

1. Introduction
Few crucial advances in the field in the past several years have markedly increased our understanding of the function of osteocytes. The evidence that sclerostin, the product of the Sost gene, is expressed and secreated primarily by osteocytes and inhibits bone formation by osteoblasts, fueled research attempting to identify regulators of this gene as well as other osteocyte products that impact the function of osteoblasts and osteoclasts. The discovery that parathyroid hormone (PTH), a central regulator of bone homeostasis, inhibits sclerostin expression generated a cascade of studies that revealed that osteocytes are crucial target cells of the actions of PTH. This review highlights these investigations and discusses their significance for advancing our understanding of the mechanisms by which osteocytes regulate bone homeostasis and for developing therapies for bone diseases targeting osteocytes.
2. Osteocytes
Osteocytes, former osteoblasts regularly distributed throughout the mineralized bone matrix, are the most abundant cells in bone comprising more than 90% of cells within the matrix or on bone surfaces [1, 2]. Entombed during the process of bone deposition, osteocytes remain highly connected with cells on the bone surface and among themselves, via cytoplasmic processes that radiate from their bodies and travel along canaliculi excavated in the mineralized matrix. Osteocyte projections reach other bone cells, cells in the bone marrow, and endothelial cells of the bone blood vessels, establishing a functional syncytium named the osteocyte network. The osteocytic lacunar-canalicular system allows the transport of proteins produced and secreted by osteocytes that exert their action on cells on other cells or tissues. An example of such molecules is sclerostin, the product of the Sost gene, highly expressed in osteocytes [3–5]. Sclerostin binds to LRP5, LPR6 and LRP4 preventing activation of Wnt signaling and also antagonizes the actions of several members of the bone morphogenetic protein (BMP) family of proteins [6–8]. Both Wnts and BMPs are critical for osteoblastogenesis and also regulate the activity of mature osteoblasts. Loss of sclerostin in humans causes the high bone mass disorders Van Buchem’s disease and sclerosteosis [9, 10]. Moreover, mice with targeted deletion of the Sost gene exhibit high bone mass and, conversely, transgenic mice overexpressing Sost exhibit low bone mass, demonstrating conservation throughout the species of the inhibitory effect of sclerostin on bone formation [3, 11–13]. In addition, an anti-sclerostin antibody has been developed to induce bone anabolism validating the high potential of targeting osteocytes for increasing bone mass and strength [14, 15].
Osteocytes are also a critical source of molecules that influence the development and activity of osteoclasts. Osteocytes directly isolated from mouse bone express higher levels of RANKL than osteoblasts and bone marrow stromal cells [16]. Moreover, mice with deletion of the RANKL gene in osteocytes exhibit an osteopetrotic phenotype and are resistant to bone loss induced by tail suspension, strongly suggesting that osteocytes are a major source of RANKL in vivo [16, 17]. Furthermore, osteocytes secrete osteoprotegerin (OPG) [18], which competes with RANKL for its receptor on osteoclast precursors and mature osteoclasts. In osteocytes, as in osteoblasts, OPG secretion is regulated by the Wnt/β-catenin pathway and mice lacking β-catenin in osteocytes are osteoporotic due to increased osteoclast numbers, whereas osteoblast function is normal. Emerging evidence also points to osteocytes as an additional source of secreted M-CSF in bone [19]. Thus, osteocytes control the bone remodeling process through direct and indirect regulation of osteoclast and osteoblast differentiation and function.
Osteocytes also express and secrete fibroblast growth factor 23 (FGF23), a hormone that regulates phosphate reabsorption in the kidney and that, by changing circulating levels of phosphate, also affects bone mineralization [20–24]. FGF23 also directly activates intracellular signaling in osteocytes and osteoblasts, mediated through binding to the FGFR1/KLOTHO receptor complex recently reported to be expressed in these bone cells [25]. FGF23 has been shown to suppress osteoblast differentiation and matrix mineralization in vitro [26, 27], suggesting a role for FGF23 not only in the regulation of systemic phosphate levels, but also in the local control of bone mineralization.
In conclusion, osteocytes produce and secrete factors (such as sclerostin, RANKL, OPG) that affect other bone cells by paracrine or autocrine mechanisms, and hormones (such as FGF23) that affect other tissues by endocrine mechanisms.
3. PTH and its receptor
PTH is an 84-amino acid polypeptide synthesized and secreted by the parathyroid glands in a calcium-regulated manner. The hormone maintains serum calcium homeostasis, controls renal phosphate reabsorption and vitamin D3 1α-hydroxylation and modulates bone turnover, through actions mediated by the PTH/PTH-related peptide (PTHrP) receptor (PPR). The receptor is expressed in bone and kidney, but is also found in a variety of other tissues not regarded as classical PTH targets.
Among bone cells, osteoblasts and chondrocytes have been regarded as the main target cells of hormonal effect, but increasing evidence identifies osteocytes as critical effectors of PTH action. Direct action of PTH upon osteocytes “in vivo” was first suggested by early experiments in which cellular retraction, mitochondrial swelling and cell death were observed in osteocytes by light electron microscopy upon administration of PTH extract [28, 29]. Increased proteolytic activity associated with enlarged osteocytic lacunae was also evident in bones of animals receiving the PTH extract for several days. The first evidence that osteocytes indeed expressed PPRs derives from autoradiography studies demonstrating binding of radiolabeled PTH in osteocytes of growing rats [30]. More recently, conclusive and definitive demonstrations that PTH indeed acts on osteocytes were provided from several in vitro and in vivo studies with osteocytic cell lines, primary osteoblastic cell preparations and genetically modified mouse models, as will be described below [5, 13, 31–33].
5. Bone re/modeling
a. Osteocytes and the bone remodeling compartment (BRC)
The ability of osteocytes to reach bone surfaces together with the recent evidence demonstrating that osteocytes express molecules that regulate osteoclast and osteoblast function, provides the basis for the long hypothesized role of the osteocyte network of coordinating the remodeling of bone. osteocyte apoptosis has been shown to precede osteoclast accumulation and resorption [34], raising the possibility that osteocytes release molecules that induce lining cell retraction facilitating the access of osteoclast precursors to bone surfaces and creating a canopy over osteoclasts and osteoblasts in the bone multicellular unit (BMU) [35]. M-CSF [19], which stimulates pre-osteoclast proliferation, and RANKL [16, 17], the master cytokine inducer of osteoclast differentiation, would then accumulate in the BRC and stimulate osteoclastogenesis. It is still uncertain whether membrane-bound RANKL in the osteocyte dendritic processes or soluble RANKL released from osteocytes through the canalicular circulation is responsible for osteoclast differentiation. Factors released from the bone matrix upon resorption, in turn, would stimulate proliferation of pre-osteoblasts and their differentiation to mature osteoblasts. It is also likely that osteocyte-derived sclerostin, reaching the BRC through the canalicular system, influences the rate of bone formation independently of resorption, providing an additional level of control of osteoblast activity. Based on these lines of evidence, the BRC might provide a supportive environment for differentiation of osteoclast and osteoblast progenitors. Within this context, as it will be discussed below, PPR actions might regulate bone remodeling by controlling the balance between resorption and formation within the BRC through the regulation of sclerostin, RANKL and OPG in osteocytes.
b. Regulation of Sost/sclerostin by PTH
PTH has profound effects on the skeleton. Elevation of the hormone in the circulation can generate both catabolic and anabolic effects on bone depending on the temporal profile of its increase. Chronic excess of PTH, as in primary hyperparathyroidism or secondary to calcium deficiency, increases the rate of bone remodeling, and can result in loss of bone. In contrast to continuous PTH elevation, intermittent increases of PTH in the circulation, as achieved by daily injections, cause bone gain, and it is the only current bone anabolic therapy. High bone remodeling rates and bone loss with chronic PTH elevation is associated with excessive production of osteoclasts coupled to increased osteoblasts, with a negative balance between bone formation and resorption within each BMU. Instead, the primary effect of intermittent PTH elevation is a rapid increase in osteoblasts and bone formation, attributed to the ability of the hormone to promote proliferation of osteoblast precursors, to inhibit osteoblast apoptosis, to reactivate lining cells to become matrix synthesizing osteoblasts, or to a combination of those effects [36, 37]. The net gain in bone obtained with intermittent PTH administration is due to enhanced osteoblast activity and bone formation. In humans, PTH stimulates bone formation by increasing the bone remodeling rate and the amount of bone formed by each remodeling unit, named “remodeling-based formation” [38]. PTH also stimulates bone formation not coupled to prior resorption, referred to as “modeling-based formation”. The latter mechanism appears to be more evident in rodents.
Recent investigations have markedly advanced our understanding of the cellular and molecular mechanisms behind the contrasting effects of the two PTH regimens on bone. Studies in mice indicate that chronic and intermittent PTH increase osteoblast number by distinct mechanisms. At least in cancellous bone in the mouse, the anabolic effect of intermittent PTH can be accounted for by attenuation of osteoblast apoptosis, whereas chronic elevation of PTH had no effect on osteoblast survival [36, 39]. Instead, the osteoblastogenic action of continuous elevation of PTH results from direct actions of the hormone on osteocytes inhibiting the expression of the Sost gene [5] (Figure 1). Continuous treatment with PTH markedly suppressed Sost mRNA and sclerostin protein expression in rodent models [5, 40]. This effect was reproduced in vitro in osteocytic and osteoblastic cell lines and in primary cultures of calvaria cells containing osteocytes, demonstrating that it results from direct effect of PTH on its receptor expressed in osteocytes, rather than arising from hormonal actions on other cells or tissues. Intermittent PTH administration also reduces Sost expression, but to a lesser extent and only transiently after each daily injection [5, 40]. However, after long-term daily PTH injection Sost levels most likely remain suppressed. These findings strongly suggest that sustained downregulation of Sost/sclerostin is not required for bone anabolism induced by intermittent PTH; however, it is likely that repetitive reductions in sclerostin could be part of the increase in bone formation induced by the hormone. These initial findings in rodents have been independently confirmed using several animal models and also validated in humans [40–46].
Figure 1. PTH downregulates Sost/sclerostin expression in osteocytes.
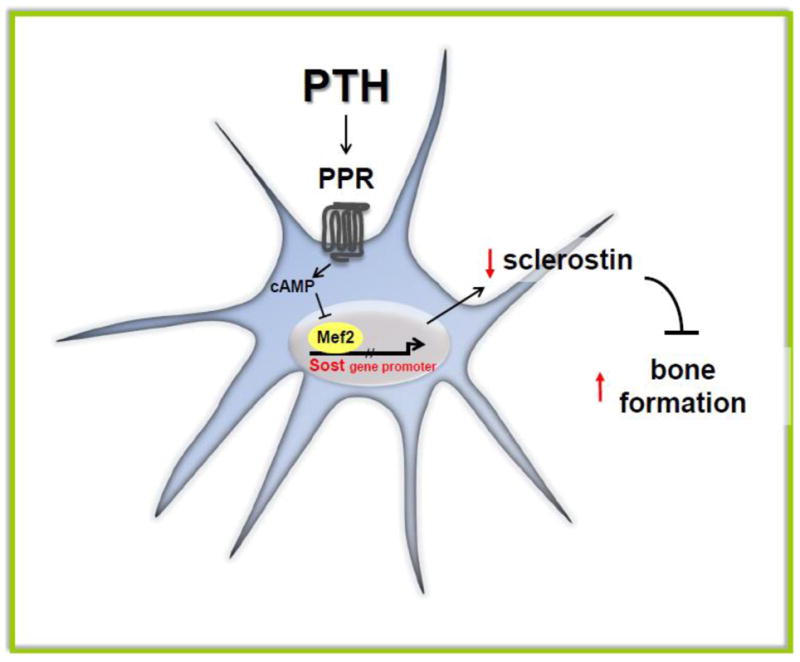
PPR activation by PTH elevates cAMP levels and inhibits Mef2-stimulated Sost promoter activity leading to decreased expression of the inhibitor of bone formation sclerostin, and elevated bone formation rate.
PTH exerts its inhibitory effect on Sost expression via the cAMP signaling pathway downstream of the PPR, as demonstrated by the fact that PTHrP, the other ligand of this receptor, and stable analogs of cAMP mimic the effects of PTH [25, 47]. However, Sost downregulation appears to not depend on transcription factors of the cAMP responsive element binding protein (CREB) family. Instead, transcription factors of the myocyte enhancer factor (MEF2) family mediate the effect of PTH on Sost expression [41] (Figure 1). Nevertheless, the exact molecular mechanism of this regulation remains unknown. Moreover, it is important to acknowledge that other mechanisms besides downregulation of Sost expression and osteoblast survival are likely to contribute to the profound skeletal effects of PTH.
c. Bone formation
Sost and Wnt signaling
In vivo activation of PPR signaling in osteocytes is sufficient for Sost inhibition as evidenced by reduced expression of Sost and sclerostin in transgenic mice expressing a constitutively active PPR under the control of an 8kb fragment of the dentin matrix protein 1 promoter (named DMP1-8kb-caPTHR1 mice) [32] (Figure 2). Bones from these transgenic mice exhibit elevated expression of Wnt target genes and high levels of beta-galactosidase activity when crossed with Wnt responsive reporter mice, indicating the activation of Wnt signaling by PPR signaling in osteocytes in vivo. DMP1-caPTHR1 mice display a remarkable increase in BMD in both the axial and appendicular skeleton, which is blunted in compound mice lacking the Wnt coreceptor LRP5 or completely abrogated in double transgenic mice overexpressing Sost in osteocytes (DMP1-8kb-caPTHR1; DMP1-8kb-Sost mice) [13, 32]. These findings demonstrate that activation of PPR signaling in osteocytes is sufficient for inhibiting Sost/sclerostin expression and for activating Wnt signaling, with the resulting concomitant increase in bone mass.
Figure 2. Converse effects of PPR activation and PPR deletion in osteocytes on bone formation and resorption.
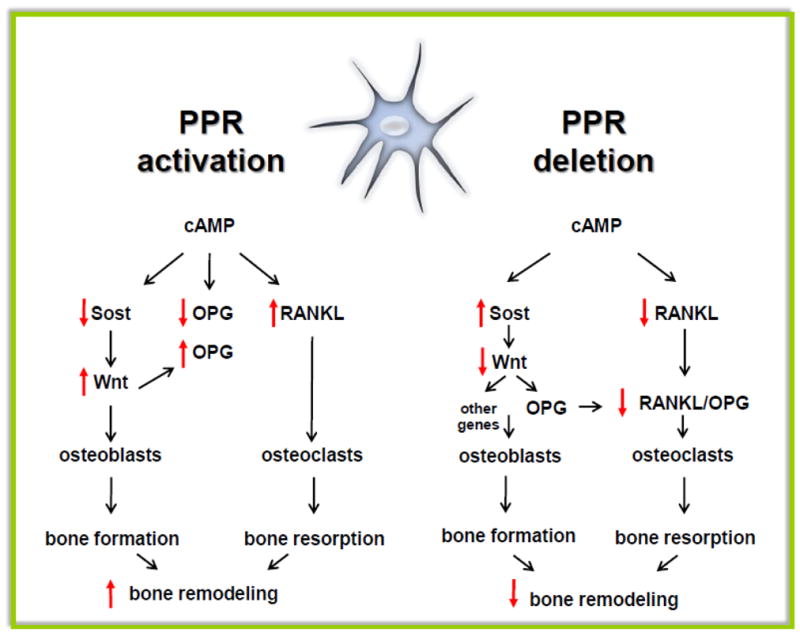
Osteocyte activation of the PPR leads to cAMP-dependent Sost downregulation and increased Wnt signaling, which in turn increases osteoblasts and stimulates bone formation. PPR activation increases RANKL expression, osteoclasts and resorption. However, OPG expression is not affected, likely resulting from opposing effects on the expression of the gene by cAMP (reduction) and Wnts (elevation). The main effect of deletion of the PPR in osteocytes is increased SOST expression, decreased RANKL expression, reduced osteoclast and osteoblast number with decreased bone resorption and bone formation, depending on the cKO model used (see text for details).
Consistent with this gain-of-function of the PPR leading to constitutive decreased in Sost and sclerostin expression, conversely, mice lacking PPR expression in osteocytes, accomplished by conditional deletion of the receptor by crossing PPRflox mice with DMP1-10kb-Cre mouse, displayed tonic elevation of Sost and sclerostin [33] (Figure. 2).
Osteocytic PTH receptor and periosteal bone formation
PTH has profound effects in cortical bone, stimulating periosteal expansion and at the same time accelerating intra-cortical bone remodeling. DMP1-8kb-caPTHR mice exhibit increased cortical bone area and elevated rate of periosteal and endocortical bone formation [13]. In addition, DMP1-8kb-caPTHR1 mice display a marked increase in intra-cortical remodeling and cortical porosity. Crossing DMP1-8kb-caPTHR1 mice with LRP5 null mice or with DMP1-Sost mice reduced or completely abolished, respectively, the increased cortical bone area, periosteal bone formation rate, and expression of osteoblast markers and Wnt target genes exhibited by the DMP1-8kb-caPTHR1 mice. In contrast, deletion of LRP5 or Sost overexpression did not decrease bone resorption induced by the DMP1-8kb-caPTHR1 transgene [13]. In fact, DMP1-8kb-caPTHR1 mice lacking LRP5 or double transgenic DMP1-caPTHR1; DMP1-Sost mice exhibit exacerbated intra-cortical remodeling and osteoclast numbers compared to the single DMP1-8kb-caPTHR1 mice, due to decreased expression of the Wnt target gene OPG (see below). These findings demonstrate that the actions of PPR signaling in osteocytes govern periosteal bone formation and cortical bone resorption. In addition, they demonstrate that Sost downregulation and the consequent Wnt activation is required for the stimulatory effect of PTH receptor signaling on periosteal bone formation. Moreover, decreased sclerostin and increased Wnt signaling restrains osteoclast development and activity.
Bone formation in PPR deletion
Conditional PPR deletion obtained by crossing the PPRflox mouse with the tamoxifen inducible 10Kb-DMP1-Cre mouse developed osteopenia, as evidenced by decreased trabecular bone compared with control animals [33]. Importantly, as described above, these mice displayed higher Sost and sclerostin expression as well as the inability to regulate this gene in response to PTH. These observations suggested a role for PPR signaling in osteocytes in trabecular bone formation.
In contrast to mice with tamoxifen-inducible deletion of the PPR, mice with constitutive loss of PPR expression achieved by using the 8 or 10Kb-DMP1-Cre mice developed higher BMD as compared to control littermates. The timing of the skeletal phenotype was dependent on the promoter used. In mice where the receptor was ablated using the 8Kb-DMP1-promoter, an increase in cancellous bone in the spine was detected at 8 weeks of age [48] whereas in mice where the 10Kb-DMP1 promoter was used, the skeletal phenotype was evident at 12 weeks of age, and micro-CT analysis at 14 weeks revealed increased trabecular BV/TV%, trabecular number, cortical thickness and bone area. Histomorphometric analysis revealed a marked reduction in osteoblast number and bone formation rate (unpublished data), indicating an important role of PPR in osteocytes in controlling bone formation (Figure 2). Future studies are required to determine whether this decrease in bone formation is secondary to reduced resorption in the absence of PPR in osteocytes (as will be discussed below). The opposite skeletal phenotype observed when the receptor was ablated with the tamoxifen-inducible model (DMP1-10kb-Cre-ERT2) versus the non-inducible DMP1 promoter (the 8kb and the 10 kb fragments) might be due to recombinase activity during development in the latter models. Alternatively, the phenotype of the inducible PPR deletion using tamoxifen could reflect a potential interaction between the PPR and the estrogen receptor in osteocytes.
Osteoblastic expansion and the hematopoietic stem cell niche
Accumulating evidence demonstrates that cells of the osteoblastic lineage create the essential niche required for self-renewal and differentiation of hematopoietic stem cells (HSC) [49]. Thus, it has been assumed that expansion of the osteoblastic cell pool would increase the number or function of HSC. Consistent with this notion, the increase in osteoblasts induced by PTH administration or by activating its receptor in osteoblastic cells (preosteoblasts, osteoblasts and osteocytes) increases the HSC niche [50]. Remarkably, however, activation of PTH receptor signaling exclusively in osteocytes in DMP1-8kb-caPTHR mice does not increase the HSC niche, despite of the striking high number of osteoblasts and increased bone formation rate exhibited by these mice [51]. These findings establish that osteocytic activation of PTH receptor signaling is not sufficient to augment bone marrow HSC and exclude osteocytes as the osteoblastic population required to initiate PTH-dependent HSC expansion. This evidence also conclusively separates the bone anabolic actions of PTH from the ability of the hormone to support HSC development and function, and strongly suggest that therapies directed to increase osteoblast number and function might not necessarily achieve HSC expansion.
d. Bone resorption
Activation of the PTH receptor in osteocytes and bone resorption
In vitro studies have shown that PTH increases osteoclast formation by upregulating RANKL and elevating RANKL/OPG ratio. Although recognized that osteoblastic lineage cells mediate osteoclastogenesis by PTH, the precise stage of differentiation of the PTH target cell is unknown. Recent evidence demonstrates that osteocytes are a major source of RANKL, as well as OPG; and mice lacking RANKL in osteocytes exhibit osteopetrosis.
As discussed earlier, activation of PPR signaling in osteocytes in DMP1-caPTHR1 mice is sufficient to increase bone formation and resorption (Figure 2). DMP1-caPTHR1 mice exhibit increased circulating CTX and osteoclasts number in cancellous and cortical bone [13, 32]; and although Sost overexpression abolishes the increase in bone formation, double TG DMP1-caPTHR1; DMP1-Sost mice still exhibit high resorption. Moreover, whereas increased expression of osteoblast markers is abolished, expression of osteoclast markers, as well as RANKL and M-CSF remains elevated in the double transgenic mice. Sost overexpression markedly decreases Wnt target genes, including OPG resulting in even higher RANKL/OPG in the double TG mice. Thus, osteocytic PPR signaling controls formation and resorption by separate mechanisms. These findings raise the possibility that PTH receptor signaling directly upregulates RANKL gene expression in osteocytes. Alternatively, activation of the PTH receptor could induce in osteocytes the production and secretion of factors, such as IL-6 type cytokines, which in turn would stimulate RANKL expression in other cells, such as stromal-osteoblastic cells. Future investigations to address this question are warranted.
Conditional deletion of the PTH receptor in osteocytes and bone resorption
The physiological role of the PTH receptor in osteocytes has been addressed by deletion of the PPR in the mouse (cKO). Mice in which the PPR was ablated constitutively, using the 8kb or the 10Kb fragments of the DMP1 promoter, or conditionally, using a tamoxifen inducible 10Kb-DMP1 promoter to drive a Cre-recombinase, have been generated in our laboratories. In these cKO mouse models, receptor expression was dramatically reduced, as assessed by semiquantitative real-time PCR in vertebra and long bones. Moreover, purified osteocytes isolated from the cKO mice expressed lower levels of PPR compared to control littermates, whereas PPR expression or function in osteoblasts remained unchanged [33, 48], further validating the DMP1-Cre models to delete genes from osteocytes. Using the 8Kb-DMP1 promoter to delete the PPR [48], RANKL and OPG expression, in tibial diaphysis (composed of cortical bone), was lower in cKO-PPR mice versus control littermates (Figure 2). In contrast, in lumbar vertebra (mostly composed of cancellous bone), only RANKL is decreased resulting in reduced RANKL/OPG ratio. These findings are consistent with osteocytes being the main source of RANKL whereas OPG is also contributed by other cells relatively more abundant in cancellous versus cortical bone. Thus, removal of the PPR from osteocytes using the DMP1-8Kb promoter leads to a site specific reduction in the RANKL/OPG ratio resulting in increased cancellous but not cortical bone. Thus, in this model, PPR actions in osteocytes are required to maintain basal levels of RANKL and OPG, and in the absence of osteocytic PPR the primary effect is a reduction in osteoclast activity in cancellous bone. Interestingly, when receptor ablation was achieved using the DMP1-10Kb promoter, mice also developed a high bone mass phenotype characterized by a significant decrease in both osteoblasts and osteoclasts number and by an increase in both cancellous and cortical bone [52] (Figure 2). Detailed analysis of the phenotypes is needed to determine the basis of these subtle differences among the different cKO mouse models and to fully elucidate the role of the receptor in osteocytes.
6. Mineral homeostasis
a. Regulation of FGF23 by PTH and its consequences for systemic and local FGF23 signaling
Osteocytes are richer than osteoblasts in genes related to mineralization and phosphate metabolism, including phosphate-regulating neutral endopeptidase (Phex), DMP1, and matrix extracellular phosphoglycoprotein (MEPE). Osteocytes also produce FGF23, a hormone produced in bone that plays a crucial role in phosphate (Pi) homeostasis by inhibiting its renal reabsorption [20–24].
High dietary Pi and the active form of Vitamin D3, 1,25-dihydroxy-vitamin D3 (1,25-D3) are major stimulators of FGF23 secretion [53–56]. FGF23 is a marker of altered phosphate metabolism that increases before detectable changes in PTH in early CKD [57]. Recent evidence suggests that PTH also regulates FGF23 production. Thus, the hyperphosphatemia exhibited by chronic kidney disease (CKD) patients is accompanied by elevation in both FGF23 and PTH [58, 59]; and parathyroidectomy prevented the increase in FGF23 in a rat model of kidney failure [60].
FGF23 is also elevated in patients and in a mouse model of primary hyperparathyroidism [61, 62]. A patient with Jansen’s metaphyseal chondrodysplasia expressing a constitutively active PTHR1 receptor mutant exhibit high FGF23 concentrations in the circulation, despite low Pi and normal 1,25-D3 levels [63]. Moreover, FGF23 production is increased in in bones and in osteocytes, but not osteoblasts, from DMP1-8kb-caPTHR1 transgenic mice, which express the same mutated PTH receptor than the patient. Circulating FGF23 is also elevated in DMP1-8kb-caPTHR1 mice; however, plasma Pi or renal Pi reabsorption is not altered. Furthermore, the FGF23 receptor complex comprising FGFR1 and KLOTHO is expressed in osteoblastic cells; and FGFR1, GALNT3, as well as downstream targets of FGF23 signaling, are increased in osteocytes but not in osteoblasts from DMP1-caPTHR1 mice. These findings demonstrate that PTH receptor signaling has the potential to modulate the endocrine and auto/paracrine functions of osteocytes by regulating FGF23 through cAMP- and Wnt-dependent mechanisms (Figure 3).
Figure 3. PPR signaling modulates the endocrine and auto/paracrine function of osteocytes by regulating FGF23 expression.
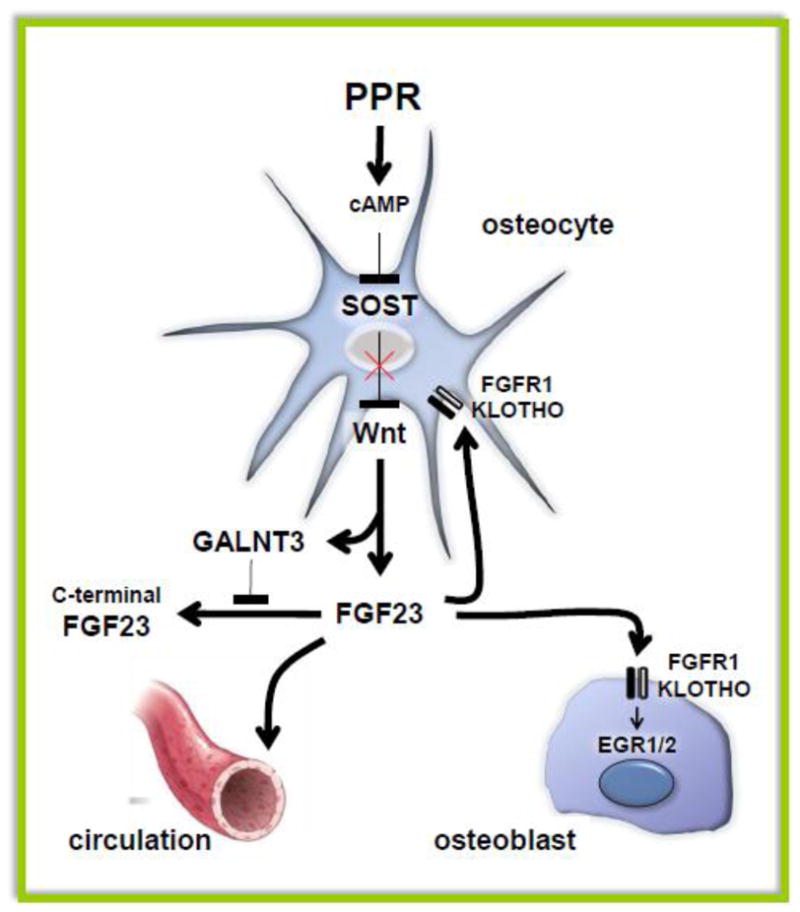
cAMP elevation in vivo by a constitutively active PPR or in vitro by the PPR ligands PTH and PTHrP increases FGF23 in a Wnt-dependent manner. A simultaneous increase in GALNT3 stabilizes FGF23, leading to increased intact FGF23 in the circulation as well as locally in bone. Binding of FGF23 to the FGFR1/KLOTHO receptor complex expressed in osteocytes and osteoblasts activates intracellular signaling.
b. Calcium
Osteocytes are also involved in calcium homeostasis. Mice in which the PPR was deleted with tamoxifen-induced DMP1-10kb-Cre demonstrated hypocalcemia when challenged with a low calcium diet [33], suggesting an important role of osteocytes in regulating calcium ion homeostasis. Under physiological conditions, the low calcium diet is easily corrected by increased PTH production and increased remodeling to bring the calcium back to normal. However, when osteocytes lack a functional PPR, this does not happen, suggesting that osteocytes are important regulators of calcium homeostasis via a PTH-dependent mechanism. The role of osteocytes in ion homeostasis however has been, and still is, quite controversial. Early studies suggested a role for osteocytes in controlling mineral homeostasis and the term “osteocytic osteolysis” was created. This concept was quickly abandoned because in vitro studies were unable to demonstrate that isolated osteocytes reabsorb dentin slices. Osteocytic osteolysis was recently revisited when new evidence was generated to support this concept. As described below, during lactation osteocytes can quickly and reversibly remodel the perilacunar space. Hyperparathyroidism [64] and lack of mechanical stimuli, such as during immobilization or space flights [65], are additional conditions in which the resorptive capability of osteocytes is evident.
7. Cross talk between the PTH receptor and mechanotransduction in osteocytes
Osteocytes are ideally positioned to integrate the responses of bone to mechanical and hormonal stimuli. Both mechanical loading and PTH promote new bone formation by downregulating the expression of sclerostin; and mice lacking the Wnt coreceptor LRP5 [66] or overexpressing Sost [67] exhibit a defective response to loading. These findings suggest a potential crosstalk between mechanotransduction and PTHR1 signaling.
Earlier studies by Chow et al. demonstrated a blunted osteogenic response to mechanical stimulation by parathyroidectomized rats compared with normal rats, which was restored after a single injection of PTH [68] Consistent with these findings, recent studies demonstrate that expression of the PTH receptor in osteocytes is indispensable for the anabolic response to mechanical loading in mice [69]. Axial loading of the ulnae caused the expected strain-dependent increase in bone formation in control mice, resulting from an increase in both mineralizing surface covered by osteoblasts (MS/BS) as well as activity of individual osteoblasts (MAR). In contrast, loading-induced bone formation was markedly reduced in mice in which the PPR was deleted from osteocytes with the DMP1-8kb-Cre, resulting mainly from lack of stimulation of MAR by loading at any strain magnitude. Loading-induced MS/BS was also reduced in the cKO mice, with significant increases induced only by medium and high strains. These findings indicate that signaling downstream of the PTHR1 in osteocytes is required for the osteogenic response induced by mechanical force, and strongly suggest that the osteocytic PTH receptor is involved in the integration of mechanical and hormonal signals leading to coordinated regulation of bone formation.
On the other hand, the role of PTH signaling in osteocytes during unloading has not been completely elucidated. Studies in unloaded rats and mice have generated conflicting evidence. Turner et al. reported that, in rats, intermittent low doses of PTH (1μg/kg/d) could prevent the reduction of bone formation and the increase in trabecular thinning induced by hindlimb unloading in the proximal tibial methaphysis, while the hormone had no effect on the cortical bone [70]. They suggested that, in rats, PTH treatment and mechanical unloading have synergistic or additive interactions depending on the dose and the bone compartment (cortical versus cancellous) analyzed. Recently, Ono et al. [71] reported that the mice expressing the constitutive active PPR in osteoblasts (Col1a1-caPPR) [72] were resistant to unloading-induced bone loss, suggesting an important role of PPR signaling in preventing bone loss due to disuse or unloading. The importance of PTH in disuse induced bone loss is further supported by the increase in serum calcium levels and suppression of PTH after 2 and 5 days of flight in Spacelab Life Science-1 crew members [73]. Taken together these results indicate PTH can prevent bone loss induced by unloading but the cellular target of this hormonal effects still awaits to be elucidated. Unloading studies using genetically modified animals in which the receptor is ablated from osteocytes will help understand the interaction between mechanical forces and PTH signaling under unloading or disuse conditions.
8. Osteocytic PTHR and lacunae/canalicular remodeling during lactation
The role of PPR signaling in osteocytes during lactation has been recently elucidated [74]. It is known that lactation, both in humans and in rodents, is associated with a rapid and reversible loss of bone mineral density, but the molecular and cellular mechanisms of this skeletal loss were still unknown. To investigate the role of the PPR and PTHrP, known to be elevated during lactation [75], Qing and colleagues studied skeletal responses to lactation in control and osteocyte-specific PTHR-KO mice. They reported that, during lactation, osteocytes express osteoclasts-specific genes such as TRAP, Cathepsin K and carbonic anidrase 2, suggesting that these cells are capable of removing, and possibly redepositing, the mineral surrounding the lacunae. Moreover, when the PTHR is ablated from osteocytes, the size of the lacunae does not increase, and TRAP and cathepsin K are not elevated, indicating that PTHrP is an important mediator of this effect. These results are in agreement with previous studies that demonstrated enlarged lacunae in animals treated with exogenous PTH, and suggest an important role for the receptor on osteocytes in controlling skeletal and mineral homeostasis (Figure 4). Additional studies are needed to better comprehend the functions of osteocytes on mineral homeostasis during lactation.
Figure 4. PTH receptor and osteocytic lacunae/canalicular remodeling during lactation.
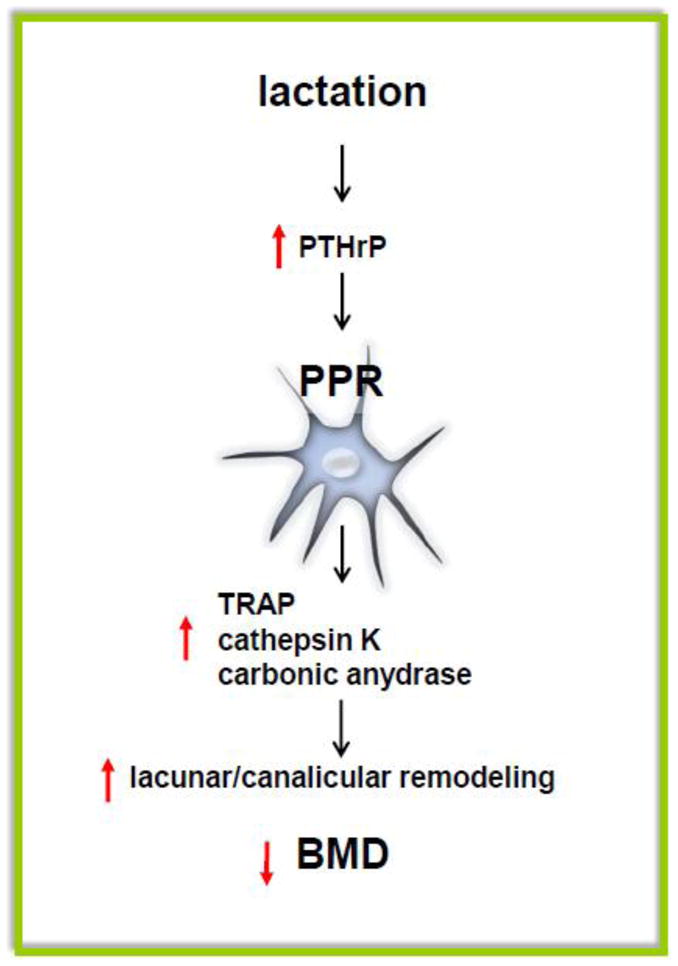
During lactation, increased circulating levels of PTHrP activate PPR expressed in osteocytes and induce increase in lacuna/canalicular remodeling and consequent bone loss. Genes such as TRAP, Cathepsin K and carbonic anydrase 2 are highly expressed in osteocytes from lactating mice and their regulation is dependent on PPR expression.
9. Closing remarks and future directions
Only few years have passed since the first reports demonstrating the regulation of sclerostin expression by PTH and it is now known that actions of the PPR in osteocytes regulate several genes greatly impacting bone homeostasis. The number of publications on osteocytes and PTH has triplicate in the last ten years, in part due to the development of in vivo tools for the analysis of these remarkable cells (addressed in the article of Kalajzic et al in this issue). Development of new and improved models that target osteocytes will increase our understanding of the complex regulation of osteocyte biology by the PPR under physiological and pathological conditions. Still remains to be determined which of the skeletal effects of PTH are mediated by direct hormonal actions on osteocytes, and to define the role of other osteocytic genes regulated by PTH in bone homeostasis. It is envisioned that these future investigations will render new candidate genes and pathways, enhancing, beyond the anti-sclerostin antibody, the therapeutic potential of targeting osteocytes to regulate bone mass and strength.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aarden EM, Burger EH, Nijweide PJ. Function of osteocytes in bone. J Cell Biochem. 1994;55:287–99. doi: 10.1002/jcb.240550304. [DOI] [PubMed] [Google Scholar]
- 2.Bonewald LF. The Amazing Osteocyte. J Bone Miner Res. 2011;26:229–38. doi: 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22:6267–76. doi: 10.1093/emboj/cdg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Bezooijen RL, Roelen BA, Visser A, Wee-Pals L, de Wilt E, Karperien M, Hamersma H, Papapoulos SE, Ten Dijke P, Lowik CW. Sclerostin ss an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805–14. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA, Manolagas SC, Jilka RL. Chronic elevation of PTH in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology. 2005;146:4577–83. doi: 10.1210/en.2005-0239. [DOI] [PubMed] [Google Scholar]
- 6.Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a WNT signaling inhibitor. J Biol Chem. 2005;280:26770–5. doi: 10.1074/jbc.M504308200. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–7. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 8.Choi HY, Dieckmann M, Herz J, Niemeier A. Lrp4, a novel receptor for dickkopf 1 and sclerostin, is expressed by osteoblasts and regulates bone growth and turnover in vivo. PLoS ONE. 2009;4:e7930. doi: 10.1371/journal.pone.0007930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, Van Den Ende J, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, Van Hul W. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001;10:537–43. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 10.Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, Skonier JE, Zhao L, Sabo PJ, Fu Y, Alisch RS, Gillett L, Colbert T, Tacconi P, Galas D, Hamersma H, Beighton P, Mulligan J. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–89. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, Kurahara C, Gao Y, Cao J, Gong J, Asuncion F, Barrero M, Warmington K, Dwyer D, Stolina M, Morony S, Sarosi I, Kostenuik PJ, Lacey DL, Simonet WS, Ke HZ, Paszty C. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–9. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 12.Loots GG, Kneissel M, Keller H, Baptist M, Chang J, Collette NM, Ovcharenko D, Plajzer-Frick I, Rubin EM. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005;15:928–35. doi: 10.1101/gr.3437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rhee Y, Allen MR, Condon K, Lezcano V, Ronda AC, Galli C, Olivos N, Passeri G, O’Brien CA, Bivi N, Plotkin LI, Bellido T. PTH receptor signaling in osteocytes governs periosteal bone formation and intra-cortical remodeling. J Bone Miner Res. 2011;26:1035–46. doi: 10.1002/jbmr.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warmington K, Morony S, Sarosi I, Gong G, Stepphens P, Winkler DG, Sutherland MK, Latham JA, Kirby H, Moore A, Robinson M, Kostenuik PJ, Simonet WS, Lacey DL, Paszty C. Sclerostin antagonism in adult rodents, via monoclonal antibody mediated blockade, increases bone mineral density and implicates sclerostin as a key regulator of bone mass during adulthood. J Bone Min Res. 2004;19:S56. [Google Scholar]
- 15.Warmington K, Ominsky M, Bolon B, Cattley R, Stephens P, Lawson A, Lightwood D, Perkins V, Kirby H, Moore A, Robinson M, Kostenuik PJ, Simonet WS, Lacey DL, Paszty C. Sclerostin monoclonal antibody treatment of osteoporotic rats completely reverses one year of ovariectomy-induced systemic bone loss. J Bone Min Res. 2005;20:S22. [Google Scholar]
- 16.Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM, Takayanagi H. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17:1231–4. doi: 10.1038/nm.2452. [DOI] [PubMed] [Google Scholar]
- 17.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235–41. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, Feng JQ, Bonewald LF, Kneissel M. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol Cell Biol. 2010;30:3071–85. doi: 10.1128/MCB.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris SE, MacDougall M, Horn D, Woodruff K, Zimmer SN, Rebel VI, Fajardo R, Feng JQ, Heinrich-Gluhak J, Harris MA, Abboud WS. Meox2Cre-mediated disruption of CSF-1 leads to osteopetrosis and osteocyte defects. Bone. 2012;50:42–53. doi: 10.1016/j.bone.2011.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshiko Y, Wang H, Minamizaki T, Ijuin C, Yamamoto R, Suemune S, Kozai K, Tanne K, Aubin JE, Maeda N. Mineralized tissue cells are a principal source of FGF23. Bone. 2007;40:1565–73. doi: 10.1016/j.bone.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 21.Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron RP. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112:683–92. doi: 10.1172/JCI18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu S, Guo R, Simpson LG, Xiao ZS, Burnham CE, Quarles LD. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem. 2003;278:37419–26. doi: 10.1074/jbc.M304544200. [DOI] [PubMed] [Google Scholar]
- 23.Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310–5. doi: 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98:6500–5. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rhee Y, Bivi N, Farrow EG, Lezcano V, Plotkin LI, White KE, Bellido T. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo, Bone. 2011;49:636–43. doi: 10.1016/j.bone.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H, Yoshiko Y, Yamamoto R, Minamizaki T, Kozai K, Tanne K, Aubin JE, Maeda N. Overexpression of fibroblast growth factor 23 suppresses osteoblast differentiation and matrix mineralization in vitro. J Bone Miner Res. 2008;23:939–48. doi: 10.1359/jbmr.080220. [DOI] [PubMed] [Google Scholar]
- 27.Sitara D, Kim S, Razzaque MS, Bergwitz C, Taguchi T, Schuler C, Erben RG, Lanske B. Genetic evidence of serum phosphate-independent functions of FGF-23 on bone. PLoS Genet. 2008;4:e1000154. doi: 10.1371/journal.pgen.1000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heller M, McLEAN FC, BLOOM W. Cellular transformations in mammalian bones induced by parathyroid extract. Am J Anat. 1950;87:315–45. doi: 10.1002/aja.1000870302. [DOI] [PubMed] [Google Scholar]
- 29.Cameron DA, Paschall HA, Robinson RA. Changes in the fine structure of bone cells after the administration of parathyroid extract. J Cell Biol. 1967;33:1–14. doi: 10.1083/jcb.33.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fermor B, Skerry TM. PTH/PTHrP receptor expression on osteoblasts and osteocytes but not resorbing bone surfaces in growing rats. J Bone Miner Res. 1995;10:1935–43. doi: 10.1002/jbmr.5650101213. [DOI] [PubMed] [Google Scholar]
- 31.Woo S, Rooser J, Dusevich V, Kalajzic I, Bonewald LF. Cell line IDG-SW3 replicates osteoblast-to-late-osteocyte differentiation in vitro and accelerates bone formation in vivo. J Bone Miner Res. 2011;26:2634–46. doi: 10.1002/jbmr.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Brien CA, Plotkin LI, Galli C, Goellner J, Gortazar AR, Allen MR, Robling AG, Bouxsein M, Schipani E, Turner CH, Jilka RL, Weinstein RS, Manolagas SC, Bellido T. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE. 2008;3:e2942. doi: 10.1371/journal.pone.0002942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Powell WF, Barry KJ, Tulum I, Kobayashi T, Harris SE, Bringhurst F, Divieti Pajevic P. Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J Endocrinol. 2011;209:21–32. doi: 10.1530/JOE-10-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bellido T. Osteocyte apoptosis induce bone resorption and impairs the skeletal response to weightlessness. BoneKEy-osteovision. 2007;4:252–6. [Google Scholar]
- 35.Eriksen EF. Cellular mechanisms of bone remodeling. Rev Endocr Metab Disord. 2010;11:219–27. doi: 10.1007/s11154-010-9153-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jilka RL. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone. 2007;40:1434–46. doi: 10.1016/j.bone.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim SW, Pajevic PD, Selig M, Barry KJ, Yang JY, Shin CS, Baek WY, Kim JE, Kronenberg HM. Intermittent PTH administration converts quiescent lining cells to active osteoblasts. J Bone Miner Res. 2012 May 23; doi: 10.1002/jbmr.1665. Ref Type: In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hodsman AB, Bauer DC, Dempster DW, Dian L, Hanley DA, Harris ST, Kendler DL, McClung MR, Miller PD, Olszynski WP, Orwoll E, Yuen CK. Parathyroid hormone and teriparatide for the treatment of osteoporosis: a review of the evidence and suggested guidelines for its use. Endocr Rev. 2005;26:688–703. doi: 10.1210/er.2004-0006. [DOI] [PubMed] [Google Scholar]
- 39.Bellido T, Ali AA, Plotkin LI, Fu Q, Gubrij I, Roberson PK, Weinstein RS, O’Brien CA, Manolagas SC, Jilka RL. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J Biol Chem. 2003;278:50259–72. doi: 10.1074/jbc.M307444200. [DOI] [PubMed] [Google Scholar]
- 40.Keller H, Kneissel M. SOST is a target gene for PTH in bone. Bone. 2005;37:148–58. doi: 10.1016/j.bone.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 41.Leupin O, Kramer I, Collette NM, Loots GG, Natt F, Kneissel M, Keller H. Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J Bone Miner Res. 2007;22:1957–67. doi: 10.1359/jbmr.070804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gooi JH, Pompolo S, Karsdal MA, Kulkarni NH, Kalajzic I, McAhren SH, Han B, Onyia JE, Ho PW, Gillespie MT, Walsh NC, Chia LY, Quinn JM, Martin TJ, Sims NA. Calcitonin impairs the anabolic effect of PTH in young rats and stimulates expression of sclerostin by osteocytes. Bone. 2010 doi: 10.1016/j.bone.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 43.Silvestrini G, Ballanti P, Leopizzi M, Sebastiani M, Berni S, Di Vito M, Bonucci E. Effects of intermittent parathyroid hormone (PTH) administration on SOST mRNA and protein in rat bone. J Mol Histol. 2007;38:261–9. doi: 10.1007/s10735-007-9096-3. [DOI] [PubMed] [Google Scholar]
- 44.Mirza FS, Padhi ID, Raisz LG, Lorenzo JA. Serum sclerostin levels negatively correlate with parathyroid hormone levels and free estrogen index in postmenopausal women. J Clin Endocrinol Metab. 2010;95:1991–7. doi: 10.1210/jc.2009-2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drake MT, Srinivasan B, Modder UI, Peterson JM, McCready LK, Riggs BL, Dwyer D, Stolina M, Kostenuik P, Khosla S. Effects of parathyroid hormone treatment on circulating sclerostin levels in postmenopausal women. J Clin Endocrinol Metab. 2010;95:5056–62. doi: 10.1210/jc.2010-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Lierop AH, Witteveen J, Hamdy N, Papapoulos S. Patients with primary hyperparathyroidism have lower circulating sclerostin levels than euparathyroid controls. Eur J Endocrinol. 2010;163:833–7. doi: 10.1530/EJE-10-0699. [DOI] [PubMed] [Google Scholar]
- 47.Kramer I, Keller H, Leupin O, Kneissel M. Does osteocytic SOST suppression mediate PTH bone anabolism? Trends Endocrinol Metab. 2010;21:237–44. doi: 10.1016/j.tem.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 48.Tu X, Edwards R, Olivos N, Benson JD, Galli C, Pellegrini G, Bivi N, Plotkin LI, Bellido T. Conditional deletion of the parathyroid hormone (PTH) receptor 1 from osteocytes results in decreased bone resorption and a progressive increase in cancellous bone mass. J Bone Miner Res. 2011;25:S16. [Google Scholar]
- 49.Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, Frenette PS. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–34. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, Milner LA, Kronenberg HM, Scadden DT. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–6. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 51.Calvi LM, Bromberg O, Rhee Y, Weber JM, Smith JN, Basil M, Frisch BJ, Bellido T. Osteoblastic expansion induced by parathyroid hormone receptor signaling in murine osteocytes is not sufficient to increase hematopoietic stem cells. Blood. 2012;119:2489–99. doi: 10.1182/blood-2011-06-360933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saini V, Barry K, Fulzele K, Feng J, Divieti Pajevic P. PTH/PTHrP (PPR) receptor signaling in osteocytes regulate bone development in temporal manner. Journal of Bone and Mineral Research. 2011;26(S1):S16. [Google Scholar]
- 53.Ito M, Sakai Y, Furumoto M, Segawa H, Haito S, Yamanaka S, Nakamura R, Kuwahata M, Miyamoto K. Vitamin D and phosphate regulate fibroblast growth factor-23 in K-562 cells. Am J Physiol Endocrinol Metab. 2005;288:E1101–E1109. doi: 10.1152/ajpendo.00502.2004. [DOI] [PubMed] [Google Scholar]
- 54.Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. 2005;146:5358–64. doi: 10.1210/en.2005-0777. [DOI] [PubMed] [Google Scholar]
- 55.Burnett SM, Gunawardene SC, Bringhurst FR, Juppner H, Lee H, Finkelstein JS. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res. 2006;21:1187–96. doi: 10.1359/jbmr.060507. [DOI] [PubMed] [Google Scholar]
- 56.Kolek OI, Hines ER, Jones MD, LeSueur LK, Lipko MA, Kiela PR, Collins JF, Haussler MR, Ghishan FK. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol. 2005;289:G1036–G1042. doi: 10.1152/ajpgi.00243.2005. [DOI] [PubMed] [Google Scholar]
- 57.Isakova T, Wahl P, Vargas GS, Gutierrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CA, Lash JP, Hsu CY, Leonard MB, Wolf M. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79:1370–8. doi: 10.1038/ki.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Silver J, Kilav R, Sela-Brown A, Naveh-Many T. Molecular mechanisms of secondary hyperparathyroidism. Pediatr Nephrol. 2000;14:626–8. doi: 10.1007/s004670000355. [DOI] [PubMed] [Google Scholar]
- 59.White KE, Larsson TE, Econs MJ. The roles of specific genes implicated as circulating factors involved in normal and disordered phosphate homeostasis: frizzled related protein-4, matrix extracellular phosphoglycoprotein, and fibroblast growth factor 23. Endocr Rev. 2006;27:221–41. doi: 10.1210/er.2005-0019. [DOI] [PubMed] [Google Scholar]
- 60.Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. PTH increases FGF23 gene expression and mediates the high FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol. 2010;299:F882–F889. doi: 10.1152/ajprenal.00360.2010. [DOI] [PubMed] [Google Scholar]
- 61.Kobayashi K, Imanishi Y, Miyauchi A, Onoda N, Kawata T, Tahara H, Goto H, Miki T, Ishimura E, Sugimoto T, Ishikawa T, Inaba M, Nishizawa Y. Regulation of plasma fibroblast growth factor 23 by calcium in primary hyperparathyroidism. Eur J Endocrinol. 2006;154:93–9. doi: 10.1530/eje.1.02053. [DOI] [PubMed] [Google Scholar]
- 62.Kawata T, Imanishi Y, Kobayashi K, Miki T, Arnold A, Inaba M, Nishizawa Y. Parathyroid hormone regulates fibroblast growth factor-23 in a mouse model of primary hyperparathyroidism. J Am Soc Nephrol. 2007;18:2683–8. doi: 10.1681/ASN.2006070783. [DOI] [PubMed] [Google Scholar]
- 63.Brown WW, Juppner H, Langman CB, Price H, Farrow EG, White KE, McCormick KL. Hypophosphatemia with elevations in serum fibroblast growth factor 23 in a child with Jansen’s metaphyseal chondrodysplasia. J Clin Endocrinol Metab. 2009;94:17–20. doi: 10.1210/jc.2008-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bonucci E, Gherardi G. Osteocyte ultrastructure in renal osteodystrophy. Virchows Arch A Pathol Anat Histol. 1977;373:213–31. doi: 10.1007/BF00432238. [DOI] [PubMed] [Google Scholar]
- 65.Rodionova NV, Oganov VS, Zolotova NV. Ultrastructural changes in osteocytes in microgravity conditions. Adv Space Res. 2002;30:765–70. doi: 10.1016/s0273-1177(02)00393-9. [DOI] [PubMed] [Google Scholar]
- 66.Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, Li J, Maye P, Rowe DW, Duncan RL, Warman ML, Turner CH. The WNT co-receptor LRP5 is essential for skeletal mechanotransduction, but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem. 2006;281:23698–711. doi: 10.1074/jbc.M601000200. [DOI] [PubMed] [Google Scholar]
- 67.Tu X, Rhee Y, Condon KW, Bivi N, Allen MR, Dwyer D, Stolina M, Turner CH, Robling AG, Plotkin LI, Bellido T. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone. 2012;50:209–17. doi: 10.1016/j.bone.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chow JW, Fox S, Jagger CJ, Chambers TJ. Role for parathyroid hormone in mechanical responsiveness of rat bone. Am J Physiol. 1998;274:E146–E154. doi: 10.1152/ajpendo.1998.274.1.E146. [DOI] [PubMed] [Google Scholar]
- 69.Tu X, Pellegrini G, Galli C, Benson JD, Condon KW, Bivi N, Plotkin LI, Robling AG, Bellido T. PTH receptor 1 expression in osteocytes is indispensable for the anabolic effect of mechanical loading in mice. J Bone Miner Res. 2011;25:S24. [Google Scholar]
- 70.Turner RT, Evans GL, Lotinun S, Lapke PD, Iwaniec UT, Morey-Holton E. Dose-response effects of intermittent PTH on cancellous bone in hindlimb unloaded rats. J Bone Miner Res. 2007;22:64–71. doi: 10.1359/jbmr.061006. [DOI] [PubMed] [Google Scholar]
- 71.Ono N, Nakashima K, Schipani E, Hayata T, Ezura Y, Soma K, Kronenberg HM, Noda M. Constitutively active parathyroid hormone receptor signaling in cells in osteoblastic lineage suppresses mechanical unloading-induced bone resorption. J Biol Chem. 2007;282:25509–16. doi: 10.1074/jbc.M610782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, Kronenberg HM, Baron R, Schipani E. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest. 2001;107:277–86. doi: 10.1172/JCI11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.LeBlanc AD, Spector ER, Evans HJ, Sibonga JD. Skeletal responses to space flight and the bed rest analog: a review. J Musculoskelet Neuronal Interact. 2007;7:33–47. [PubMed] [Google Scholar]
- 74.Qing H, Ardeshirpour L, Pajevic PD, Dusevich V, Jahn K, Kato S, Wysolmerski J, Bonewald LF. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J Bone Miner Res. 2012;27:1018–29. doi: 10.1002/jbmr.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.VanHouten JN, Wysolmerski JJ. Low estrogen and high parathyroid hormone-related peptide levels contribute to accelerated bone resorption and bone loss in lactating mice. Endocrinology. 2003;144:5521–9. doi: 10.1210/en.2003-0892. [DOI] [PubMed] [Google Scholar]