchored cell surface protein, has been shown to drive ERK phosphorylation and the MEK/ERK axis promotes both myeloid differentiation and proliferation (11–13). Therefore, Sca-1 may signal though ERK to drive both proliferation and differentiation of eGMP. Interestingly, cells sharing the phenotype of eGMP have been observed in Ehrlichia muris and Plasmodium chabaudi infection and have been shown to be type II IFN dependent (9, 10). Thus, our work is the first to define the role of type I IFN in driving eGMP generation.
Here we link chronic TLR7 signaling to type I IFN production, which can then act at the level of hematopoiesis to drive emergency myelopoiesis and peripheral expansion of inflammatory monocytes and neutrophils. This may be the mechanism underlying a number of previously published models of TLR7-driven inflammation. For example, SLE-prone BXSB mice and mice serially treated with TLR7/8 agonist display selective increases in inflammatory monocytes and neutrophils (14, 15). Unc93b1D34A mutant mice, in which TLR7 signaling is increased, exhibit increases in spleen size and expansion of CD11b+CD11c+ cells, inflammatory monocytes and neutrophils, similar to TLR7.1 mice (16). The etiology of these observations was unclear, and our results would suggest that type I IFN-driven emergency myelopoiesis is occurring.
Our results are particularly interesting given that TLR7.1 mice are a model of SLE. SLE patients have gene signatures in their blood reflective of both type I IFN signaling and granulopoiesis (17, 18). SLE pathogenesis is driven in part by immune complexes, formed by anti-nucleic acid antibodies and their antigens provided by dying neutrophils undergoing type I IFN-driven NETosis. These immune complexes are recognized by pDC and elicit TLR-dependent type I IFN secretion, generating an autoimmune circuit (19,20). Our data suggest that type I IFN produced by pDC due to chronic TLR7 signaling promotes development of neutrophils in the BM, which may then go on to participate in the chronic feed-forward loop between neutrophils and pDC; thereby, providing a mechanistic link between the granulopoiesis and type I IFN signatures in SLE. In support of this model, TLR7.1 mice require B cells for myeloid expansion (21), suggesting that RNA/anti-RNA immune complexes drive pDC type I IFN production in these mice. Our observations linking type I IFN to myelopoiesis may also have implications for other situations of increased type I IFN, including acute and chronic viral infections as well as type I IFN treatment of infections, such as hepatitis C virus.
Supplementary Material
Acknowledgments
We thank Griff Gessay and Dr. Xizhang Sun for assistance with mouse breeding, and Drs. Daniel Campbell, Natalia Giltiay, Edward Clark, Steven Ziegler and members of the Hamerman laboratory for helpful discussions and critical reading of the manuscript.
Abbreviations
- BM
bone marrow
- LT-HSC
long-term hematopoietic stem cell
- qPCR
quantitative real time PCR
- pDC
plasmacytoid dendritic cell
- GMP
granulocyte/macrophage progenitor
- CDP
common dendritic cell progenitor
- ST-HSC
short-term hematopoietic stem cell
- MPP
multipotent progenitor
- LSK
Lineage−Sca1+c-Kit+ surface phenotype
- CMP
common myeloid progenitor
- eGMP
emergency granulocyte/macrophage progenitor
- CLP
common lymphoid progenitor
- IFNARKO
common lymphoid precursor
- SLE
systemic lupus erythematous
Footnotes
This work was supported in part by NCI grant T32CA009537 (MBB), NSF GRFP grant DGE-0718124 (MBB), NIH grants R01 AI081948 (JAH), R01 AI073441 (JAH), AR48796 (KBE) and an investigator award from the Cancer Research Institute (JAH).
The authors have no conflicting financial interests.
References
- 1.Baldridge MT, King KY, Goodell MA. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 2011;32:57–65. doi: 10.1016/j.it.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood. 2012;119:2991–3002. doi: 10.1182/blood-2011-12-380113. [DOI] [PubMed] [Google Scholar]
- 3.Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, Takatsu K, Kincade PW. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24:801–812. doi: 10.1016/j.immuni.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 5.Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S. Control of Toll-like Receptor 7 Expression Is Essential to Restrict Autoimmunity and Dendritic Cell Proliferation. Immunity. 2007;27:801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Santer DM, Wiedeman AE, Teal TH, Ghosh P, Elkon KB. Plasmacytoid Dendritic Cells and C1q Differentially Regulate Inflammatory Gene Induction by Lupus Immune Complexes. J Immunol. 2012;188:902–915. doi: 10.4049/jimmunol.1102797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, Klinakis A, Charo IF, Jung S, Gommerman JL, Ivanov II, Liu K, Merad M, Reizis B. Notch2 Receptor Signaling Controls Functional Differentiation of Dendritic Cells in the Spleen and Intestine. Immunity. 2011;35:780–791. doi: 10.1016/j.immuni.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwasaki H, Akashi K. Myeloid Lineage Commitment from the Hematopoietic Stem Cell. Immunity. 2007;26:726–740. doi: 10.1016/j.immuni.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Macnamara KC, Oduro K, Martin O, Jones DD, McLaughlin M, Choi K, Borjesson DL, Winslow GM. Infection-induced myelopoiesis during intracellular bacterial infection is critically dependent upon IFN-γ signaling. J Immunol. 2011;186:1032–1043. doi: 10.4049/jimmunol.1001893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belyaev NN, Brown DE, Diaz AIG, Rae A, Jarra W, Thompson J, Langhorne J, Potocnik AJ. Induction of an IL7-R+c-Kithi myelolymphoid progenitor critically dependent on IFN-γ signaling during acute malaria. Nat Immunol. 2010;11:477–485. doi: 10.1038/ni.1869. [DOI] [PubMed] [Google Scholar]
- 11.Melvan JN, Siggins RW, Stanford WL, Porretta C, Nelson S, Bagby GJ, Zhang P. Alcohol Impairs the Myeloid Proliferative Response to Bacteremia in Mice by Inhibiting the Stem Cell Antigen-1/ERK Pathway. J Immunol. 2012;188:1961–1969. doi: 10.4049/jimmunol.1102395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geest CR, Buitenhuis M, Groot Koerkamp MJA, Holstege FCP, Vellenga E, Coffer PJ. Tight control of MEK-ERK activation is essential in regulating proliferation, survival, and cytokine production of CD34+-derived neutrophil progenitors. Blood. 2009;114:3402–3412. doi: 10.1182/blood-2008-08-175141. [DOI] [PubMed] [Google Scholar]
- 13.Hsu CL, Kikuchi K, Kondo M. Activation of mitogen-activated protein kinase kinase (MEK)/extracellular signal regulated kinase (ERK) signaling pathway is involved in myeloid lineage commitment. Blood. 2007;110:1420–1428. doi: 10.1182/blood-2007-02-071761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amano H, Amano E, Santiago-Raber ML, Moll T, Martinez-Soria E, Fossati-Jimack L, Iwamoto M, Rozzo SJ, Kotzin BL, Izui S. Selective expansion of a monocyte subset expressing the CD11c dendritic cell marker in the Yaa model of systemic lupus erythematosus. Arthritis Rheum. 2005;52:2790–2798. doi: 10.1002/art.21365. [DOI] [PubMed] [Google Scholar]
- 15.Baenziger S, Heikenwalder M, Johansen P, Schlaepfer E, Hofer U, Miller RC, Diemand S, Honda K, Kundig TM, Aguzzi A, Speck RF. Triggering TLR7 in mice induces immune activation and lymphoid system disruption, resembling HIV-mediated pathology. Blood. 2009;113:377–388. doi: 10.1182/blood-2008-04-151712. [DOI] [PubMed] [Google Scholar]
- 16.Fukui R, Saitoh SI, Kanno A, Onji M, Shibata T, Ito A, Onji M, Matsumoto M, Akira S, Yoshida N, Miyake K. Unc93B1 Restricts Systemic Lethal Inflammation by Orchestrating Toll-like Receptor 7 and 9 Trafficking. Immunity. 2011;35:69–81. doi: 10.1016/j.immuni.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 17.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197:711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, Bassett R, Amuro H, Fukuhara S, Ito T, Liu YJ, Gilliet M. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra19. doi: 10.1126/scitranslmed.3001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, Barrat FJ, Banchereau J, Pascual V. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walsh ER, Pisitkun P, Voynova E, Deane JA, Scott BL, Caspi RR, Bolland S. Dual signaling by innate and adaptive immune receptors is required for TLR7-induced B-cell-mediated autoimmunity. Proc Natl Acad Sci USA. 2012 doi: 10.1073/pnas.1209372109. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.