

Abstract
The aggregation and deposition of the neuronal protein α-Synuclein in the substantia nigra region of the brain is a key pathological feature of Parkinson’s disease. α-Synuclein assembles from a monomeric state in solution, which lacks stable secondary and tertiary contacts, into highly structured fibrillar aggregates through a pathway which involves the population of multiple oligomeric species over a range of timescales. These features make α-Synuclein well-suited for study with single molecule techniques, which are particularly useful for characterizing dynamic, heterogeneous samples. Here we review the current literature featuring single molecule fluorescence studies of α-Synuclein and discuss how these studies have contributed to our understanding of both its function and its role in disease.
Introduction
There is strong evidence to support a central role for the protein α-Synuclein (AS) in the pathogenesis of Parkinson’s disease (PD). It is the primary component of Lewy bodies, dense cytoplasmic amyloid inclusions, which are associated with selective loss of dopaminergic neurons in the substantia nigra region of the brain. Although its precise role in the progression of PD remains unclear, point mutants of AS (A30P, E46K, and A53T) and multiplication of the AS gene linked to familial forms of PD established that it is critical to disease development [1–4].
AS is a small (140 amino acids), abundant neuronal protein. Its sequence is divided into three domains: a positively charged N-terminal region that contains repeats of the highly conserved KTKEGV sequence, a central, hydrophobic region known as the non-amyloid-β component (NAC), and an acidic C-terminal region (Figure 1). AS is generally thought to be an intrinsically disordered monomer in solution, although there is some recent evidence that it may be capable of forming a partially α-helical tetramer under some conditions [5, 6]. The N-terminal ~95 residues form an amphipathic α-helix upon binding to lipid membranes, while the C-terminal region remains largely unstructured [7]. The proposed native functions of AS include a role in synaptic vesicle trafficking[8, 9], regulation of the synaptic vesicle pool[10], and maintenance of neuronal plasticity[11]. Defects in vesicle trafficking are observed when AS is overexpressed in model organisms from yeast to rodents [8, 12–14]. A potential link between membrane association and disease has been established by the finding A30P and E46K show altered membrane binding properties [15, 16]. Moreover, it has been suggested that toxicity in PD may be mediated through interaction of toxic, oligomeric forms of AS and cell membranes [17–21]
Figure 1.
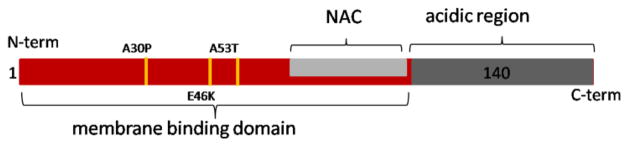
Schematic of AS. Shown in red is the N-terminal membrane binding region (residues 1–95), light gray is the central hydrophobic domain, and dark gray is the negatively charged C-terminal region. PD-associated disease mutations are marked in yellow.
The ability of AS to exchange between many different conformational states – disordered monomer, partially structured oligomer, α-helical membrane-bound, and -sheet fibrillar aggregate - may be central both to its native function as well as to its role in PD. Single molecule approaches offer a number of advantages for the study of a dynamic, conformationally heterogeneous protein like AS. One of their greatest advantages is the ability to analyze signals from different conformational states without the need to sort the molecular populations. Single molecule approaches provide complementary information to higher resolution methods, such as NMR; however, they can be applied to study of AS interacting with more physiological relevant model membranes, including vesicles and planar bilayers, that are not accessible to solution NMR. While single molecule approaches that have been used to study AS include force-probe techniques [22, 23] and computational simulations [24–26], in this review we focus on the application of single molecule fluorescence methods. We will discuss how these methods have been applied to AS to gain insight into both functional and dysfunctional states.
Folding
The Deniz group has used smFRET (single molecule Förster resonance energy transfer: Box 1) to map the folding landscape of AS in detail as a function of sodium dodecyl sulfate (SDS) concentration [27–30]. In all of this work, AS was labeled at residues 7 and 84 to probe its membrane-binding region (Figure 1). In an initial equilibrium study [27], it was found that at SDS concentrations below its critical micelle concentration (CMC), monomer SDS molecules bound to AS, resulting in its exchange between three distinct conformational states. At very low SDS concentrations (0.25 mM), disordered AS (ETeff ≈0.4; U-state) transitioned to a folded conformation (ETeff ≈0.85; I-state). Further increasing the SDS to 1.2 mM resulted in binding of additional monomer SDS molecules and population of a more extended, folded conformation (ETeff<0.2; F-state). Based on previous circular dichroism (CD) measurements [31], these two liganded states, I and F, were known to be α-helical and the distances calculated from the mean ETeff were consistent with two different α-helical conformations, a horse-shoe shaped structure composed of two α-helical domains (I) and a single, elongated α-helix (F) (Figure 2). In subsequent work, PD-associated point mutants of AS were characterized by a combination of CD and smFRET [28]. While E46K and A53T displayed very similar behavior to wild-type AS, folding in three states from U through I to F, A30P was found to never populate the extended α-helical F-state at sub-CMC SDS concentrations. The impact of this study is that it provides direct evidence of a difference in the conformational ensembles populated by wild-type and a disease-associated point mutant of AS, which may have implications for understanding pathogenic protein aggregation and why this particular mutant leads to disease. Moreover, studies of AS folding in the presence of SDS show that the amino acid sequence of AS is compatible with secondary structure formation, which could be important for its ordering preceding aggregation to toxic species on pathway to disease.
Box 1. SmFRET (single molecule Förster resonance energy transfer).
FRET occurs via a non-radiative dipole – dipole interaction between the donor and acceptor fluorophore. The efficiency of transfer is strongly dependent upon the distance between the two fluorophores making it a powerful tool for measuring inter- and intramolecular distances. In a typical ensemble experiment, the average energy transfer efficiency (ETeff) is measured for an entire population of molecules. One limitation is that subpopulations can be obscured due to ensemble averaging or the mean ETeff can be skewed by the presence of improperly labeled or photophysically damaged molecules. However, on the single molecule level, subpopulations can be directly resolved and improperly labeled molecules can be discriminated easily in data processing. This allows for the analysis of several conformational states simultaneously present within a population or access to very rarely populated states. Moreover, for disordered proteins like AS, it can be used to gain structural information in the absence of canonical secondary or tertiary structure. Studies of AS have relied on a confocal instrument configuration, whereby a laser is focused into a very dilute (10–100 pM) solution of fluorescently labeled protein, such that on average much less than one protein is present in the observation volume at any given time. When the proteins diffuse through the observation volume, a stream of donor and acceptor photons are emitted. For each transit event, an ETeff is calculated and these values are compiled into a histogram. A mean ETeff is determined from fitting the histogram and this value can then be converted, using an appropriate model, to a distance between the fluorophores. A number of recent reviews that focus on smFRET studies of disordered protein states provide excellent background to the technique and its strengths and challenges that will not be covered in this contribution [86–88]. Relevant to this review, smFRET has been used to determine the conformations sampled by monomer AS in solution and when associated with various types of membrane mimetics.
Figure 2.

The conformational states of AS. Cartoons in green show three of the most well studied conformational states of AS. The unfolded state (U) is largely disordered, though the hydrodynamic radius is not as large as expected for a fully random coil. The U-state transitions to an intermediate state (I) upon binding of SDS monomers or micelles. The I-state is also known as the hairpin or horseshoe conformational state. The folded state (F) can be populated via either the I-state, upon additional SDS monomer binding, or from the U-state upon binding to large unilamellar vesicles (LUV) or extended SDS structures. The SDS concentrations given are sub-CMC, where monomer SDS binds to AS. The F-state is also known as the extended helical conformation when bound to LUVs. The U, I, F nomenclature is from Reference 27.
In collaboration with the Groisman group, Deniz and colleagues have used microfluidic devices to characterize the equilibrium folding landscape of AS with higher spatial resolution [30] as well as to resolve time-dependent folding features [29]. In the time-resolved measurements, both folding and unfolding reactions were followed. In the folding measurements, the U state found in the absence of SDS collapsed to a more compact (ETeff ~0.6) state within the first 500 μs of adding SDS. By 1 ms, the high ETeff I-state was present, and over the next 11 ms the folded F-state formed in a two-state transition from the I-state. Interestingly, in the unfolding studies where the protein was rapidly diluted over 100 fold into SDS-free buffer, the protein transitioned from the F-state to the U-state within 1 ms without ever populating the intermediate I-state. This apparent difference in the folding and unfolding pathways suggests that all binding sites for the detergent molecules may not be equally accessible in the unfolded state.
Aggregation
In our own lab, the conformations of AS relevant to the initiation of aggregation have been characterized by smFRET [32]. By placing the donor and acceptor probes at nine different sets of locations throughout AS, we probed the response of specific domains to different aggregation-promoting conditions, namely low pH and the presence of molecular aggregation-inducers. Our studies showed that the C-terminus of AS compacts substantially at pH 3.0 relative to pH 7.4. The C-terminus has a very high negative charge density at pH 7.4 that is neutralized at pH 3.0, resulting in collapse of this region. This may allow the formation of hydrophobic patches which, combined with decreased intramolecular repulsions, result in accelerated aggregation. In contrast to the C-terminus, the conformational ensemble of the N-terminal and central regions compensate for the compaction of the C-terminus, such that the overall hydrodynamic radius of the protein is very similar under both conditions. To investigate whether the collapsed state observed at low pH is common to other aggregation pathways, smFRET measurements of AS were made in the presence of heparin and spermine, polyions previously shown to accelerate AS aggregation [32]. However, in contrast to the pH 3.0 measurements, heparin and spermine resulted in only minor conformational changes in any regions probed. This suggests that enhanced aggregation by these molecules may primarily be the result of shielding of repulsive charges rather that by populating specific conformations and emphasizes that that there may be diverse initial steps leading to aggregation. Moreover, the results of this study illustrate one of the unique advantages of single molecule approaches; they allow access to aggregation-prone conformations under conditions that strongly disfavor protein aggregation and subsequent complications due to heterogeneity in, or loss of, signal.
Further investigation of the C-terminus included monitoring its response to chemical denaturation at low pH. The ETeff histograms showed a single peak that shifted to lower mean values with increasing denaturant concentration. This behavior is the hallmark of a random coil expansion in the presence of chemical denaturant [33], indicating that while the low pH state of AS makes tertiary contacts, and perhaps even forms a molten globule-like structure, it is not a canonical globular fold. The hydrodynamic radius and intrachain distances of AS measured by smFRET fall somewhere between a typical globular protein and a statistical random coil. This suggests that although AS lacks stable secondary and compact tertiary contacts in solution, it exhibits non-random structural features. In contrast to the SDS-induced folding into a canonical α-helix, these results highlight a different type of conformational change in AS, one that impacts its ability to self-associate and thus may provide insight into the earliest stages of pathogenic aggregation.
Conformation of membrane-bound monomer
Because direct interactions between AS and cell membranes are thought to be important to its function, there is a great deal of interest in characterizing this interaction. CD established that AS becomes partially α-helical upon binding to SDS micelles and lipid vesicles while NMR provided structural details of micelle-bound AS [34–36]. However, these studies left the particular conformation of the α-helix, proposed to be either a single continuous extended helix [37–40] or two helices arranged in a hairpin or horseshoe shape [36, 41–43], a subject of debate. A paper from the Subramaniam group was the first publication of a smFRET investigation of the conformation of AS bound to SDS micelles[44]. To probe the membrane binding domain, AS was labeled at residues 9 and 69. While no conformational changes for SDS concentrations less 0.5 mM were detected, at greater than 0.5 mM SDS, a shift to a high ETeff state was observed which was attributed to binding to SDS micelles and was consistent with the hairpin α-helical structure reported by NMR (Figure 2).
Both we and the Deniz group used smFRET to show that AS adopts an extended α-helix upon binding to lipid bilayers of a more physiological size, namely ~50–100 nm diameter large unilamellar vesicles (LUVs) [27, 45]. Building upon their folding work described above, the Deniz group showed that upon exceeding the CMC of SDS, a high ETeff state was observed, which was similar to the I-state observed during folding and consistent with the hairpin structure (Figure 2). However, when the SDS concentration was further increased to 450 mM, a concentration where it no longer forms spherical micelles, but instead forms elongated tubular structures, they found a very low ETeff state that resembled the folding F-state and suggested an extended α-helical conformation (Figure 2). They confirmed that the same extended structure was observed in the presence of anionic LUVs. Our own study focused on AS binding to LUVs where we showed that AS formed an extended α-helix upon binding. In order to unambiguously differentiate between the extended and hairpin α-helical structures, AS was labeled at two different sets of positions within the membrane-binding domain to provide independent, self-consistent measurements of this domain. We have also observed the extended α-helical conformation on lipid nanodiscs [46]. In the presence of an excess of lipid nanodiscs, the mean ETeff of AS was comparable to that of AS bound to LUVs, suggesting that the extended helical conformation is populated on both membrane surfaces. Together, these studies have shown that the helical structure that AS populates is highly dependent on the curvature of the membrane or membrane mimetic surface to which it is associated.
The similarity of AS states induced by SDS monomer binding in solution (the I and F states) and states populated upon micelle and vesicle binding is notable. This suggests that despite its general lack of structure, AS has a propensity for specific α-helical structures in solution. Moreover, low concentrations of SDS [47] and some lipid compositions [43, 48–51] have been used very effectively to accelerate AS aggregation, suggesting that some α-helical conformations of AS are on-pathway to amyloid formation. Thus mutations or modifications to AS that favor this particular α-helical state may predispose the protein towards aggregation.
Regulation of membrane-binding
In addition to studies of native AS membrane interaction, single molecule studies have also been useful in probing potentially pathological modifications to AS which alter its interactions with lipid bilayers [52]. Oxidative modification and damage to proteins has been broadly implicated in neurodegenerative diseases. Disruption of normal AS membrane binding is thought to be one cause of aggregation in PD, and there is building evidence that specifically nitration of tyrosine residues in AS may disrupt native membrane interactions [53]. We recently investigated the effects of tyrosine nitration on AS conformational state and membrane binding [52]. With smFRET, we were able to probe multiple regions of AS and localize conformational changes conferred by nitration specifically to the C-terminus of the protein. This allowed us to propose a model whereby changes in the C-terminal region of AS allosterically alter the membrane binding behavior of the N-terminal region of the protein. The C-terminus of AS has been proposed to interact with a variety of cellular binding partners, and our findings suggest that binding to or modifying the C-terminus is a viable mechanism for modulating interactions with cellular membranes.
Oligomers
The interest in pre-fibrillar oligomeric species of AS is motivated both by in vitro measurements of differences in oligomerization rates of wild-type and PD-associated mutants, as well as by evidence that the presence of fibrillar AS aggregates does not necessarily correlate with cell death and disease severity. Dependent upon experimental conditions, oligomers may be stably or transiently populated [54–59], as well as on-pathway or off-pathway to fibrillar aggregate formation [60–63]. The toxic effects of some AS oligomers on cultured cells [9, 19, 64] resulted in the proposal that for AS [64, 65], as well as other amyloid-forming proteins [66–68], soluble oligomeric species may be the toxic species responsible for cellular damage. Furthermore, it has been suggested that the toxic effects are a consequence of interactions between oligomers and cellular membranes [17–19]. AS has been extracted from cells, transgenic mouse brains, and PD human brains in the form of soluble oligomers [69, 70], providing evidence that AS may exist in oligomeric forms in vivo as well. Various cofactors and protein modifications have been found to affect oligomerization of AS, including oxidative modifications to specific residues [71–73], metal ions [74–76], and dopamine [54, 77, 78], as well as other small molecules [63]. Single molecule approaches are exceptionally useful for the study of AS oligomeric species given their rarity and transience. Furthermore, they allow for the study of low concentrations of molecules that kinetically limit the aggregation process, providing access to transient populations present at the beginning stages of aggregation.
Collaborative efforts from the Kostka and Giese labs have relied on a variety of diffusion and brightness-based techniques, including FCS, FIDA, and SIFT (fluorescence correlation spectroscopy, fluorescence intensity distribution analysis, and scanning for intensely fluorescent targets; Box 2), to classify and characterize AS oligomers. Their initial work [79] demonstrated that SIFT useful method for following protein aggregation. In this study, an increase in the SIFT signal was found to parallel that of an ensemble aggregation assay, although the SIFT assay was shown to be significantly more sensitive. Wild-type AS and the A30P mutant were labeled with different colored fluorophores which allowed for monitoring of co-aggregation using dual-color SIFT. While their measurements did provide evidence that aggregates contained both protein variants, the most surprising observation was that specific molar ratios of the two variants resulted in an increase in the number (although not the sizes) of the aggregates formed. This led the authors to hypothesize that AS oligomers formed at these specific protein ratios were more likely to seed aggregate formation or be incorporated into growing fibers.
Box 2. FCS (fluorescence correlation spectroscopy), SIFT(scanning for intensity fluorescence targets), and FIDA(fluorescence intensity distribution analysis).
Although FCS, SIFT, and FIDA are not strictly single molecule approaches, they can be carried out at the single molecule level and they are most sensitive for studies of <100 molecules. As with smFRET (Box 1), each of these techniques uses a highly focused laser to form an observation volume to measure low concentrations of fluorescent molecules. While these methods reliably detect low nanomolar concentrations of fluorescent molecules, a small amount of labeled AS can be doped into a large excess of unlabeled protein, such that only one or two fluorescent molecules are present per aggregate. This makes these techniques experimentally useful over a broad range of protein concentrations. In FCS, the temporal autocorrelation of spontaneous fluctuations in fluorescence intensity are fit with a variety of models to yield quantitative parameters associated with concentration, molecular size, and chemical or photophysical kinetics [89–91]. For studies of AS aggregation, typically the number of fluorescent molecules and their hydrodynamic properties (as measured by diffusion coefficient or diffusion time) are monitored, as these properties are expected to change as monomer protein is consumed by growing aggregates. Closely related to FCS is fluorescence cross-correlation spectroscopy (FCCS). In FCCS, two populations of molecules are labeled with spectrally separable fluorophores and both molecules are directly excited. A cross-correlation signal arises when two different fluorophores co-diffuse, signaling that they have been incorporated into the same molecular species. Thus it can be used as a measure of co-aggregation of the two protein populations. More details on FCS and FCCS can be found in several recent reviews [92–94]. SIFT was developed as a more sensitive measurement of relatively rare aggregated species [95]. This method counts the number of photons detected in a certain time interval and histograms the intensity distribution to examine aggregates which contain multiple labeled molecules and are consequently very bright relative to monomers. Since molecular diffusion is not measured in this analysis, the laser is scanned through the sample to enable detection of slow-diffusing species that might otherwise not encounter a stationary observation volume. Two-color SIFT follows the same principle, but similar to FCCS, two differently labeled molecules are used such that co-aggregation can be monitored. Lastly, FIDA also utilizes photon counting over defined time intervals and measures molecular size based on a brightness analysis [96]. In FIDA, however, an expected brightness distribution for variously sized molecular species is calculated to account for the fact that illumination in an observation volume is not uniform. For example an intrinsically bright species at the edge of the observation volume will not yield as many photons as a less bright species diffusing through the center of the observation volume. By accounting for this, a more accurate determination of the molecule size distribution can be obtained, making FIDA useful for measuring the relative population of various sized aggregates within a sample. In this review, we discuss how these approaches have been used to characterize AS aggregation in real time as well as stable oligomeric forms of AS.
AS aggregation has also been followed in real time using FCS, both in the presence [80] and absence [60] of aggregation inducers. In an initial study, Engelborghs and colleagues simply measured an increase in diffusion time to follow the aggregation of AS and found that aggregation was accelerated upon the addition of an FK506 binding protein (FKBP). By labeling AS and the FKBP with differently colored fluorophores, they showed that FKBP was not incorporated into the AS fibers. This led the authors to suggest that FKBP, which has peptidyl prolyl isomerase activity, impacted AS aggregation by facilitating cis to trans isomerization of C-terminal prolines, allowing population of aggregation-prone conformations. In a later study from the same group, the authors used a more sophisticated approach to characterizing aggregation by plotting distributions of measured diffusion coefficients as a function of time. Over the course of hours, the diffusion coefficient of the measured species shifted from an initial narrow distribution of quickly diffusing species, characteristic of monomer protein, through a transient population of small oligomers which were gradually replaced by large oligomers, defined by a broad distribution of more slowly diffusing species. Moreover, it was demonstrated the aggregation kinetics could be monitored by following the loss of the fast-diffusing monomer species. This assay showed that loss of monomer occurred during the aggregation lag-phase, as measured by turbidity, suggesting that oligomeric species formed during this phase and implicating these species as on-pathway intermediates to amyloid formation. Moreover, ensemble intramolecular FRET showed that conformational changes occurred upon oligomerization which proceeded the conversion to β-sheet fibrillar aggregates. These species are of interest as they represent a population critical for amyloid formation that are not experimentally accessible by most conventional means given their low abundance and transience.
While FKBP appears to alter AS aggregation through transient interactions, a number of other proteins have been found to either accelerate or inhibit AS aggregation by more direct mechanisms. One example of this are the matrix metalloproteases; these proteases can cleave AS at multiple different locations in it sequence and there is also evidence of matrix metalloprotease (MMP) de-regulation in PD and other neurodegenerative diseases. FCCS and SIFT were used to characterize the effect of MMP cleavage on AS aggregation [81]. Intermediate concentrations of certain MMPs were found to accelerate AS aggregation by cleaving the protein between the NAC region and the C-terminus. Removing this highly charged portion of the protein decreases intermolecular electrostatic repulsion and has previously been shown to accelerate AS aggregation.
More recent efforts from the Giese and Kostka labs focused primarily on the study of stable oligomers of AS [82]. They developed three protocols for oligomer formation that differed by the absence or presence of FeCl3, the absence (Type A) or presence (Type B) of stirring during incubation, and ultracentrifugation concentration prior to analysis (Type C). By FIDA, while no systematic differences caused by the addition of FeCl3 were observed, stirring was found to cause a greater fraction of the protein to convert to small oligomers. Interestingly, only Type A oligomers were found to be capable of causing calcium influx and cell death in cultured SH-SY5Y cells. By contrast, Types B & C were both taken up by the cells, whereupon they seeded aggregation of endogenous AS. This work provided direct evidence that oligomeric species with physical properties differentiable by fluorescence methods also had different cellular phenotypes. Correlation of toxicity with the structural features measured by FIDA is an important step towards understanding the underlying mechanism for the differential effects of specific oligomer species on cells.
In a related study [76], the authors used SIFT and FCS to study oligomers formed in the presence of EtOH, which has been shown to accelerate AS aggregation. Oligomer produced at EtOH concentrations between 5 and 20% differed significantly from those formed at higher EtOH concentrations. Namely, the low EtOH oligomers (intermediate I): (1) formed reversibly, such that they could be dissociated into monomers upon dilution, (2) could be blocked by the presence of detergent or BSA, and (3) were on-pathway to amyloid fiber formation in that they were capable of seeding AS aggregation. When Fe3+ was added along with the low concentrations of EtOH, much larger, detergent-resistant oligomers were formed (intermediate II), that otherwise shared the properties listed above of the intermediate I oligomers. However, only the intermediate II oligomers were capable of causing electrical conductivity changes to planar bilayers, consistent with permanently open pore-like species bound to the bilayer. Conductivity could be blocked using antibodies specific for AS monomers or oligomers, suggesting pore-like behavior derived from specific AS species and not simply generic effects from oligomers binding the membrane surface or other artifacts. These findings lead the authors to suggest that the intermediate II oligomers are formed from the intermediate I oligomers and both are potentially relevant model drug targets for developing therapeutic approaches for PD.
In that same study, the authors showed that a small molecule that inhibits oligomer formation as seen by SIFT also inhibited toxic effects of AS on PC12 cells. They expanded upon this theme in subsequent work where they focused on small molecules that were N′-benzylidene-benzohydrazide (NBB) derivatives [83] or natural products [84]. By SIFT, NBB derivatives were found to inhibit both intermediate I and intermediate II oligomer formation. Cell models of aggregation also showed a decrease in higher MW species in the presence of NBB compounds and a decrease in cell toxicity. In the natural products study, the authors observed that molecules, such as trihydroxy-polyphenols and black tea extract, which were most effective at preventing aggregation, were also most effective at disaggregating pre-formed oligomers. Overall, the compound potency was found to be correlated with the number of vicinal hydroxyl groups on a single phenyl ring, leading the authors to suggest that such a structure could serve as a general scaffold for drug development. Given that fluorescence methods are compatible with high throughput drug screening, these studies also illustrate the potential of single molecule fluorescence as a novel, potentially powerful means for drug discovery.
Conclusions
Over the past almost twenty years, experiments using single molecule fluorescence methods have evolved from ‘proof-of-principle’ demonstrations to those which provide valuable insight into biological molecules and complexes. This evolution is very well illustrated by the studies of AS described here. Going forward, single molecule fluorescence is a natural complement to computational studies that have the ability to provide high resolution structural and dynamic information about AS. In that vein, we have recently developed a computational method whereby distances derived from smFRET measurements of AS were used as constraints to guide Monte Carlo simulations[85]. These simulations were able to accurately determine physical features of AS derived from conditions favoring aggregation. In the future, this combination of single molecule experimental and computational approaches could prove a versatile tool for probing interactions between AS and small molecules and thus be a novel approach for identifying potential drugs for the treatment of PD.
Abbreviations
- AS
α-Synuclein
- PD
Parkinson’s disease
- NAC
non-amyloid beta component
- FRET
Förster resonance energy transfer
- SDS
sodium dodceyl sulfate
- CMC
critical micelle concentration
- CD
circular dichroism
- LUV
large unilamellar vesicle
- FCS
fluorescence correlation spectroscopy
- FIDA
fluorescence intensity distribution analysis
- SIFT
scanning for intensely fluorescent targets
- FCCS
fluorescence cross-correlation spectroscopy
- FKBP
FK506 binding protein
- MMP
matrix metalloprotease
- NBB
N′-benzylidene-benzohydrazide
- ETeff
energy transfer efficiency
References
- 1.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55 (2):164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 2.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18 (2):106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 3.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276 (5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 4.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302 (5646):841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 5.Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477 (7362):107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, Ju S, Cookson MR, Asturias FJ, Agar JN, Webb BN, Kang C, Ringe D, Petsko GA, Pochapsky TC, Hoang QQ. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci USA. 2011;108 (43):17797–17802. doi: 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307 (4):1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 8.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu KN, Xu KX, Strathearn KE, Liu F, Cao SS, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, LaBaer J, Rochet JC, Bonini NM, Lindquist S. alpha-synuclein blocks ER- Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313 (5785):324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, Hyman BT, McLean PJ. Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS One. 2008;3 (4):e1867. doi: 10.1371/journal.pone.0001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20 (9):3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahle PJ, Neumann M, Ozmen L, Haass C. Physiology and pathophysiology of alpha-synuclein. Cell culture and transgenic animal models based on a Parkinson’s disease-associated protein. Ann N Y Acad Sci. 2000;920:33–41. doi: 10.1111/j.1749-6632.2000.tb06902.x. [DOI] [PubMed] [Google Scholar]
- 12.Gitler AD, Bevis BJ, Shorter J, Strathearn KE, Hamamichi S, Su LJ, Caldwell KA, Caldwell GA, Rochet JC, McCaffery JM, Barlowe C, Lindquist S. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci USA. 2008;105 (1):145–150. doi: 10.1073/pnas.0710685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased Expression of alpha-Synuclein Reduces Neurotransmitter Release by Inhibiting Synaptic Vesicle Reclustering after Endocytosis. Neuron. 2010;65 (1):66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thayanidhi N, Helm JR, Nycz DC, Bentley M, Liang YJ, Hay JC. alpha-Synuclein Delays Endoplasmic Reticulum (ER)-to-Golgi Transport in Mammalian Cells by Antagonizing ER/Golgi SNAREs. Mol Biol Cell. 2010;21 (11):1850–1863. doi: 10.1091/mbc.E09-09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jensen PH, Nielsen MS, Jakes R, Dotti CG, Goedert M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J Biol Chem. 1998;273 (41):26292–26294. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- 16.Bodner CR, Maltsev AS, Dobson CM, Bax A. Differential phospholipid binding of alpha-synuclein variants implicated in Parkinson’s disease revealed by solution NMR spectroscopy. Biochemistry. 2010;49 (5):862–871. doi: 10.1021/bi901723p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8 (2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 18.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300 (5618):486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 19.Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT., Jr Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol. 2002;322 (5):1089–1102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- 20.Giehm L, Svergun DI, Otzen DE, Vestergaard B. Low-resolution structure of a vesicle disrupting -synuclein oligomer that accumulates during fibrillation. Proc Natl Acad Sci USA. 2011:3246–3251. doi: 10.1073/pnas.1013225108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Rooijen BD, Claessens MMAE, Subramaniam V. Membrane Permeabilization by Oligomeric -Synuclein: In Search of the Mechanism. PLoS One. 2010;5:e14292. doi: 10.1371/journal.pone.0014292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu J, Lyubchenko YL. Early stages for Parkinson’s development: alpha-synuclein misfolding and aggregation. J Neuroimmune Pharmacol. 2009;4 (1):10–16. doi: 10.1007/s11481-008-9115-5. [DOI] [PubMed] [Google Scholar]
- 23.Sandal M, Valle F, Tessari I, Mammi S, Bergantino E, Musiani F, Brucale M, Bubacco L, Samori B. Conformational equilibria in monomeric alpha-synuclein at the single-molecule level. PLoS Biol. 2008;6 (1):e6. doi: 10.1371/journal.pbio.0060006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Losasso V, Pietropaolo A, Zannoni C, Gustincich S, Carloni P. Structural role of compensatory amino acid replacements in the alpha-synuclein protein. Biochemistry. 2011;50 (32):6994–7001. doi: 10.1021/bi2007564. [DOI] [PubMed] [Google Scholar]
- 25.Tsigelny IF, Sharikov Y, Miller MA, Masliah E. Mechanism of alpha-synuclein oligomerization and membrane interaction: theoretical approach to unstructured proteins studies. Nanomedicine. 2008;4 (4):350–357. doi: 10.1016/j.nano.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoon J, Jang S, Lee K, Shin S. Simulation studies on the stabilities of aggregates formed by fibril-forming segments of alpha-Synuclein. J Biomol Struct Dyn. 2009;27 (3):259–270. doi: 10.1080/07391102.2009.10507314. [DOI] [PubMed] [Google Scholar]
- 27.Ferreon AC, Gambin Y, Lemke EA, Deniz AA. Interplay of alpha-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc Natl Acad Sci USA. 2009;106 (14):5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferreon AC, Moran CR, Ferreon JC, Deniz AA. Alteration of the alpha-synuclein folding landscape by a mutation related to Parkinson’s disease. Angew Chem. 2010;49 (20):3469–3472. doi: 10.1002/anie.201000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gambin Y, VanDelinder V, Ferreon AC, Lemke EA, Groisman A, Deniz AA. Visualizing a one-way protein encounter complex by ultrafast single-molecule mixing. Nat Methods. 2011;8 (3):239–241. doi: 10.1038/nmeth.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vandelinder V, Ferreon AC, Gambin Y, Deniz AA, Groisman A. High-resolution temperature-concentration diagram of alpha-synuclein conformation obtained from a single Forster resonance energy transfer image in a microfluidic device. Anal Chem. 2009;81 (16):6929–6935. doi: 10.1021/ac901008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferreon AC, Deniz AA. Alpha-synuclein multistate folding thermodynamics: implications for protein misfolding and aggregation. Biochemistry. 2007;46 (15):4499–4509. doi: 10.1021/bi602461y. [DOI] [PubMed] [Google Scholar]
- 32.Trexler AJ, Rhoades E. Single molecule characterization of alpha-synuclein in aggregation-prone states. Biophys J. 2010;99 (9):3048–3055. doi: 10.1016/j.bpj.2010.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sherman E, Haran G. Coil-globule transition in the denatured state of a small protein. Proc Natl Acad Sci USA. 2006;103 (31):11539–11543. doi: 10.1073/pnas.0601395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273 (16):9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 35.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35 (43):13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 36.Ulmer TS, Bax A, Cole NB, Nussbaum RL. Structure and dynamics of micelle-bound human alpha-synuclein. J Biol Chem. 2005;280 (10):9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 37.Bussell R, Jr, Eliezer D. A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins. J Mol Biol. 2003;329 (4):763–778. doi: 10.1016/s0022-2836(03)00520-5. [DOI] [PubMed] [Google Scholar]
- 38.Georgieva ER, Ramlall TF, Borbat PP, Freed JH, Eliezer D. Membrane-bound alpha-synuclein forms an extended helix: long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J Am Chem Soc. 2008;130 (39):12856–12857. doi: 10.1021/ja804517m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jao CC, Der-Sarkissian A, Chen J, Langen R. Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc Natl Acad Sci USA. 2004;101 (22):8331–8336. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc Natl Acad Sci USA. 2008;105 (50):19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borbat P, Ramlall TF, Freed JH, Eliezer D. Inter-helix distances in lysophospholipid micelle-bound alpha-synuclein from pulsed ESR measurements. J Am Chem Soc. 2006;128 (31):10004–10005. doi: 10.1021/ja063122l. [DOI] [PubMed] [Google Scholar]
- 42.Chandra S, Chen X, Rizo J, Jahn R, Sudhof TC. A broken alpha -helix in folded alpha -Synuclein. J Biol Chem. 2003;278 (17):15313–15318. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
- 43.Drescher M, van Rooijen BD, Veldhuis G, Subramaniam V, Huber M. A stable lipid-induced aggregate of alpha-synuclein. J Am Chem Soc. 132(12):4080–4082. doi: 10.1021/ja909247j. [DOI] [PubMed] [Google Scholar]
- 44.Veldhuis G, Segers-Nolten I, Ferlemann E, Subramaniam V. Single-molecule FRET reveals structural heterogeneity of SDS-bound alpha-synuclein. ChemBioChem. 2009;10 (3):436–439. doi: 10.1002/cbic.200800644. [DOI] [PubMed] [Google Scholar]
- 45.Trexler AJ, Rhoades E. Alpha-synuclein binds large unilamellar vesicles as an extended helix. Biochemistry. 2009;48 (11):2304–2306. doi: 10.1021/bi900114z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nath A, Trexler AJ, Koo P, Miranker AD, Atkins WM, Rhoades E. Single-molecule fluorescence spectroscopy using phospholipid bilayer nanodiscs. Methods Enzymol. 2010;472:89–117. doi: 10.1016/S0076-6879(10)72014-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giehm L, Oliveira CL, Christiansen G, Pedersen JS, Otzen DE. SDS-induced fibrillation of alpha-synuclein: an alternative fibrillation pathway. J Mol Biol. 2010;401 (1):115–133. doi: 10.1016/j.jmb.2010.05.060. [DOI] [PubMed] [Google Scholar]
- 48.Lee HJ, Choi C, Lee SJ. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem. 2002;277 (1):671–678. doi: 10.1074/jbc.M107045200. [DOI] [PubMed] [Google Scholar]
- 49.Zhu M, Fink AL. Lipid binding inhibits alpha-synuclein fibril formation. J Biol Chem. 2003;278 (19):16873–16877. doi: 10.1074/jbc.M210136200. [DOI] [PubMed] [Google Scholar]
- 50.Zhu M, Li J, Fink AL. The association of alpha-synuclein with membranes affects bilayer structure, stability, and fibril formation. J Biol Chem. 2003;278 (41):40186–40197. doi: 10.1074/jbc.M305326200. [DOI] [PubMed] [Google Scholar]
- 51.Comellas G, Lemkau LR, Zhou DH, George JM, Rienstra CM. Structural intermediates during alpha-synuclein fibrillogenesis on phospholipid vesicles. J Am Chem Soc. 2012;134 (11):5090–5099. doi: 10.1021/ja209019s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sevcsik E, Trexler AJ, Dunn JM, Rhoades E. Allostery in a disordered protein: oxidative modifications to alpha-synuclein act distally to regulate membrane binding. J Am Chem Soc. 2011;133 (18):7152–7158. doi: 10.1021/ja2009554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290 (5493):985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 54.Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294 (5545):1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 55.Kaylor J, Bodner N, Edridge S, Yamin G, Hong DP, Fink AL. Characterization of oligomeric intermediates in alpha-synuclein fibrillation: FRET studies of Y125W/Y133F/Y136F alpha-synuclein. J Mol Biol. 2005;353 (2):357–372. doi: 10.1016/j.jmb.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 56.Volles MJ, Lee SJ, Rochet JC, Shtilerman MD, Ding TT, Kessler JC, Lansbury PT., Jr Vesicle permeabilization by protofibrillar alpha-synuclein: implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry. 2001;40 (26):7812–7819. doi: 10.1021/bi0102398. [DOI] [PubMed] [Google Scholar]
- 57.Zakharov SD, Hulleman JD, Dutseva EA, Antonenko YN, Rochet JC, Cramer WA. Helical alpha-synuclein forms highly conductive ion channels. Biochemistry. 2007;46 (50):14369–14379. doi: 10.1021/bi701275p. [DOI] [PubMed] [Google Scholar]
- 58.Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283 (44):29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kayed R, Pensalfini A, Margol L, Sokolov Y, Sarsoza F, Head E, Hall J, Glabe C. Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J Biol Chem. 2009;284 (7):4230–4237. doi: 10.1074/jbc.M808591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nath S, Meuvis J, Hendrix J, Carl SA, Engelborghs Y. Early aggregation steps in alpha-synuclein as measured by FCS and FRET: evidence for a contagious conformational change. Biophys J. 2010;98 (7):1302–1311. doi: 10.1016/j.bpj.2009.12.4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bisaglia M, Mammi S, Bubacco L. Structural insights on physiological functions and pathological effects of alpha-synuclein. FASEB J. 2009;23 (2):329–340. doi: 10.1096/fj.08-119784. [DOI] [PubMed] [Google Scholar]
- 62.Fink AL. The aggregation and fibrillation of alpha-synuclein. Acc Chem Res. 2006;39 (9):628–634. doi: 10.1021/ar050073t. [DOI] [PubMed] [Google Scholar]
- 63.Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, Wanker EE. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc Natl Acad Sci USA. 2010;107 (17):7710–7715. doi: 10.1073/pnas.0910723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Conway KA, Lee SJ, Rochet JC, Ding TT, Harper JD, Williamson RE, Lansbury PT., Jr Accelerated oligomerization by Parkinson’s disease linked alpha-synuclein mutants. Ann N Y Acad Sci. 2000;920:42–45. doi: 10.1111/j.1749-6632.2000.tb06903.x. [DOI] [PubMed] [Google Scholar]
- 65.Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004;11 (3):213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- 66.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416 (6880):507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 67.Kagan BL, Hirakura Y, Azimov R, Azimova R, Lin MC. The channel hypothesis of Alzheimer’s disease: current status. Peptides. 2002;23 (7):1311–1315. doi: 10.1016/s0196-9781(02)00067-0. [DOI] [PubMed] [Google Scholar]
- 68.Ono K, Condron MM, Teplow DB. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc Natl Acad Sci USA. 2009;106 (35):14745–14750. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sharon R, Bar-Joseph I, Frosch MP, Walsh DM, Hamilton JA, Selkoe DJ. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron. 2003;37 (4):583–595. doi: 10.1016/s0896-6273(03)00024-2. [DOI] [PubMed] [Google Scholar]
- 70.Sharon R, Goldberg MS, Bar-Josef I, Betensky RA, Shen J, Selkoe DJ. alpha-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc Natl Acad Sci USA. 2001;98 (16):9110–9115. doi: 10.1073/pnas.171300598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamin G, Uversky VN, Fink AL. Nitration inhibits fibrillation of human alpha-synuclein in vitro by formation of soluble oligomers. FEBS Lett. 2003;542 (1–3):147–152. doi: 10.1016/s0014-5793(03)00367-3. [DOI] [PubMed] [Google Scholar]
- 72.Uversky VN, Yamin G, Munishkina LA, Karymov MA, Millett IS, Doniach S, Lyubchenko YL, Fink AL. Effects of nitration on the structure and aggregation of alpha-synuclein. Brain Res Mol Brain Res. 2005;134 (1):84–102. doi: 10.1016/j.molbrainres.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 73.Uversky VN, Yamin G, Souillac PO, Goers J, Glaser CB, Fink AL. Methionine oxidation inhibits fibrillation of human alpha-synuclein in vitro. FEBS Lett. 2002;517 (1–3):239–244. doi: 10.1016/s0014-5793(02)02638-8. [DOI] [PubMed] [Google Scholar]
- 74.Cole NB, Murphy DD, Lebowitz J, Di Noto L, Levine RL, Nussbaum RL. Metal-catalyzed oxidation of alpha-synuclein: helping to define the relationship between oligomers, protofibrils, and filaments. J Biol Chem. 2005;280 (10):9678–9690. doi: 10.1074/jbc.M409946200. [DOI] [PubMed] [Google Scholar]
- 75.Nielsen MS, Vorum H, Lindersson E, Jensen PH. Ca2+ binding to alpha-synuclein regulates ligand binding and oligomerization. J Biol Chem. 2001;276 (25):22680–22684. doi: 10.1074/jbc.M101181200. [DOI] [PubMed] [Google Scholar]
- 76.Kostka M, Hogen T, Danzer KM, Levin J, Habeck M, Wirth A, Wagner R, Glabe CG, Finger S, Heinzelmann U, Garidel P, Duan W, Ross CA, Kretzschmar H, Giese A. Single particle characterization of iron-induced pore-forming alpha-synuclein oligomers. J Biol Chem. 2008;283 (16):10992–11003. doi: 10.1074/jbc.M709634200. [DOI] [PubMed] [Google Scholar]
- 77.Pham CL, Leong SL, Ali FE, Kenche VB, Hill AF, Gras SL, Barnham KJ, Cappai R. Dopamine and the dopamine oxidation product 5,6-dihydroxylindole promote distinct on-pathway and off-pathway aggregation of alpha-synuclein in a pH-dependent manner. J Mol Biol. 2009;387 (3):771–785. doi: 10.1016/j.jmb.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 78.Zhou W, Gallagher A, Hong DP, Long C, Fink AL, Uversky VN. At low concentrations, 3,4-dihydroxyphenylacetic acid (DOPAC) binds non-covalently to alpha-synuclein and prevents its fibrillation. J Mol Biol. 2009;388 (3):597–610. doi: 10.1016/j.jmb.2009.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Giese A, Bader B, Bieschke J, Schaffar G, Odoy S, Kahle PJ, Haass C, Kretzschmar H. Single particle detection and characterization of synuclein co-aggregation. Biochem Biophys Res Commun. 2005;333 (4):1202–1210. doi: 10.1016/j.bbrc.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 80.Gerard M, Debyser Z, Desender L, Kahle PJ, Baert J, Baekelandt V, Engelborghs Y. The aggregation of alpha-synuclein is stimulated by FK506 binding proteins as shown by fluorescence correlation spectroscopy. FASEB J. 2006;20 (3):524–526. doi: 10.1096/fj.05-5126fje. [DOI] [PubMed] [Google Scholar]
- 81.Levin J, Giese A, Boetzel K, Israel L, Hogen T, Nubling G, Kretzschmar H, Lorenzl S. Increased alpha-synuclein aggregation following limited cleavage by certain matrix metalloproteinases. Exp Neurol. 2009;215 (1):201–208. doi: 10.1016/j.expneurol.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 82.Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27 (34):9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hillmer AS, Putcha P, Levin J, Hogen T, Hyman BT, Kretzschmar H, McLean PJ, Giese A. Converse modulation of toxic alpha-synuclein oligomers in living cells by N′-benzylidene-benzohydrazide derivates and ferric iron. Biochem Biophys Res Commun. 2010;391 (1):461–466. doi: 10.1016/j.bbrc.2009.11.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Caruana M, Hogen T, Levin J, Hillmer A, Giese A, Vassallo N. Inhibition and disaggregation of alpha-synuclein oligomers by natural polyphenolic compounds. FEBS Lett. 2011;585 (8):1113–1120. doi: 10.1016/j.febslet.2011.03.046. [DOI] [PubMed] [Google Scholar]
- 85.Nath A, Sammalkorpi MT, DeWitt D, Schreck C, Trexler AJ, Elbaum-Garfinkle S, O’Hern CS, Rhoades E. The Conformational Ensembles of α-Synuclein and Tau: Combining Single-Molecule FRET and Simulations. Biophys J. 2012 doi: 10.1016/j.bpj.2012.09.032. Manuscript under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen H, Rhoades E. Fluorescence characterization of denatured proteins. Curr Opin Struct Biol. 2008;18 (4):516–524. doi: 10.1016/j.sbi.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ferreon AC, Moran CR, Gambin Y, Deniz AA. Single-molecule fluorescence studies of intrinsically disordered proteins. Methods Enzymol. 2010;472:179–204. doi: 10.1016/S0076-6879(10)72010-3. [DOI] [PubMed] [Google Scholar]
- 88.Schuler B, Eaton WA. Protein folding studied by single-molecule FRET. Curr Opin Struct Biol. 2008;18 (1):16–26. doi: 10.1016/j.sbi.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Elson EL, Magde D. Fluorescence Correlation Spectroscopy. 1. Conceptual Basis and Theory. Biopolymers. 1974;13:1–27. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- 90.Magde D, Elson EL, Webb WW. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers. 1974;13 (1):29–61. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- 91.Magde D, Webb WW, Elson E. Thermodynamic Fluctuations in a Reacting System - Measurement by Fluorescence Correlation Spectroscopy. Physical Review Letters. 1972;29:705–708. [Google Scholar]
- 92.Elson EL. Fluorescence correlation spectroscopy: past, present, future. Biophys J. 2011;101 (12):2855–2870. doi: 10.1016/j.bpj.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bacia K, Schwille P. Practical guidelines for dual-color fluorescence cross-correlation spectroscopy. Nat Protoc. 2007;2 (11):2842–2856. doi: 10.1038/nprot.2007.410. [DOI] [PubMed] [Google Scholar]
- 94.Haustein E, Schwille P. Fluorescence correlation spectroscopy: novel variations of an established technique. Annu Rev Biophys Biomol Struct. 2007;36:151–169. doi: 10.1146/annurev.biophys.36.040306.132612. [DOI] [PubMed] [Google Scholar]
- 95.Bieschke J, Giese A, Schulz-Schaeffer W, Zerr I, Poser S, Eigen M, Kretzschmar H. Ultrasensitive detection of pathological prion protein aggregates by dual-color scanning for intensely fluorescent targets. Proc Natl Acad Sci USA. 2000;97 (10):5468–5473. doi: 10.1073/pnas.97.10.5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kask P, Palo K, Ullmann D, Gall K. Fluorescence-intensity distribution analysis and its application in biomolecular detection technology. Proc Natl Acad Sci USA. 1999;96 (24):13756–13761. doi: 10.1073/pnas.96.24.13756. [DOI] [PMC free article] [PubMed] [Google Scholar]